Crystallization-induced switching of the morphology of poly(ethylene oxide)-block-polybutadiene micelles
Adriana M.
Mihut
a,
Arnaud
Chiche†
a,
Markus
Drechsler
b,
Holger
Schmalz
*b,
Emanuela
Di Cola
c,
Georg
Krausch
d and
Matthias
Ballauff
*a
aPhysical Chemistry I, University of Bayreuth, 95440, Bayreuth, Germany. E-mail: Matthias.Ballauff@uni-bayreuth.de
bMacromolecular Chemistry II, University of Bayreuth, 95440, Bayreuth, Germany. E-mail: Holger.Schmalz@uni-bayreuth.de
cEuropean Synchrotron Radiation Facility (ESRF), 38043, Grenoble, France
dJohannes Gutenberg University, 55128, Mainz, Germany
First published on 15th October 2008
Abstract
We studied the morphology of micelles formed by a well-defined poly(1,2-butadiene)-block-poly(ethylene oxide) diblock copolymer (PB-b-PEO). Dissolved in n-heptane at 70 °C, that is, above the melting point of PEO, spherical micelles are formed due to the selectivity of the solvent for the PB-block. If the solutions are cooled down to low temperatures, the liquid PEO-block crystallizes within the cores of the spherical micelles that remain stable. If, however, the solutions are quenched to 30 °C, the spherical micelles aggregate to a novel meander-like structure within several minutes. In its final state, the meander-like super-structure is crystalline, as revealed by time-resolved wide-angle X-ray scattering. The super-structure is shown to result from crystallization-induced aggregation of spherical micelles. Moreover, crystallization leads to well-defined angles between subsequent aggregating units. A quantitative Avrami-type analysis of the crystallization kinetics demonstrates that the formation of the meander-type structure resembles a 2D growth process combined with a breakout crystallization, showing an Avrami-exponent of 2.5. In contrast to this, crystallization at low temperatures resembles a confined crystallization with a low Avrami-exponent of 0.7. All data demonstrate that the morphology of block copolymers having a crystallizable block can be switched by the competition of aggregation and crystallization.
I. Introduction
Amphiphilic block copolymers can assemble to micellar structures that extend over many length scales. Hence, amphiphilic block copolymers can self-assemble in selective solvents for one block to form well-defined micellar structures such as spheres,1,2 cylinders,3 or more complex architectures.4–12 The interest in these types of materials is largely stimulated by their potential in various applications from nanotechnology4,13 to drug-delivery systems.14 It has been shown that specific morphologies can be controlled through the selection of the monomer, the adjustment of the chain length and architecture,5,15–17 temperature,18 and through solution conditions2,7,19–22 (quality of the solvent, pH, and salinity).Control of the self-assembly of block copolymers can also be achieved by crystallization when the insoluble block is able to crystallize. While crystallization of polymers and block copolymers has been intensively studied in good solvents23,24 or in bulk,25–28 less work has been done for crystalline block copolymers in selective solvents for the amorphous block. Lotz, Kovacs and co-workers23,24 were the first to study the morphology of polystyrene-block-poly(ethylene oxide) block copolymers crystallized from solution. Square platelets with crystalline regions having the same structures as PEO homopolymers were observed. Gast and co-workers29,30 obtained large stable crystalline lamellae of polystyrene-block-poly(ethylene oxide) (PS-b-PEO) in cyclopentane. They have also shown that the PEO crystallization and the resulting shaped lamellae can be switched off by the addition of a small amount of water that swells the PEO block, and results in spherical micelles with an amorphous core. Richter et al.31 and Gast et al.32 studied the chain folding in micelles of polyethylene-block-poly(ethylene-alt-propylene) (PE-b-PEP) block copolymers in n-decane. Cheng et al.33,34 studied the interaction changes of the polystyrene (PS) chains on the poly(ethylene oxide) (PEO) or poly(L-lactic acid) (PLLA) platelet basal surface. Xuet al.35,36 reported on a strong effect of molecular weight on the micellar structure for poly(ethylene oxide)-block-poly(butylene oxide) (PEO-b-PBO, 55 wt% PEO) diblock copolymers in solution and blends with low-molecular-weight PBO. High-molecular-weight PEO-b-PBO diblock copolymers formed spherical micelles, whereas samples with identical composition but lower molecular weight formed platelet-like structures.
Recently, Winnik, Manners and co-workers37–41 have reported that block copolymers containing crystalline blocks can form cylindrical micelles. They showed that crystallization is the main driving force behind the cylindrical micelles formation of poly (ferrocenyldimethylsilane)-block-polydimethylsiloxane (PFDMS-b-PDMS). In addition, a reversible transition was observed from cylindrical micelles to hollow nanotubes. This work has demonstrated that the interplay between aggregation and crystallization can lead to a time-dependent reorganization in micellar systems.
Here, we study the size and shape of semicrystalline block copolymer micelles using a well-defined poly(1,2-butadiene)-block-poly(ethylene oxide) diblock copolymer. We demonstrate that the thermally controlled crystallization of the confined poly(ethylene oxide) (PEO) core in a selective solvent leads to different but well-defined morphologies, that can be explained by concomitant aggregation and crystallization.
II. Experimental
A. Materials and methods
The poly(1,2-butadiene)-block-poly(ethylene oxide) diblock copolymer was synthesized via sequential anionic polymerization of butadiene and ethylene oxide in tetrahydrofuran using the phosphazene base t-BuP4, as described elsewhere.42,43The composition of the diblock copolymers is B52EO485.6 (subscripts denote the mass fraction in percent and the superscript gives the overall number average molecular weight Mn in kg mol−1). The molecular weights of the B and EO blocks are 2900 and 2700 g mol−1, respectively; the polydispersity index of the diblock copolymer is 1.02. The amount of 1,2-units within the polybutadiene block is 92 mol%.
The polymer was dried for 2 days at 70 °C under vacuum, until residual traces of water were removed. The samples were prepared from 1 wt% and 20 wt% n-heptane solutions at different crystallization temperatures for the PEO block.
B. Cryogenic transmission electron microscopy (cryo-TEM)
Samples for cryo-TEM were prepared by adding a 2 µl droplet of a 1 wt% solution of B52EO485.6 in n-heptane on a lacey carbon-coated copper grid, where most of the liquid was removed with blotting paper, leaving a thin film stretched over the lace. The specimens were prepared by vitrification in liquid nitrogen and then cooled to approximately 77 K in a temperature-controlled freezing unit (Zeiss Cryobox, Zeiss NTS GmbH, Oberkochen, Germany). After freezing, the specimen was placed into a cryo-transfer holder (CT 3500, Gatan, München, Germany) and transferred to a Zeiss 922 OMEGA EFTEM (Zeiss NTS GmbH, Oberkochen, Germany). Due to the fact that the contrast provided by the electron-density differences between the block copolymer and the solvent is very low, the embedding n-heptane was heated in situ at a temperature of 163 K for 15 min and sublimated during this time.44 After sublimation of n-heptane, the objects could be identified on the supporting “lacey” carbon membrane. Subsequently, the sample was cooled to a temperature of 97 K for image recording. The TEM was operated at an acceleration voltage of 200 kV. A CCD camera system (Ultrascan 1000, Gatan) was used for image recording and the images were processed with a digital image processing system (Gatan Digital Micrograph 3.15 for GMS 1.5).C. Transmission electron microscopy (TEM)
Samples were prepared by placing a drop of the B52EO485.6 solution (0.1 wt% in n-heptane) on a carbon-coated copper grid. After a few seconds, the excess solution was removed by blotting with filter paper. Subsequently, bright-field TEM was performed on a Zeiss CEM 902 operating at 80 kV. Staining was performed with OsO4 vapor for 60 min. OsO4 is known to selectively stain PB; i.e., PB domains are expected to appear darker compared to PEO domains, which enables us to distinguish between the two polymers.D. Dynamic light scattering (DLS)
Dynamic light scattering was carried out on an ALV DLS/SLS-SP 5022F compact goniometer system with an ALV 5000/E correlator and a He-Ne laser (λ = 632.8 nm). All measurements were performed on a 0.1 wt% solution of B52EO485.6 in n-heptane at a scattering angle of 90 °. A CONTIN analysis was taken for the measured intensity correlation function. For the temperature-dependent measurements the toluene bath of the instrument was thermostatted and the target temperatures were equilibrated at least 20 min before the experiments. A second cumulant analysis was used in order to investigate the morphology formation at different temperatures.E. Wide-angle X-ray scattering (WAXS)
Time-resolved wide-angle X-ray scattering (WAXS) experiments were performed using the ID2 beamline at the European Synchrotron Radiation Facilities (ESRF, Grenoble, France). The operating wavelength of the X-ray was λ = 0.1 nm. The intensity is represented as a function of the scattering vector q = (4π sin θ)/λ, 2θ being the scattering angle. The beam size was 0.3 × 0.3 µm and the sample–detector distance was 2 m. The detector was a fibre optically coupled FReLoN (Fast-Readout, Low Noise) CCD with a readout rate of 5 frames per second. Prior to data analysis, background scattering was subtracted from the data and corrections were made for spatial distortion, the detector efficiency and beamstop. Typical data acquisition time was 1 s per frame separated by a waiting-time of 2.75 s. The temperature-dependent WAXS experiments were performed using a Linkam THMS600 temperature-controller system as a sample holder. The hot stage was equipped with a liquid-nitrogen cooling accessory and enabled fast temperature ramps with 80 °C min−1 (nominal stability of 1 °C). The 20 wt% solutions of the diblock copolymer in n-heptane were first heated above the melting temperature at 70 °C and quenched to the desired crystallization temperature (−30 and 30 °C) and held until isothermal crystallization was completed.III. Results and discussion
A. Morphologies of B52EO485.6 in n-heptane
To study the effect of crystallization on self-assembly with a given block ratio of a block copolymer, we prepared samples having two distinct thermal histories. The dried polymer was dissolved in n-heptane at 70 °C, first. The hot n-heptane solution (70 °C) was then quenched to different crystallization temperatures: The first solution was quenched into liquid nitrogen and will correspond to pathway A. The second solution which was quenched to 30 °C corresponds to pathway B. All solutions were equilibrated at room temperature for at least 1 day after quenching. The solution corresponding to pathway A was transparent after quenching. Solutions corresponding to pathway B were turbid. The whole process is reversible as soon as the solutions are heated up again to 70 °C.Pathway A. Fig. 1 shows the obtained morphologies by cryogenic transmission electron microscopy (cryo-TEM), where the samples have been prepared by in situfreeze-drying. Spherical core–corona micelles with crystalline PEO domains were observed. The darker cores correspond to the PEO block (higher electron density) embedded in the PB matrix (lower electron density). DLS at 70 °C revealed the presence of spherical micelles with a molten PEO core, as will be discussed later (see Fig. 3). Thus, upon quenching from 70 °C into liquid nitrogen the spherical morphology is retained. The spherical objects appeared to be mono-disperse in size with a mean core radius of 13 nm. Fig. 1(b) shows that the spherical micelles are regularly packed forming a hexagonal structure over the whole film. The packing is induced during the freeze-drying sample-preparation process. This points to a rather narrow size distribution of the micelles. The size estimated from Fig. 1 corresponds well to the hydrodynamic radius of 18 nm measured by DLS of micellar solution after quenching in liquid nitrogen. The micellar solutions are stable over months. No aggregation was observed even after a few months by DLS at room temperature.
 | ||
| Fig. 1 Cryo-TEM micrographs of structures formed by B52EO485.6 in n-heptane: (a), (b) spherical crystalline micelles obtained from pathway A and (e), (f) meander-like structures obtained from pathway B. Images (c)–(f) represent the evolution of meander-like structure over time upon cooling at 30 °C after: (c) 1 min, (d) 3 min (e, f) 24 h. The inset picture from Fig. 1(a) represents the solution corresponding to pathway A, and the picture from Fig. 1(f) corresponds to pathway B. | ||
Pathway B . Fig. 1(e, f) show the morphologies obtained through pathway B, namely, by quenching the hot solution from 70 to 30 °C. Pathway B leads to the formation of an novel meander-like morphology. The length of the objects is in the range of 100 nm to 1 µm, enabling a bridging over the lacey carbon grids. The structure exhibits lateral growth and forms simultaneously two or four new branches of equivalent width. By tilting the samples during the image-recording process the width of these objects has been shown to vary (from a minimum of 20 nm to a maximum of 35 nm). Tilted objects can be visualized in Fig. 1(e) as the darker structures. This excludes a cylindrical morphology. The structure of the core resembles more of a ribbon-like or rectangular prismatic structure having ellipsoidal endings.
Selective staining of the PB domains with OsO4 was used to obtain a closer insight into the morphology of meander-like structures. The sample was first drop-coated onto a carbon-coated copper grid (0.1 wt% solution in n-heptane), followed by staining with OsO4 vapor for 60 min. The crystalline PEO domains in the core appear bright because of the preferential staining of the PB blocks. Well-separated darker areas (higher scattering contrast), corresponding to stained PB domains, can be detected along the PEO crystalline domains that formed the meander-like structure (Fig. 2). The average width of the crystalline PEO domains was 17 nm surrounded by an amorphous PB layer of 16 nm.
 | ||
| Fig. 2 TEM micrograph of B52EO485.6 in n-heptane: meander-like structures obtained from pathway B. The sample was prepared by dip-coating a 0.1 wt% solution onto a carbon-coated copper grid, followed by staining with OsO4 vapor for 60 min. | ||
Fig. 1(c) and (d) display images of the B52EO485.6 structures evolved upon cooling after different times. After 1 min the amorphous micelles, which are present in the hot solution, aggregate upon cooling to 30 °C. The radius of these aggregates is ca. 26 nm, which is twice the original radius of the micelles in hot n-heptane. The micrograph (Fig. 1(d)) taken after 3 min demonstrates clearly the aggregation of the micelles to larger objects. Moreover, the round endings of the meanders point to an aggregation of spherical micelles.
The apparent hydrodynamic radius of the meander-like structure obtained from pathway B was determined by DLS. The radius was determined to be 150–200 nm. Moreover, the evolution of the meander-like structure over time upon cooling from 70 to 30 °C could be monitored by DLS as well. Fig. 3 shows the dependence of the hydrodynamic radius on time at 70 °C, and after quenching from 70 to 30 °C for a 0.1 wt% B52EO485.6n-heptane solution. At 70 °C the DLS measurements revealed that B52EO485.6 self-assembles in n-heptane, a selective solvent for the PB block, into a stable micellar structure with an average hydrodynamic radius of 12 nm (see Fig. 3). Upon quenching to 30 °C (pathway B) a marked increase of the intensity is observed after an induction period of t ∼ 400 s. This is accompanied by an increase of the Rh from 12 nm to over 140 nm, indicating rearrangement of micelles and growth into larger structures. This rearrangement is relatively fast, occurring in only a few minutes and the resulting structures are stable for months.
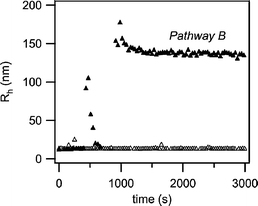 | ||
| Fig. 3 Time-resolved hydrodynamic radius of B52EO485.6 in n-heptane solution measured by DLS at 70 °C (△) and after quenching at 30 °C (▲), pathway B. The quenching at 30 °C is followed by an aggregation process into a larger crystalline structure. | ||
B. Time-dependent WAXS: crystallization kinetics
Fig. 4 shows the WAXS profiles as a function of time collected during isothermal crystallization experiments performed at T = −30 °C (a) and T = 30 °C (b) on a 20 wt% solution. The samples have been previously equilibrated at 70 °C, i.e. above the melting temperature of PEO (60 °C), for 15 min before quenching to the crystallization temperature. | ||
| Fig. 4 The evolution of the WAXS patterns of 20 wt% B52EO485.6 in n-heptane quenched at a rate of 80 °C min−1 from 70 to −30 °C (a) and to 30 °C (b), forming micelles and meander-like structure, respectively. The figure shows the time-resolved WAXS profiles from the moment the quenching temperatures were reached. | ||
Pathway A. The PEO crystal reflections (monoclinic unit cell; cf.ref. 45–48) appeared immediately after quenching to −30 °C (Fig. 4(a)). The strongest PEO reflections were observed at q = 13.41 and 16.34 nm−1. These peaks are assigned to the crystallographic reflection of the planes indexed by (120), and (032 + 112) of the monoclinic unit cell of the PEO crystals.
Pathway B . The isothermal crystallization at 30 °C is shown in the Fig. 4(b). Comparing with the isothermal crystallization at −30 °C (Fig. 4(a)), the WAXS profiles measured at Tc = 30 °C show only an amorphous halo scattering in the earlier stage, until the first appearance of discernable crystalline peaks at t ∼ 300 s. This is consistent with the observed induction period in DLS (cf.Fig. 3). Between 300 and 600 s the PEO crystals are forming the meander-like structure. The WAXS patterns show the same reflections of the monoclinic unit cell of PEO as observed in Fig. 4(a) for the crystalline micelles. In addition, PEO shows a monoclinic crystal modification in B52EO485.6 bulk samples, too.
C. Mechanism of self-assembly
All data obtained so far demonstrate that the meander-like structure is formed via a crystallization-induced aggregation of spherical micelles upon cooling. Fig. 6 displays the tentative mechanism of the formation of micelles along both pathways. The selective solvent n-heptane leads to formation of micelles having a liquid core at elevated temperatures. In the case of pathway A, supercooling to low temperatures leads to rapid crystallization of the cores. Thus, the structure of the micelles is fixed by crystallization and no further growth can occur.However, if the solution is cooled down to only 30 °C, an induction period was observed by DLS and WAXS, followed by a sudden growth of the meander-like structures. Approximately at this point, crystallization starts and clear WAXS-peaks can be seen. Moreover, the ramifications seen in the later stage point to the fact that the overall shape of the meanders must be related to crystallization. Obviously, crystallization favors well-defined angles between two subsequent micelles. Thus, it seems that growth of the meanders starts around a micellar structure in which nucleation of the crystalline phase has taken place. Further micelles will aggregate and immediately become crystalline upon merging with this primary nucleus. Evidently, this is a rather fast process, as shown by DLS, which may be followed by a depletion of micelles around the rapidly growing core micelle. This may explain the formation of a meander-like structure, which is reminiscent of fractal growth of particles. Similar fractal growth processes have been observed for diblock copolymers in thin films, too.49 Hence, crystallization triggers the aggregation and formation of a stable super-structure. In a later stage, crystallinity within the meanders is increasing.
D. Degree of crystallinity
The degree of crystallinity was determined from the WAXS data where the reflection profiles were separated into the crystalline PEO reflections and an amorphous halo by using Vonk's method.50 The crystallinity of the samples was calculated as the ratio between the diffracted areas under the deconvoluted crystalline peaks over the total diffracted area after subtraction of the continuous background. The shape of the amorphous halo due to the amorphous fraction was estimated from the diffraction pattern of the amorphous samples at temperatures above their melting points. Crystallinity increased to 22% in the case of the spherical micelles and up to 27% for the meander-like structures.Further information can be obtained from an Avrami analysis. The Avrami equation can be expressed as:
| 1 − Xc = exp(−ztn) | (1) |
The information about the early stage crystallization of B52EO485.6 at Tc = −30 °C and Tc = 30 °C are given in Fig. 5. The Avrami exponent of 0.7 shows that a confined crystallization takes place in B52EO485.6 at low temperature (Tc = −30 °C). Here, in each micelle nucleation and crystallization occurs independent from all other spheres. Avrami exponents (n = 0.5) were also found by Lotz and Kovacs52 for block copolymers with a glassy matrix and a crystallizable minority block (PEO-b-PS), or by Shiomi et al.53 for polytetrahydrofuran-block-polystyrene (PTHF-b-PS). The small exponent seems to reflect the constraints imposed by the confinement by a glassy matrix. Thus, Xuet al.54 also observed an Avrami exponent of n = 0.5 for PS-b-PEO-b-PS, which was attributed to confined crystallization. Therefore, we conclude that the Avrami exponent of n = 0.7 found for pathway A is related to confined crystallization where a homogeneous nucleation exclusively takes place within each individual spherical microdomain, i.e., kinetics are determined by nucleation. The Avrami exponent thus reflects the rate at which PEO microdomains nucleate.52 Predominant homogeneous nucleation was also observed for PEO confined in miniemulsion droplets.56
 | ||
| Fig. 5 Avrami plots derived from WAXS data for B52EO485.6 isothermally crystallized at different Tc: (▽) spherical micelles, Tc = −30 °C, (■) meander-like structures, Tc = 30 °C. The dotted line and the full line represent the first degree fits from which the Avrami exponents n are obtained. The crystallization process for the meander-like structures (pathway B) has a 300 s delay compared with the micellar structures after the temperature of crystallization was reached. | ||
 | ||
| Fig. 6 Schematic representation of different morphologies formed by B52EO485.6 in n-heptane driven by crystallization. The morphologies that emerge depend on the thermal history of the two different pathways A and B. Amorphous micelles are formed at 70 °C. After quenching from 70 °C by liquid nitrogen crystalline spherical micelles are formed (pathway A), whereas a quenching at 30 °C (pathway B) results in a branched crystalline morphology denoted as a meander-like structure with squeezed ellipsoidal endings. | ||
The situation changes when the crystals are no longer confined to the microdomains in which they nucleate, i.e., breakout occurs upon crystallization.28,55Fig. 5 shows that a value of n = 2.5 is found for the early stage of the crystallization within the meanders (pathway B) at Tc = 30 °C. This Avrami exponent is consistent with the observed 2D growth of the meander-like structure as revealed by cryo-TEM (Fig. 1(e, f)). Furthermore, comparable Avrami exponents were found for crystallization in bulk under comparable conditions.51
Our results showed that the observed meander-like structures are formed by crystallization-induced aggregation of spherical micelles upon quenching from 70 to 30 °C. The present results can hence be compared qualitatively to recent data obtained by Winnik, Manners and co-workers.41 The Avrami exponent of 2.5 indicates that crystallization and aggregation of spherical micelles occurs side-by-side, rather then step-wise as reported for PEO-b-PBO blends with low-molecular-weight PBO.35Crystallization may hence be used to adjust the morphology of micellar systems.
IV. Conclusion
We have observed the formation of meander-like structures and of spherical crystalline micelles for a PB-b-PEO diblock copolymerviacrystallization upon cooling. As shown in Fig. 6, rapid supercooling leads to crystallization of the liquid PEO-cores and to fixation of the single spherical micelles (pathway A). In case of pathway B, i.e., by cooling down the solution to 30 °C, crystalline meanders are formed. The rapid aggregation process leads to a depletion of the micelles around this growing core leading to the ramificated meander-like structure of the micelles. Hence, crystallization-induced aggregation can lead to a novel type of micellar super-structure.Acknowledgements
The authors acknowledge helpful discussions with Alejandro Müller and Günter Reiter. A.M.M. and A.C. acknowledge the financial support from the European Community's “Marie-Curie Actions” under contract no. MRTN-CT-2004-504052 [POLYFILM]. M.D acknowledge the financial support from the SFB 481 project.References
- A. Harada and K. Kataoka, Science, 1999, 283, 65–67 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- J. S. Pedersen, I. W. Hamley, C. Y. Ryu and T. P. Lodge, Macromolecules, 2000, 33, 542–550 CrossRef CAS.
- S. A. Jenekhe and X. L. Chen, Science, 1999, 283, 372–375 CrossRef CAS.
- Y. Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- Y. Y. Won, A. K. Brannan, H. T. Davis and F. S. Bates, J. Phys. Chem. B, 2002, 106, 3354–3364 CrossRef CAS.
- L. Yang and P. Alexandridis, Curr. Opin. Colloid Interface Sci., 2000, 5, 132–143 CrossRef CAS.
- K. Yu, L. F. Zhang and A. Eisenberg, Langmuir, 1996, 12, 5980–5984 CrossRef CAS.
- Y. S. Yu, L. F. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144–1154 CrossRef CAS.
- M. Svensson, P. Alexandridis and P. Linse, Macromolecules, 1999, 32, 637–645 CrossRef CAS.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- M. Malmsten, Soft Matter, 2006, 2, 760–769 RSC.
- E. Minatti, P. Viville, R. Borsali, M. Schappacher, A. Deffieux and R. Lazzaroni, Macromolecules, 2003, 36, 4125–4133 CrossRef CAS.
- N. Ouarti, P. Viville, R. Lazzaroni, E. Minatti, M. Schappacher, A. Deffieux and R. Borsali, Langmuir, 2005, 21, 1180–1186 CrossRef CAS.
- Y. Zheng, Y. Y. Won, F. S. Bates, H. T. Davis, L. E. Scriven and Y. Talmon, J. Phys. Chem. B, 1999, 103, 10331–10334 CrossRef CAS.
- P. Bhargava, Y. F. Tu, J. X. Zheng, H. M. Xiong, R. P. Quirk and S. Z. D. Cheng, J. Am. Chem. Soc., 2007, 129, 1113–1121 CrossRef CAS.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- A. Choucair and A. Eisenberg, Eur. Phys. J. E, 2003, 10, 37–44 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- L. F. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CAS.
- B. Lotz and A. J. Kovacs, Kolloid Z. Z. Polym., 1966, 209, 97 CAS.
- B. Lotz, A. J. Kovacs, G. A. Bassett and A. Keller, Kolloid Z. Z. Polym., 1966, 209, 115 CAS.
- A. J. Ryan, I. W. Hamley, W. Bras and F. S. Bates, Macromolecules, 1995, 28, 3860–3868 CrossRef CAS.
- Y. L. Loo, R. A. Register and A. J. Ryan, Phys. Rev. Lett., 2000, 84, 4120–4123 CrossRef CAS.
- G. Reiter, G. Castelein, J. U. Sommer, A. Rottele and T. Thurn-Albrecht, Phys. Rev. Lett., 2001, 87, 226101-1–226101-4.
- A. J. Müller, V. Balsamo, M. L. Arnal, T. Jakob, H. Schmalz and V. Abetz, Macromolecules, 2002, 35, 3048–3058 CrossRef.
- K. A. Cogan and A. P. Gast, Macromolecules, 1990, 23, 745–753 CrossRef CAS.
- A. P. Gast, P. K. Vinson and K. A. Cogan-Farinas, Macromolecules, 1993, 26, 1774–1776 CrossRef CAS.
- D. Richter, D. Schneiders, M. Monkenbusch, L. Willner, L. J. Fetters, J. S. Huang, M. Lin, K. Mortensen and B. Farago, Macromolecules, 1997, 30, 1053–1068 CrossRef CAS.
- E. K. Lin and A. P. Gast, Macromolecules, 1996, 29, 4432–4441 CrossRef CAS.
- W. Y. Chen, J. X. Zheng, S. Z. D. Cheng, C. Y. Li, P. Huang, L. Zhu, H. M. Xiong, Q. Ge, Y. Guo, R. P. Quirk, B. Lotz, L. F. Deng, C. Wu and E. L. Thomas, Phys. Rev. Lett., 2004, 93, 028301-1–028301-4.
- J. X. Zheng, H. M. Xiong, W. Y. Chen, K. M. Lee, R. M. Van Horn, R. P. Quirk, B. Lotz, E. L. Thomas, A. C. Shi and S. Z. D. Cheng, Macromolecules, 2006, 39, 641–650 CrossRef CAS.
- J. T. Xu, W. Jin, G. D. Liang and Z. Q. Fan, Polymer, 2005, 46, 1709–1716 CrossRef CAS.
- J. T. Xu, J. P. A. Fairclough, S. M. Mai and A. J. Ryan, J. Mater. Chem., 2003, 13, 2740–2748 RSC.
- G. Guerin, J. Raez, I. Manners and M. A. Winnik, Macromolecules, 2005, 38, 7819–7827 CrossRef CAS.
- J. Raez, R. Barjovanu, J. A. Massey, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2000, 39, 3862–3865 CrossRef CAS.
- J. Raez, J. P. Tomba, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2003, 125, 9546–9547 CrossRef CAS.
- I. Korczagin, M. A. Hempenius, R. G. Fokkink, M. A. C. Stuart, M. Al-Hussein, P. H. H. Bomans, P. M. Frederik and G. J. Vancso, Macromolecules, 2006, 39, 2306–2315 CrossRef CAS.
- L. Shen, H. Wang, G. Guerin, C. Wu, I. Manners and M. Winnik, Macromolecules, 2008, 41, 4380–4389 CrossRef CAS.
- H. Schmalz, M. G. Lanzendörfer, V. Abetz and A. H. E. Müller, Macromol. Chem. Phys., 2003, 204, 1056–1071 CrossRef CAS.
- H. Schmalz, A. Knoll, A. J. Müller and V. Abetz, Macromolecules, 2002, 35, 10004–10013 CrossRef CAS.
- R. Borsali, E. Minatti, J.-L. Putaux, M. Schappacher, A. Deffieux, P. Viville, R. Lazzaroni and T. Narayanan, Langmuir, 2003, 19, 6–9 CrossRef CAS.
- H. Tadokoro, Y. Chatani, T. Yoshihara, S. Tahara and S. Murahashi, Makromol. Chem., 1964, 73, 109–127 CrossRef CAS.
- Y. Takahash, I. Sumita and H. Tadokoro, J. Polym. Sci., Part B: Polym. Phys., 1973, 11, 2113–2122 Search PubMed.
- Y. Takahash and H. Tadokoro, Macromolecules, 1973, 6, 672–675 CrossRef CAS.
- L. Zhu, P. Huang, W. Y. Chen, Q. Ge, R. P. Quirk, S. Z. D. Cheng, E. L. Thomas, B. Lotz, B. S. Hsiao, F. J. Yeh and L. Z. Liu, Macromolecules, 2002, 35, 3553–3562 CrossRef CAS.
- G. Reiter, P. Hörner, G. Hurtrez, G. Riess, J. U. Sommer and J. F. Joanny, J. Surf. Sci. Technol., 1998, 14, 93–103 Search PubMed.
- C. G. Vonk, J. Appl. Crystallogr., 1973, 6, 148–152 CrossRef CAS.
- J. T. Xu, J. P. A. Fairclough, S. M. Mai, A. J. Ryan and C. Chaibundit, Macromolecules, 2002, 35, 6937–6945 CrossRef CAS.
- B. Lotz and A. Kovacs, Am. Chem. Soc., Div. Polym. Chem., 1969, 10, 820 CAS.
- T. Shiomi, K. Imai, K. Takenaka, H. Takeshita, H. Hayashi and Y. Tezuka, Polymer, 2001, 42, 3233–3239 CrossRef CAS.
- J. T. Xu, J. J. Yuan and S. Y. Cheng, Eur. Polym. J., 2003, 39, 2091–2098 CrossRef CAS.
- A. Müller, V. Balsamo and M. L. Arnal, Adv. Polym. Sci., 2005, 190, 1–63 CAS.
- A. Taden and K. Landfester, Macromolecules, 2003, 36, 4037–4041 CrossRef CAS.
Footnote |
| † Present address: DSM Research, Material Science Centre, P.O. Box 18, 6160 MD Geleen, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2009 |