Asymmetric lipid bilayer sandwiched in polyelectrolyte multilayer films through layer-by-layer assembly†
Jiangshan
Chen
*a,
Ralf
Köhler
ab,
Thomas
Gutberlet‡
c,
Helmuth
Möhwald
a and
Rumen
Krastev
*a
aDepartment of Interfaces, Max-Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: rumen.krastev@mpikg.mpg.de; jiangshan.chen@mpikg.mpg.de
bHelmholtz Centre Berlin for Materials and Energy GmbH (former Hahn-Meitner Institute, Berlin), 14109 Berlin, Germany
cLaboratory for Neutron Scattering, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland
First published on 16th October 2008
Abstract
Lipid bilayer of 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE) was formed on the surface of a linearly growing polyanion/polycation polyelectrolyte multilayer (PEM) film prepared by the layer-by-layer (LbL) technique. The PEM films were prepared with two strong polyelectrolytes—poly(sodium 4-styrenesulfonate) (PSS) as the polyanion and poly-(diallyldimethylammonium chloride) (PDADMAC) as the polycation. The DMPE was always deposited onto the PEM cushion terminated with positively charged PDADMAC. The surface charge of the DMPE-coated film depends on the pH of the buffer solution. Formation of PEM could be continued on the top of the lipid bilayer by tuning the headgroup charge of the DMPE. An asymmetric structure of polycation/lipid/polyanion “sandwich” was created in the case where anionic PSS was used as the first layer of the subsequent PEM. In this system, the DMPE lipid membrane was constructed as a charge asymmetric barrier sandwiched in the interior of the PEM films. The formation of PEM architectures incorporated with an asymmetric DMPE bilayer was confirmed by ζ potential, UV-vis absorption, quartz crystal microbalance (QCM) and neutron reflectometry experiments.
1. Introduction
Since Decher1 introduced the method for preparing multilayer films through consecutively depositing oppositely charged polyelectrolytes (PE) from dilute aqueous solution, polyelectrolyte multilayer (PEM) films self-assembled by this layer-by-layer (LbL) process have been intensively studied. The popularity of the LbL procedure is due to its simplicity, versatility, and control over the structure and the thickness of the resulting films. Moreover, a large variety of materials have been used in LbL build-up process studies ranging from small organic molecules2,3 and inorganic compounds4,5 to biomacromolecules such as proteins and DNA.6–11Assemblies of amphiphilic molecules and polyelectrolytes are interesting systems for various biomimetic applications, such as cell-membrane models, biosensors and drug carriers.12–16Lipid membranes on polymer cushions can be created directly viavesicle fusion. The electrostatic interaction between lipid molecules and polymers plays an important role in film formation.17–19 The assembly of lipid bilayer membranes alternated with polyelectrolytes has been demonstrated.18,20–24 In these systems, lipid membranes act as internal barriers to separate two compartments of the PEM. Usually, this structure is charge symmetric, i.e., the lipid bilayer is sandwiched between polyelectrolytes with the same charge because of the symmetric structure of the bilayer. The preparation of asymmetric lipid bilayers is challenging, and there are a few reports about it. E.g. Katagiri and Caruso25 used the LbL method to prepare monodisperse vesicles comprising asymmetric lipid bilayers supported on colloid particles coated with polyelectrolytes.
In the present work, we show the formation of a charge asymmetric lipid bilayer of 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE) sandwiched between cationic poly-(diallyldimethylammonium chloride) (PDAMDAC) and anionic poly(sodium 4-styrenesulfonate) (PSS). The structure of the prepared PEM was confirmed by ζ potential, UV-vis absorption, quartz crystal microbalance (QCM) and neutron reflectometry experiments.
2. Experimental section
Materials
Poly-(diallyldimethylammonium chloride) (PDADMAC, Mw = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–350000 g mol−1), poly(sodium 4-styrenesulfonate) (PSS, Mw = 70000 g mol−1), poly(ethylenimine) (PEI, Mw = 75000 g mol−1), poly(allylamine hydrochloride) (PAH, Mw = 70000 g mol−1), 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DOTC), and deuterium oxide (D2O, min. 99.9% isotope enrichment) were purchased from Sigma-Aldrich. Pyrene-labeled poly(sodium 4-styrenesulfonate) (PSS-Py) was a present from the group of Prof. Lidong Li (University of Science and Technology, and Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, China). It was synthesized according to their previous work.26 Perdeuterated poly(sodium 4-styrenesulfonate) (d-PSS, Mw = 80300 g mol−1) was purchased from Polymer Standards Service (Mainz, Germany). 1,2-Dimyristoyl-sn-glycero-3- phosphoethanolamine (DMPE) was purchased from Avanti Polar Lipids Inc. Fig. 1 shows the chemical structures of the polyelectrolytes and lipids used.
000–350000 g mol−1), poly(sodium 4-styrenesulfonate) (PSS, Mw = 70000 g mol−1), poly(ethylenimine) (PEI, Mw = 75000 g mol−1), poly(allylamine hydrochloride) (PAH, Mw = 70000 g mol−1), 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DOTC), and deuterium oxide (D2O, min. 99.9% isotope enrichment) were purchased from Sigma-Aldrich. Pyrene-labeled poly(sodium 4-styrenesulfonate) (PSS-Py) was a present from the group of Prof. Lidong Li (University of Science and Technology, and Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, China). It was synthesized according to their previous work.26 Perdeuterated poly(sodium 4-styrenesulfonate) (d-PSS, Mw = 80300 g mol−1) was purchased from Polymer Standards Service (Mainz, Germany). 1,2-Dimyristoyl-sn-glycero-3- phosphoethanolamine (DMPE) was purchased from Avanti Polar Lipids Inc. Fig. 1 shows the chemical structures of the polyelectrolytes and lipids used.
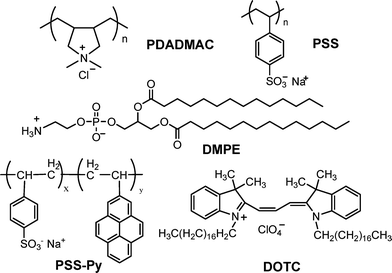 | ||
| Fig. 1 Chemical structures of the used PDADMAC, PSS, DMPE, PSS–Py and DOTC. | ||
All chemicals were used without further purification. The aqueous solutions were prepared with ultrapure water from a Milli-Q Plus 185 water generation system (Millipore, resistivity >18.2 MΩ cm).
Substrates
Weakly cross-linked melamine formaldehyde particles (MF particles, diameter = 5.0 ± 0.2 µm) were obtained from Microparticles GmbH, Germany. Silicon blocks from Siliciumbearbeitung Andrea Holm (Tann/Ndb., Germany) with two sides polished (dimensions 8 × 5 × 1.5 cm3, orientation <100>), were used for the NR experiments.Preparation of lipid small unilamellar vesicles (SUV)
10 mg of DMPE was dissolved in chloroform–methanol (2 : 1 v/v) in a glass vessel. The organic solvent was evaporated by a N2 stream. The lipid film obtained on the walls of the vessel was dried under vacuum overnight. Hydration of the lipid film was then performed at room temperature with 2 mL phosphate buffer (10 mM, pH = 10 adjusted by 0.5 M NaOH). Large multilamellar vesicles (LMV) were generated using a vortex mixer. These LMV were ultrasonicated with a bath-type sonicator for 30 min at a temperature of 60 °C. The resulting suspension was extruded 21 times through a 100 nm pore diameter polycarbonate filter and then diluted to 0.1 mg mL−1 to obtain the final lipid SUV suspension.SUV containing lipophilic DOTC were used in the UV-adsorption experiments. It was included in the SUV by adding it at 3% (w/w) to the DMPE solution in chloroform : methanol mixture.
Coating of colloid particles
The adsorption of the polyelectrolytes onto the MF microparticles (1% w/w in suspension) was conducted in 0.5 M NaCl solutions containing 1 mg mL−1PE for 10 min followed by three washings in water. The excess polyelectrolytes were removed by centrifugation at 2000 rpm for 3 min. After assembly of the PE multilayer (PDADMAC being the terminating layer), the coated particles were incubated in the lipid SUV suspension for 30 min at temperature of 60 °C. The suspension was centrifuged, followed by three washing steps with phosphate buffer (10 mM, pH = 10) in order to remove the excess nonadsorbed DMPE. Subsequent adsorption of PE layers onto DMPE-terminated MF particles was performed following the same procedure as above.Coating of planar substrates
Silicon blocks, quartz and QCM crystals were treated with “piranha” solution for 1 h and were thoroughly rinsed with water (caution: piranha solution, contains 3 : 7 H2O2 : H2SO4, and reacts violently with many organic materials. It must be used with extreme care, and it should not be stored in sealed containers). The planar substrates were always dipped for 20 min in PEI solution (1 mg mL−1, salt free) and washed with water to form a precursor layer before the deposition of the PEM. The pretreated substrates were sequentially immersed into polyelectrolyte solutions (1 mg mL−1PE, in 0.5 M NaCl) for 10 min, with three rinsing steps in water for 2 min after each PE deposition step. The substrates were incubated in the lipid SUV suspension for 30 min at 60 °C after assembly of the PE multilayer and then rinsed three times with phosphate buffer. The following PE and lipid layers were adsorbed by the same method. All further measurements were performed only at room temperature.ζ potential
ζ potential measurements were performed with a Zetasizer Nano ZS instrument (ZEN3500, Malvern Instruments Ltd). An average of five measurements at the stationary level was taken for each data point. The ζ potential measurements were conducted in pure Milli-Q water for PE and in phosphate buffer for lipid covered particles. The solution pH was adjusted by adding HCl or NaOH for the pH-dependent studies.UV-vis spectroscopy
The UV-vis absorption spectra were recorded by a Cary 50 UV-visible spectrophotometer (Varian Inc., USA). All measurements were performed in water after PE or lipid assembly on the quartz slide. The background spectra of the sample support quartz/PEI/(PSS/PDADMAC)2/PSS was directly subtracted from the measured spectra.Quartz crystal microbalance (QCM)
QCM measurements were carried out using a Q-Sense E4 device equipped with flow modules (Q-Sense AB, Vastra Frolundra, Sweden). An AT-cut Au covered quartz crystal with a resonance frequency of 5 MHz was used.Neutron reflectometry
The change in the film thickness and composition was followed in situ using neutron reflectometry (NR), NR is a powerful technique for studying the structure and the properties of thin layers on solid supports.18,27,28 It allows the measurement of the film thickness and composition profiles along the z direction normal to the interface over a length scale of up to 500 nm, with a resolution down to about 0.2 nm. The use of compounds with different isotopes of the same chemical element allows NR measurements to be focused on particular parts of a thin layer without perturbing the whole chemical structure. The specular neutron reflectivity method is based on the variation of the specular reflection R at the interface between two phases with the variation of the wave vector transfer Q = (4π/λ)sinθ, where θ is the angle of incidence of the neutron beam and λ is the neutron wavelength. The variation depends on the interfacial composition perpendicular to the layer interface characterized by the neutron scattering length density (SLD) (ni is the number density of the element i, and bi is its scattering length). Different isotopes are characterized by different values of the coefficient bi. The SLD can be easily transformed into the material density or composition of the PEM. Thus, the method is suitable for estimating the changes in the PEM thickness (swelling) and the amount of absorbed water (water uptake) in a single experiment.
(ni is the number density of the element i, and bi is its scattering length). Different isotopes are characterized by different values of the coefficient bi. The SLD can be easily transformed into the material density or composition of the PEM. Thus, the method is suitable for estimating the changes in the PEM thickness (swelling) and the amount of absorbed water (water uptake) in a single experiment.
A home-built cell used in the NR experiments allowed the liquid (typical volume ≈ 10 mL) in contact with the solid interface to be exchanged. The deposition was always performed from the respective H2O solution. Then, the sample was thoroughly washed with pure H2O and finally the H2O was exchanged with excess of D2O which was used as the lower phase against the Si blocks on top during the NR experiments. In this case the lower medium has a higher neutron scattering length density than the upper one and assures high resolution of the experiments and the formation of a well-pronounced critical edge.18 The experimentally obtained reflectivity curves were analyzed by applying the standard fitting routine, Parratt 3229 which determines the optical reflectivity of neutrons from planar surfaces using a calculation based on Parratt's recursion scheme for stratified media.30 The film is modeled as consisting of layers of specific thickness, SLD, and roughness, which are the fitting parameters. The model reflectivity profile is calculated and compared to the measured one, and then the model is adjusted by a change in the fitting parameters to best fit the data.
3. Results and discussions
Fig. 2 shows the step by step build-up of PEM on MF. The ζ potential changes as a function of PE and lipid layer number. The PSS-coated MF particles yielded a ζ potential of −50 mV in pure water. The presence of PDADMAC on the particle surface causes a reversal in ζ potential to about +40 mV. Alternating ζ potentials were obtained with the subsequent deposition of anionic PSS and cationic PDADMAC on the particles. DMPE was adsorbed onto PDADMAC-terminated particles from 10 mM phosphate buffer with pH = 10 at layer number 5. PDADMAC was used to assure strongly positive charge of the terminating layer because its charge density is not pH-dependent. The DMPE-coated particles yielded a ζ potential of about −35 mV (the same buffer was used for a dispersant in this case). When the pH value of the buffer was adjusted to 6 (see the shaded area in Fig. 2), the ζ potential of the particles shifts to a positive value of +23 mV. | ||
| Fig. 2 ζ potential as a function of layer number for MF particles coated with PSS, PDADMAC and DMPE layers. | ||
A detailed dependence of the charge of MF particles coated with PE multilayer and DMPE bilayer on pH was performed. The change in the ζ potential as a function of pH is shown in Fig. 3. It changes from about +25 to −40 mV for MF/(PSS/PDADMAC)2/DMPE particles when the medium pH changes from 4 to 12. DMPE is a typical zwitterionic lipid and the charge of its phosphoethanolamine head group depends on the pH of the buffer solution. The particles have a large negative value at base condition, reflecting the deprotonation of both phosphate and amine groups (see the inset in Fig. 3). In acid conditions, the ζ potential of the DMPE-terminated particles exhibits a positive value, which can be interpreted as the formation of a cationic amine and the polar orientation of the zwitterionic headgroup towards the outer phase. It should be mentioned that a further decrease in pH leads to the aggregation of the coated particles and the dissolution of the colloid cores. The particle aggregation should be due to the formation of hydrogen bonds between phosphate and amine, and the core dissolution was caused by the weak cross-linking of MF.
 | ||
| Fig. 3 ζ potential as a function of pH for MF particles coated with (PSS/PDADMAC)2/DMPE. | ||
The dependence of the surface charge of the DMPE covered particles on pH indicates that the lipid can be used to form a charge asymmetric structure by tuning the pH conditions during the deposition of the lipid and consequently the deposition of further PE layers. The use of a strong PE whose charge does not depend on the pH conditions is a necessary requirement for a successful build-up process. The deposition of further PE layers on the top of the DMPE-terminated MF particles followed (see Fig. 2). Under acid conditions, anionic PSS was successfully adsorbed onto the obtained particles, which is seen by the sharp change in the ζ potential of the particles. The further deposition of PDADMAC/PSS layers was possible after this step. Alternating ζ potentials were obtained by further deposition of PSS/PDADMAC on the DMPE-terminated particles. Thus, the architecture of PE multilayers embedded with DMPE was built on the colloid particles. In this structure, the lipid layer of DMPE was sandwiched between the polycation PDADMAC and the polyanion PSS to form a component of the polycation/lipid/polyanion trilayer, which is an asymmetric structure.
Further studies with planar samples were performed to clarify the structure of the already obtained asymmetric samples. The formation of a PEM asymmetric sandwich-like structure on a quartz substrate was examined by UV-vis spectroscopy. In order to clearly demonstrate the formation of the DMPE lipid layer and the subsequent PSS layer, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DOTC) and PSS-Py were employed as probes for the lipid and PSS layers, respectively. Fig. 4a shows the UV-vis spectra of quartz slides covered with PEM/(PDADMAC/DMPE:DOTC/PSS/Py)m multilayers [here PEM = PEI/(PSS/PDADMAC)2/PSS, and the repetition number m = 0, 1, 2, 3, 4, 5]. The absorption peaks at 520 and 556 nm indicate the presence of a lipid layer because the peaks correspond to the characteristic visible absorption wavelengths of the lipophilic DOTC molecules inserted in the DMPE layer.31,32 The appearance of absorption peaks at 334 and 350 nm prove that PSS-Py was successfully deposited on top of the DMPE layer. The absorption band intensities at 556 and 350 nm increase gradually with the increasing number of PDADMAC/DMPE:DOTC/PSS-Py trilayers, demonstrating that the DMPE has been consecutively sandwiched between PDADMAC and PSS-Py. The linear increase in absorbance (see Fig. 4b) indicates that equal amounts of lipid and PE were assembled in each deposition of PDADMAC/DMPE:DOTC/PSS-Py, confirming that the film grows uniformly.
 | ||
| Fig. 4 (a) UV-vis spectra of PEI/(PSS/PDADMAC)2/PSS/(PDADMAC/DMPE:DOTC/PSS-Py)m multilayers (m = 0, 1, 2, 3, 4, 5). (b) UV-vis absorption intensity increased with the number of repetition m at 556 and 350 nm. | ||
To quantify the amount of adsorbed PE and lipid in the multilayer system, the asymmetric sandwiched structure was built on Au covered QCM crystals. The frequency of oscillation of the QCM crystal (ΔF) decreases proportionally to the mass increase of deposited films. The relation between ΔF and Δm is described by the Sauerbrey equation:33
 | (1) |
Fig. 5 shows the values for ΔF and Δm obtained after each PE and lipid layer deposition. The bare sample has the structure Au/PEI/(PSS/PDADMAC)2/PSS. The average frequency change ΔF measured for the DMPE adsorption was about 29 Hz and the calculated Δm value was 0.51 × 10−6 g cm−2, which are in agreement with the values for a lipid bilayer reported by Decher and co-workers.34,35 This suggests that the adsorbed DMPE in our system could have a bilayer structure.
 | ||
| Fig. 5 QCM frequency decrease and corresponding mass increase for the deposition of PDADMAC, PSS and DMPE. | ||
NR experiments were performed to analyze the structure of the PEM sandwiched with asymmetric DMPE lipid bilayer. A PEM of PEI/(d-PSS/PAH)4/d-PSS/PDADMAC was assembled prior to the NR experiments as a supporting cushion for the DMPE membrane. Perdeuterated d-PSS was used to build the PEM and assure suitable contrast at the border between the lipid covered PEM cushion and the D2O subphase (see ref. 18 for a detailed description of the contrast variation during deposition of lipids onto the PEM cushion). PDADMAC was used to guarantee strong positive charge of the terminating layer at different pH conditions because of the independence of its charge on the pH.
Considering the high incubation temperature for DMPE vesicle fusion (about 60 °C) one could expect changes in the PEM thickness or SLD during the deposition step. Results which demonstrate the strong influence of temperature on the thickness of PSS/PDADMAC multilayers have been published.36–38 The effect is negligible in our case where PEM is prepared from PSS/PAH polyelectrolytes. No change in the reflectivity curves after heating the samples up to 60 °C was observed (see Fig. S2 in the ESI†).
The build-up process was followed in situ. The reflectivity curves at different deposition steps are shown in Fig. 6a. The NR curve 1 corresponds to the PEM cushion of Si/PEI/(d-PSS/PAH)4/d-PSS/PDADMAC. The well-pronounced fringes in the NR curve suggest a good contrast between the D2O subphase and the PDADMAC-terminated PEM. It was possible to fit the NR result using a three-layer model. The best fit curve is shown as line 1 in Fig. 6a and the corresponding SLD distribution along the distance z is shown as line 1′ in Fig. 6b.
 | ||
| Fig. 6 (a) NR curves from the samples: (1) Si/PEI/(d-PSS/PAH)4/d-PSS/PDADMAC, (2) after DMPE deposition, (3) after PSS deposition, and (4) after subsequent PDADMAC/PSS deposition. The solid lines show the best fit to the experimental data. The curves are shifted for clarity. (b) SLD profiles for the corresponding samples. | ||
The whole PEM block can be divided into three sub-layers (A, B, C) each with a characteristic SLD composition. Such a complex structure of the PEM was already reported in the literature 39,40. Close to the Si–PEM interface there is a layer with low density (sub-layer A) which is a result of the interaction with the solid support. This first layer is followed by a block with high SLD which represents the bulk of the film (sub-layer B). The third block (sub-layer C) which borders the D2O subphase has a relatively low SLD. The low SLD should be due to the terminated layer of PDADMAC which has theoretically predicted low SLD. The overall thickness of the PEM film is 20.4 nm. Curve 2 in Fig. 6a was obtained after the deposition of DMPE. The thickness and SLD of the DMPE layer were calculated as 4.5 nm and 2.8 × 10−6 Å−2, respectively. The obtained thickness of the lipid layer demonstrates the formation of a bilayer of DMPE. 18,41 Combining the thickness with the absorbed mass per unit area determined by QCM, the density of the DMPE bilayer can be calculated to be 1.13 g cm−3. Curve 3 shows the NR of the system after further deposition of a single PSS layer on top of the DMPE bilayer. The thickness of the overall film is 27.3 nm, the thickness of the single layer of PSS was determined as 2.5 nm. Curve 4 shows the NR after the subsequent deposition of the PDADMAC/PSS bilayer. The overall thickness is 34.1 nm.
It is shown in Fig. 6b that the SLD of the PEM cushion decreases after the deposition of the lipid layer. It is known that the measured SLD of the PEM cushion mainly consists of two individual components of PE and D2O.18 The decrease in the measured SLD should result from the reduction of D2O in the PEM, which demonstrates that the formed lipid layer can regulate the penetration of D2O into the PEM cushion. This behavior might be similar to the already discussed “odd–even” effect.40,42 The NR results of the formation of the PEM1/DMPE/PEM2 structure are summarized in Table 1. The SLD profiles shown in Fig. 6b demonstrate the successful formation of a sandwich-like asymmetric architecture of a lipid bilayer bound between the polycation (PDADMAC) and the polyanion (PSS) through LbL self-assembly.
| Deposition step | PEM and lipid internal sub-layer | |||||
|---|---|---|---|---|---|---|
| PEM1 | DMPE | PEM2 | ||||
| PEM sub-layers | A | B | C | D | ||
| 1 | Thickness/nm | 3.6 | 11.5 | 5.3 | ||
| SLD/10−6 Å−2 | 4.0 | 5.3 | 3.8 | |||
| 2 | Thickness/nm | 3.6 | 11.5 | 5.3 | 4.5 | |
| SLD/10−6 Å−2 | 2.9 | 4.6 | 3.9 | 2.8 | ||
| 3 | Thickness/nm | 3.5 | 11.5 | 5.4 | 4.5 | 2.5 |
| SLD/10−6 Å−2 | 3.0 | 4.7 | 4.0 | 2.9 | 5.1 | |
| 4 | Thickness/nm | 3.5 | 11.5 | 5.4 | 4.5 | 9.2 |
| SLD/10−6 Å−2 | 3.1 | 4.9 | 3.9 | 2.8 | 4.8 | |
4. Conclusions
The formation of a model lipid membrane was achieved by fusing DMPE vesicles from a phosphate buffer on top of layer-by-layer prepared polyelectrolyte cushions. The charge of the phosphoethanolamine group depends on the pH of the surrounding phase and allows for a change in the surface charge of the DMPE from positive to negative during the deposition process. The process can only be performed when strong PEs were used because their charge does not depend on the pH of the surroundings. The build-up process was characterized by ζ potential, UV-vis spectrometry, QCM and in situneutron reflectometry experiments. A 4.5 nm thick DMPE layer was successfully “constructed” after deposition onto PDADMAC-terminated PEM. Its thickness implies that DMPE forms a bilayer membrane. The ζ potential measurements show that the surface charge of the DMPE bilayer is strongly dependent on the pH of the surrounding phase. Anionic polyelectrolyte PSS was also successfully deposited on the lipid bilayer when the surface was treated with an acid buffer. The successive PE multilayer and lipid bilayer can be continued based on the same processes. Thus, asymmetric lipid bilayer was embedded in the interior of PE films by self-assembly. The obtained structure is depicted in Fig. 7. | ||
| Fig. 7 A schematic structure of the PE multilayer architecture of sandwiched asymmetric lipid bilayers. | ||
Acknowledgements
J. C. is grateful to the Alexander von Humboldt Foundation for a research fellowship. J. C. also thanks Prof. Lidong Li for the present of PSS–Py, and H. Zastrow for the assistance with ζ potential measurements. BENSC at HMI, Berlin, Germany is acknowledged for beam time at the neutron reflectometer V6. The Swiss spallation neutron source SINQ at the Paul Scherrer Institute, Villigen, Switzerland is acknowledged for the beam time allocated at the neutron reflectometer AMOR and the good infrastructure needed for the experiments.The experiments performed at the SINQ have been supported by the European Commission under the 6th Framework Programme (Strengthening the European Research Area, Research Infrastructures. Contract nr: RII3-CT-2003-505925).
References
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- K. Ariga, Y. Lvov and T. Kunitake, J. Am. Chem. Soc., 1997, 119, 2224–2231 CrossRef CAS.
- C. Tedeschi, F. Caruso, H. Möhwald and S. Kirstein, J. Am. Chem. Soc., 2000, 122, 5841–5848 CrossRef CAS.
- M. Y. Gao, B. Richter, S. Kirstein and H. Möhwald, J. Phys. Chem. B, 1998, 102, 4096–4103 CrossRef CAS.
- Y. W. Lin, W. L. Tseng and H. T. Chang, Adv. Mater., 2006, 18, 1381–1386 CrossRef CAS.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117–6123 CrossRef CAS.
- Y. Lvov, H. Haas, G. Decher, H. Möhwald, A. Michailov, B. Mtchedlishvily, B. Margunova and B. Vainshtain, Langmuir, 1994, 10, 4232 CrossRef CAS.
- M. K. Beissenhirtz, F. W. Scheller, W. F. M. Stöcklein, D. G. Kurth, H. Möhwald and F. Lisdat, Angew. Chem., Int. Ed., 2004, 43, 4357–4360 CrossRef CAS.
- F. Caruso and H. Möhwald, J. Am. Chem. Soc., 1999, 121, 6039–6046 CrossRef CAS.
- F. Caruso, K. Niikura, D. N. Furlong and Y. Okahata, Langmuir, 1997, 13, 3427–3433 CrossRef CAS.
- Y. J. Ma, W. F. Dong, M. A. Hempenius, H. Möhwald and G. J. Vancso, Angew. Chem., Int. Ed., 2007, 46, 1702–1705 CrossRef CAS.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656–663 CrossRef CAS.
- E. Sackmann and M. Tanaka, Trends Biotechnol., 2000, 18, 58–64 CrossRef CAS.
- L. Duan, Q. He, K. W. Wang, X. H. Yan, Y. Cui, H. Möhwald and J. B. Li, Angew. Chem., Int. Ed., 2007, 46, 6996–7000 CrossRef CAS.
- T. W. McBee, L. Y. Wang, C. H. Ge, B. M. Beam, A. L. Moore, D. Gust, T. A. Moore, N. R. Armstrong and S. S. Saavedra, J. Am. Chem. Soc., 2006, 128, 2184–2185 CrossRef CAS.
- Z. H. An, G. Lu, H. Möhwald and J. B. Li, Chem.–Eur. J., 2004, 10, 5848–5852 CrossRef CAS.
- K. Katagiri and F. Caruso, Macromolecules, 2004, 37, 9947–9953 CrossRef CAS.
- C. Delajon, T. Gutberlet, R. Steitz, H. Möhwald and R. Krastev, Langmuir, 2005, 21, 8509–8514 CrossRef CAS.
- G. Krishna, T. Shutava and Y. Lvov, Chem. Commun., 2005, 2796–2798 RSC.
- J. B. Li, H. Möhwald, Z. H. An and G. Lu, Soft Matter, 2005, 1, 259–264 RSC.
- A. M. Pilbat, Z. Szegletes, Z. Kóta, V. Ball, P. Schaaf, J. C. Voegel and B. Szalontai, Langmuir, 2007, 23, 8236–8242 CrossRef CAS.
- Z. H. An, H. Möhwald and J. B. Li, Biomacromolecules, 2006, 7, 580–585 CrossRef CAS.
- L. Q. Ge, H. Möhwald and J. B. Li, Biochem. Biophys. Res. Commun., 2003, 303, 653–659 CrossRef CAS.
- L. Q. Ge, H. Möhwald and J. B. Li, Chem.–Eur. J., 2003, 9, 2589–2594 CrossRef CAS.
- K. Katagiri and F. Caruso, Adv. Mater., 2005, 17, 738–743 CrossRef CAS.
- L. D. Li, C. Tedeschi, D. G. Kurth and H. Möhwald, Chem. Mater., 2004, 16, 570–573 CrossRef CAS.
- J. Y. Wong, J. Majewski, M. Seitz, C. K. Park, J. N. Israelachvili and G. S. Smith, Biophys. J., 1999, 77, 1445–1457 CrossRef CAS.
- R. Kügler, J. Schmitt and W. Knoll, Macromol. Chem. Phys., 2002, 203, 413 CrossRef CAS.
- C. Braun, Parratt 32 Program for Reflectivity Fitting, Hahn-Meitner Institut, Berlin, 1999 Search PubMed.
- L. G. Parratt, Phys. Rev., 1954, 95, 359–369 CrossRef.
- R. D. Klausner and D. E. Wolf., Biochemistry, 1980, 19, 6199–6203 CrossRef CAS.
- M. F. Ethier, D. E. Wolf and D. L. Melchior., Biochemistry, 1983, 22, 1178–1182 CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 Search PubMed.
- B. Seantier, C. Breffa, O. Félix and G. Decher, Nano Lett., 2004, 4, 5–10 CrossRef CAS.
- B. Seantier, C. Breffa, O. Félix and G. Decher, J. Phys. Chem. B, 2005, 109, 21755–21765 CrossRef CAS.
- C. Y. Gao, S. Leporatti, S. Moya, E. Donath and H. Möhwald, Chem.–Eur. J., 2003, 9, 915–920 CrossRef CAS.
- K. Köhler, D. G. Shchukin, H. Möhwald and G. B. Sukhorukov, J. Phys. Chem. B, 2005, 109, 18250–18259 CrossRef.
- K. Köhler, H. Möhwald and G. B. Sukhorukov, J. Phys. Chem. B, 2006, 110, 24002–24010 CrossRef.
- J. E. Wong, F. Rehfeldt, P. Hänni, M. Tanaka and R. V. Klitzing, Macromolecules, 2004, 37, 7285–7289 CrossRef CAS.
- M. Schönhoff, V. Ball, A. R. Bausch, C. Dejugnat, N. Delorme, K. Glinel, R. V. Klitzing and R. Steitz, Colloids Surf. A: Physicochemical and Engineering Aspects, 2007, 303, 14–29 Search PubMed.
- R. Kügler and W. Knoll, Bioelectrochemistry, 2002, 56, 175–178 CrossRef CAS.
- D. Carièrre, R. Krastev and M. Schönhoff, Langmuir, 2004, 20, 11465–11472 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: A graph showing the dependence of the decreased QCM frequency on the overtone at DMPE deposition, and NR curves of PSS/PAH multilayers before and after temperature treatment. See DOI: 10.1039/b808572b |
| ‡ Present address: Forschungszentrum Jülich GmbH, Jülich Centre for Neutron Science, Lichtenbergstr. 1, 85747 Garching, Germany |
| This journal is © The Royal Society of Chemistry 2009 |