Interaction of human serum albumin with charge transfer probe ethyl ester of N,N-dimethylamino naphthyl acrylic acid: An extrinsic fluorescence probe for studying protein micro-environment
Received
12th August 2008
, Accepted 2nd October 2008
First published on 6th November 2008
Abstract
The charge transfer (CT) probe ethyl ester of N,N-dimethylamino naphthyl acrylic acid (EDMANA) bound to Human Serum Albumin (HSA) serves as an efficient reporter of the polarity and conformational changes of protein in aqueous buffer (Tris-HCl buffer, pH= 7.03) and in presence of denaturant, quencher and reverse micelles. The change in fluorescence intensity and the position of emission maxima of EDMANA in presence of HSA well reflect the nature of binding and location of the probe inside the proteinous environment. The increase in steady state anisotropy values with increase of protein concentration indicate restriction imposed on the mobility of the probe molecules in the proteinous medium. The results of fluorescence quenching of EDMANA by acrylamide, Fluorescence Resonance Energy Transfer (FRET) and Red Edge Excitation Shift (REES) studies throw light on the accessibility to the probe bound to HSA and hence indicate the probable location of the probe within the hydrophobic cavity of HSA. The complicated nature of protein unfolding in presence of urea is well studied by change in the fluorescence properties of EDMANA bound to HSA protein.
Introduction
Serum albumins are best known proteins that serve as physiological carriers of different substances in the blood stream and are of remarkable in terms of the number of available binding sites1–4 to variety of non-polar ligands like fatty acids,5surfactants,6–8 enzymes9 and to an impressive variety of drugs.10,11 The most widely studied serum albumins are Human Serum Albumin (HSA) and Bovine Serum Albumin (BSA).
The protein HSA is the most abundant (0.6 mmol dm−3) and important protein found in the circulatory system. The molecule HSA is composed of a single polypeptide chain with 585 amino acids and seventeen disulfide bridges distributed regularly.1–4 It has three domains - a large double loop with a short connecting segment, a small double loop with a long connecting segment and another large loop connected to the next segment. The three-dimensional crystallographic structure of HSA at resolution of 0.28 nm reveals that the three domains assemble to form a heart-shaped molecule. The protein molecule HSA has a single tryptophan residue (Trp-214) and exists in a similar location to that of Trp-212 of BSA.2,12 It serves mainly as transport protein for lipophilic plasma components especially fatty acids which bind to different binding sites.5 Furthermore, it also helps for the transportation of a variety of drugs through the circulatory system and has a great impact on drug pharmacokinetics.10,11 The carrier function of HSA for endogenous and exogenous substances is explained by the existence of different but specific binding sites.13 Kragh-Hausen proposed the existence of at least six different binding sites on albumin molecule located in sub-domains II-A and III-A. However, the most important sites are I and II which are also called warfarin binding site and the indole or benzodiazepine binding site, respectively.14
It is seen that fluorescence techniques have been well utilized for the study of polarity of protein cavities and the binding of foreign species using extrinsic probes.15–23 Apart from this, the use of extrinsic probes for elucidation of protein structure and dynamics using both steady state and time-resolved fluorescence techniques has attracted the attention of many research groups.24,25 A number of studies show that some synthetic and therapeutically active naturally occurring flavonols can serve as excellent monitors of the microenvironments of biologically relevant systems.22,23 Charge transfer fluorescent probes such as Prodan13 and Nile Red19 that exhibit both changes in fluorescence intensity and spectral shift have been successively used for protein binding and displacement studies and analysis of protein-probe complexation equilibrium. Extrinsic probes are more successfully applied for studying conformational changes of the protein structure with variation of its solvent characteristics etc. instead of direct monitoring of the tryptophanyly fluorescence.15–25 Direct studies are often difficult and the fluorescence from other foreign species present in the solvent often interference with the tryptophanyl fluorescence in the range of 310–350 nm.26,27 It is found that charge transfer probes have longer wavelength fluorescence and are relatively unaffected by impurities and the results are reliable.13,18–20
The molecule EDMANA exhibits the phenomenon of intramolecular charge transfer reaction in its excited state in polar solvents. The charge transfer (CT) band is sensitive to solvent polarity and shifts to higher wavelength with increasing polarity of the solvent.28 Thus it can serve as an efficient fluorescent reporter of its surrounding microenvironment and is suitable for the study of polarity of the protein cavities. The binding between protein HSA and EDMANA, the micropolarity of the binding site and the probable location of the probe inside the protein have been investigated by monitoring the intensity and position of emission maxima of the polarity dependent CT emission band of the probe molecule. Steady state anisotropy values, Red Edge Excitation Shift, acrylamide induced fluorescence quenching and Fluorescence Resonance Energy Transfer provide valuable information regarding the binding nature of this complexation process. The denaturation of HSA by urea which leads to exposure of the probe molecules to more polar environment has been explored by monitoring the shift of the CT emission band of the probe. Studies of fluorescence characteristics of protein-probe complex in water pools of AOT/n-heptane reverse micelles also bring out interesting results.
Materials and methods
The synthetic procedure of EDMANA (Scheme 1) has been described in our previous publication.28Tris buffer purchased from SRL, India was used to prepare Tris-HCl buffer (0.01 M) of pH = 7. Human Serum Albumin and AOT purchased from Aldrich Chemicals were used as supplied. Triple distilled water was used for the preparation of all solutions. Spectroscopic grade n-heptane purchased from Spectrochem was used after vacuum distillation. The purity of all solvents in the wavelength range used for fluorescence measurements was checked before preparation of solutions.
 |
| | Scheme 1 Structure of charge transfer probe EDMANA. | |
The absorption and emission measurements were done by Hitachi UV-Vis U-3501 spectrophotometer and Perkin Elmer LS-50B spectrophotometer, respectively. In all measurements, the concentration of probe EDMANA was maintained in the range of 10−6 mol dm−3 in order to avoid aggregation and reabsorption effects.
Fluorescence quantum yield (Φf) was determined using β-naphthol as the secondary standard (Φf = 0.23 in methylcyclohexane) using the following equation:29
where Φ
S and Φ
R are the quantum yield,
AS and
AR are the integrated fluorescence area, and (
Abs)
S and (
Abs)
R are absorbance for the sample and reference, respectively.
nS and
nR are the refractive index for the sample and reference solution, respectively.
Steady state anisotropy measurements were carried out using the same fluorescence spectrophotometer. The steady state anisotropy r is defined as
| r = (IVV−GIVH)/(IVV− 2GIVH) |
where
IVV and
IVH are the emission intensities when the excitation polarizer is vertically oriented and the emission polarizer is oriented vertically and horizontally, respectively. G is the correction factor. The terms
IVH and
IHH are the emission intensity when the excitation polarization is horizontally oriented and the emission polarization is oriented vertically and horizontally respectively.
The experimental setup for picosecond TCSPC includes a picosecond diode laser at 408 nm (IBH, UK, NanoLED-07, s/n 03931), which was used as light source. The fluorescence signal was detected in magic angle (54.7°) polarization using Hamamatsu MCP PMT (3809U).30 The typical system response of the laser system was 90ps. The decays were analyzed using IBH DAS-6 decay analysis software. The fluorescence decay curves were analyzed by multi-exponential fitting program of IBH. The average lifetimes were calculated using the equation:
where
τi and
ai are the fluorescence lifetime and its coefficient of the
ith component, respectively.
Results and discussion
The molecule EDMANA shows a single broad absorption band at ∼350 nm and dual fluorescence bands - a higher energy band at ∼430 nm and a lower energy but relatively intense emission band at ∼521 nm in water.28 The low energy emission band is found to be sensitive to the changes in solvent polarity and it gradually shifts to higher wavelength with increasing polarity of solvents. Hence, the large Stokes-shifted band at 521 nm has been ascribed to CT emission of EDMANA. The position, intensity, quantum yields and fluorescence lifetime of the CT band is found to be sensitive to the nature of the medium. It is pertinent to mention here that theoretical calculations by considering twisted intramolecular charge transfer model support the ICT phenomena and the solvent polarity dependent red shifted emission spectra.28
As seen in Fig. 1, addition of HSA to a solution of EDMANA in aqueous buffer (Tris buffer, 0.01 M, pH = 7.03) results in gradual shift of the absorption maxima to the red side from ∼350 nm to ∼361 nm.15,19 However, the position of the emission maxima of EDMANA with addition of HSA shows a blue shift of the lower energy CT band from 521 nm in aqueous buffer to 478 nm in presence of 146 μM HSA (Fig. 2a).15,18,19 The low energy CT band also shows an appreciable enhancement of emission intensity (Fig. 2b). The blue shift of the emission maxima of the CT band indicates encapsulation of the probe molecules inside the hydrophobic cavity of the protein. As can be seen in Fig. 2b, the intensity of the blue shifted CT band increases sharply and reaches a saturation value where no more probe molecules left for binding. The increase in intensity further indicates towards the fact that as more and more binding of the probe to the protein occurs, the non-radiative channels generally present in aqueous solution become less operative.15 In general, protic solvents through hydrogen bonding facilitate non-radiative path of the CT emission band and hence fluorescence quantum yields and lifetime values of the probe reduces in protic solvents. It is found that the fluorescence quantum yields (see later) obtained in presence of HSA are much higher compared to that obtained in aqueous phase. Thus, the funneling out of energy via non-radiative pathway decreases to a great extent when the probe remains encapsulated within the hydrophobic cavity of the protein.
 |
| | Fig. 1 Effect of increasing concentration of HSA (0 μM, 22 μM, 30 μM, 38 μM, 42 μM, 58 μM, 80 μM and 124 μM) on the absorption spectra of EDMANA (conc. of probe= 6.4 × 10−6 M). | |
 |
| | Fig. 2 (a) Effect of increasing concentration of HSA on the fluorescence emission (λext. = 350 nm) spectra of EDMANA; (b) plot of variation of the intensity of emission maxima of EDMANA with increasing concentration of HSA; (c) variation of emission maxima of EDMANA in dioxane-water mixture against ET(30) values: open circle indicates the polarity in HSA medium calculated using EDMANA. | |
The knowledge of the micropolarity of biological systems is very crucial and can be determined using fluorescence probe techniques due to relative ease of the determination of micropolarity using this technique. The common method used is the one using different percentages of dioxane-water mixture with known ET(30) values. The standard ET(30) values of the dioxane-water mixture was determined based on the intramolecular charge transfer transition absorption of the betaine dye 2,6-diphenyl-4-(2,4,6 triphenyl-1-pyridino) phenolate.18 The standard plot of ET(30) vs. emission maxima (λmax) of the probe molecule dissolved in these mixtures is shown in Fig. 2c. The polarity or ET(30) value of the proteinous microenvironment surrounding the fluorophore can hence be determined based on the position of the emission maxima in these environments. As seen in Fig. 2c, the emission maxima of the ICT state of EDMANA shows an excellent correlation with the empirical solvent polarity parameter ET(30) in dioxane-water mixtures. From Fig. 2c the micropolarity (i.e.ET(30) value) of the surrounding of the probe molecules solubilized in HSA is found to be 38.4. This clearly indicates that the polarity at the binding site of protein HSA is less polar compared to that of pure water. Therefore, the CT emission band of the probe shifted to the blue side in proteinous environment.
Determination of binding constant of protein-probe complex
Though the changes of spectral characteristics with addition of HSA are indicative of interaction between probe and protein, a quantitative estimate or the extent of binding of EDMANA to the hydrophobic cavity of HSA is determined using the Benesi-Hildebrand relation.31,32
The binding or complexation equilibrium for the 1:1 complex can be expressed as
where
K is the binding constant of the complex and can be written in terms of concentration of the components at the equilibrium position.
| K = [EDMANA:HSA]/[EDMANA][HSA] |
Benesi-Hildebrand relation for these types of complexation processes is expressed in terms of fluorescence intensity with the assumption that [HSA] >> [EDMANA:HSA], i.e., concentration of the complex is very low compared to that of the free protein. Hence, free protein concentration can be replaced by total protein concentration.31,32 The above equation for binding constant can be expressed by replacing the concentration of the components in terms of fluorescence intensity as
rearranging the above equation we have
where
I0,
I and
I1 are the emission intensities in absence of, at intermediate and infinite concentration of HSA, respectively. As seen in
Fig. 3, the plot of 1/[
I−
I0]
vs. 1/[HSA] shows a straight line indicating 1:1 complexation between EDMANA and HSA. From the ratio of intercept and slope of Benesi-Hildebrand plot of the emission intensity the extent of binding (
K) is determined to be 6.63 × 10
3 M
−1 (
Table 1). The free energy change (Δ
G) for such probe-
protein complexation is calculated to be −21.8 kJ mole
−1 using the value of K (6.63 × 10
3 M
−1). The values of
K and Δ
G are well in agreement with that obtained for such complexation processes studied earlier.
15,18 The large binding constant indicates strong binding of the CT probe with HSA
protein and the change of free energy change favours spontaneous complexation process.
Table 1 Different parameters obtained from spectral study of EDMANA bound to HSA
| Proteinous environment |
Binding constant K/M−1 |
Free energy change, ΔG/kJ mole−1 |
Stern–Volmer quenching constant, KSV/mol−1 L |
Energy transfer tfficiency, E (%) |
| HSA |
6.63 × 103 |
−21.8 |
1.02 |
70.88 |
![Benesi-Hildebrand plot of 1/[I−I0] vs. 1/[protein] (M−1) for binding of EDMANA with HSA (22 μM, 26 μM, 30 μM, 34 μM, 38 μM, 42 μM, 46 μM, 58 μM, 80 μM and 124 μM).](/image/article/2009/PP/b814050b/b814050b-f3.gif) |
| | Fig. 3 Benesi-Hildebrand plot of 1/[I−I0] vs. 1/[protein] (M−1) for binding of EDMANA with HSA (22 μM, 26 μM, 30 μM, 34 μM, 38 μM, 42 μM, 46 μM, 58 μM, 80 μM and 124 μM). | |
 |
| | Fig. 4 Variation of steady state anisotropy (λem = 480 nm and λext. = 350 nm) of EDMANA with increasing concentration of HSA. (b) Variation of steady state anisotropy (λem = 480 nm) of EDMANA with change in excitation wavelength at definite HSA concentrations in the medium. | |
A wavelength sensitive tool often used to establish the location of the probe in an organized media is the measurement of excitation anisotropy.33 If shifting the excitation wavelength towards red end results in increase in anisotropy value at a particular protein concentration, then the probe is expected to exist in a motionally restricted environment. The shifting of the excitation wavelength towards red end of the absorption spectra selectively excites the solvent stabilized fluorophore population. These give a more vivid picture of the surrounding atmosphere while emitting from the excited state. Since for biological systems, solvent is water and hydration plays a predominant role in most of the cellular processes, these techniques come in handy to monitor directly the environment and dynamics around a fluorophore in a complex biological system.33 A linear increase of anisotropy values with excitation wavelength is observed in case of EDMANA-HSA binding (Fig. 4b). This arises due to slow rates of reorientation of the solvent molecules around the probe fluorophore in the excited state in the organized media, i.e., at the hydrophobic binding site of the protein which is basically a motionally restricted environment.
Red Edge Excitation Effect
The Red Edge Excitation effect is a set of wavelength sensitive tools for directly monitoring the solvation dynamics within the organized media.33 The shifting of the emission maxima to the red end upon shifting of the excitation wavelength to the red end of the absorption spectrum of the probe is termed as the ‘Red Edge Excitation Shift’ or REES. This effect is observed in case of fluorophores with increased dipole moment values in the excited state than in their ground state. Shifting the excitation wavelength to the red end of the absorption spectra selectively excites the fluorophore population that is more solvent stabilized in the ground state. In an organized media, the solvent dipoles experience greater restriction on their mobility and hence take longer time to reorient themselves around the excited fluorophore. The time scale lies within or is greater than the lifetime of the excited fluorophore. In pure solvents, we get emission from the solvent relaxed state of the excited fluorophore. But, for the organized media, this Red Edge Effect is observed due to slower relaxation rates of the solvent dipoles.33 Solvatochromism measurement of EDMANA supports high dipole moment for the CT state and the calculated dipole moment for the ground and excited states are found to be 4.69D and 13.67D, respectively.28 It is found that the probe molecule EDMANA exhibits a shift of the emission maxima towards the red end with shift in the excitation wavelength towards red end. As can be seen in Fig. 5, the REES increase with increase of HSA concentration and reaches maxima shifts to 4 nm at 10 μM HSA. An increase in REES values with increase of HSA concentration indicates greater restriction on the mobility of the probe bound within the hydrophobic cavity of the protein.33
 |
| | Fig. 5 Plot of REES values vs. concentration of HSA (in μM). | |
Time-resolved measurements
Fluorescence lifetimes also throw further light on the microenvironment surrounding the excited probe molecules in the proteinous media.12,15,18,19 The fluorescence decay curves for such binding processes in heterogeneous media are generally multiexponential.15,18,19 For our system, the fluorescence decay curves were fitted best to triple exponential function with acceptable values for χ2 (Table 2). As seen in the decay curve (Fig. 6), it is found that the fluorescence lifetime of EDMANA increases with increasing HSA concentration. The origin of this triple exponential decay is not very clear. But, in analogy to other such studies, it can be said that different components may arise due to binding of the probe to different binding sites of the protein.19 The increase in lifetime is due to decrease in non-radiative decay channels by encapsulation of the probe molecules in a hydrophobic cavity where the probe is less exposed to water.18 Though the lifetime of the probe molecules in aqueous buffer could not be obtained due to the ultrafast nature of the CT process, the lifetimes of the probe bound to HSA is within the resolution limit (90ps) of the instrument. Higher fluorescence lifetime of the probe with increasing HSA concentration (Table 2) indicates that as more and more the protein molecules go into the medium, the number of free or unbound EDMANA molecules decreases. This also explains the enhancement of emission intensity of the probe with increase of protein concentration.
Table 2 Fluorescence lifetimes of the CT band of EDMANA with increasing HSA concentration
| Environment |
a1 |
τ
1/ns |
a2 |
τ
2/ns |
a3 |
τ
3/ns |
χ2 |
〈τ〉/ns |
| HSA (4 μM) |
0.070 |
0.518 |
0.086 |
0.141 |
0.021 |
1.62 |
1.11 |
0.465 |
| HSA (100 μM) |
0.062 |
0.704 |
0.053 |
0.259 |
0.036 |
1.84 |
1.58 |
0.818 |
 |
| | Fig. 6
Fluorescence decay curves of EDMANA with increasing HSA concentration (λext = 408 nm). | |
Though the non-radiative pathways become less operative in the proteinous environment, they do not cease completely as is evident from the quantum yield values (Table 3). Radiative (kr) and non-radiative (knr) rate constants were calculated using the following relations15,18 (Table 3):
where Φ
f and
τf are the fluorescence quantum yield and average fluorescence lifetime, respectively. The value of
knr indicates the presence of non-radiative channels to a lesser extent than in pure aqueous medium. Stronger the binding, lesser is the probability of deactivation
via non-radiative pathways as is observed in case of aqueous solution due to hydrogen bonding deactivating channels. Hence, radiative rate constant increases and non-radiative rate constant decreases with increase of HSA concentration.
Table 3 Quantum yields and rate constant for radiative and non-radiative decay in different HSA concentrations
| Environment |
Quantum yield Φf |
k
r/s−1 |
k
nr/s−1 |
| Aqueous buffer |
0.003 |
— |
— |
| HSA (4 μM) |
0.015 |
0.032 × 109 |
2.12 × 109 |
| HSA (100 μM) |
0.052 |
0.064 × 109 |
1.16 × 109 |
Steady state acrylamide quenching of EDMANA-HSA fluorescence
The well-known quencher acrylamide is used to obtain an idea about the probable location of the probe within the hydrophobic cavity of HSA.19Acrylamide shows an unusually high degree of static quenching of tryptophanyly fluorescence of Serum Albumins.34Fig. 7 shows that addition of acrylamide to EDMANA-HSA mixture results in decrease of fluorescence intensity of ∼478 nm band (HSA concentration maintained at 100 μM). Acrylamide releases probe molecules from the hydrophobic sites by approaching close to the site.19 A blue shift of the emission maxima at higher concentration of acrylamide (by ∼3 nm at 2 M acrylamide concentration) indicates that as more and more quencher molecule is added to the medium, the probe molecules lying in more exposed locations within the hydrophobic cavity is expelled and the spectral shift to the blue side is due to fluorescence from the deeply buried probe molecules. However, from this we cannot predict the exact binding site of the probe in the hydrophobic interiors of the protein. The quencher concentration is increased upto 2 M and the plot of I0/I vs. [Acrylamide] gives a straight line (Fig. 7 inset). The linearity is maintained even at higher concentrations of quencher unlike the case of Nile Red bound to HSA where heterogeneity is reported in the quenching mechanism at higher values of quencher concentration.19 The Stern–Volmer quenching constant, KSV is determined to be 1.02 mol−1 L from the slope of the plot (Table 1).
Study of urea denaturation of HSA using EDMANA
Urea is a well known denaturant which disrupts the tertiary structure of proteins.35–37 The process of protein unfolding is a complicated one and occurs through a diverse array of mechanisms.38–40 It can be studied by various methods such as far-UV circular dichroism spectroscopy or by monitoring the intrinsic fluorescence of protein tryptophan. Several studies have reported that the denaturant-induced unfolding of multidomain protein HSA occurred through co-operative two-step process involving only the native and unfolded states.38,39 However, monitoring the spectral characteristics of an extrinsic probe bound to HSA can also serve as an effective tool for studying the denaturation process. As seen in Fig. 8a, the position of emission maxima of EDMANA bound to HSA gradually shifts to the red side with increasing concentration of urea from 0–10 M. The spectral shift is about 33 nm and this indicates a drastic increase in polarity of the microenvironment of the probe. As seen in Fig. 8b, the plot of emission maxima of the probe bound to protein vs. urea concentration shows a sharp change in peak position at unfolding transition region between 4–7 M urea which is in agreement with the literature data.37 The peak of the emission maxima of the probe stands at ∼516 nm at 10 M urea concentration and this is quite close to the position of the emission maxima of the probe observed in aqueous buffer (521 nm). This indicates that the probe molecules bound to the hydrophobic interiors of the protein are gradually exposed to the aqueous phase with increasing concentration of urea. As the protein structure gets disrupted, the probe molecules are no more within the hydrophobic cavity where exposure to water molecules was less. As water leads to fluorescence quenching of the probe, the shift of emission maxima is accompanied by a decrease in intensity. The polarity experienced by the probe molecules at highest urea concentration is close to that of aqueous medium as is evident from the peak position of the emission maxima and the anisotropy value at 10 M urea (Fig. 8a inset). Anisotropy value obtained at final concentration of urea is very close to that obtained in pure aqueous buffer solution. This spectral behavior well point out that the probe binds and remains encapsulated within the protein cavity and urea induced denaturation leads to greater exposure of the probe to aqueous phase.
 |
| | Fig. 8 (a) Variation in fluorescence emission spectra of EDMANA-HSA complex and (inset) variation of steady state anisotropy values with increasing concentration of urea (0–10 M); (b) Plot of emission maxima of EDMANA-HSA complex at different concentrations of urea (0–10 M). | |
Conformational changes of HSA in AOT reverse micelles
Reverse micelles, which are formed by dissolving surfactant in organic solvent, provide an artificial system that mimics the biological systems.41 In the reverse micelles, the non-polar tails are directed towards the organic phase and the polar head groups cluster together enclosing a water pool. Recently, a lot of attention is paid towards the study of protein solubilized in these reverse micelles formed in the non-polar media.42,43 There is a direct relationship between w0 and size of the reverse micelles. The structure of the water molecules within the micelle is determined by the micellar size; if w0 increases, the amount of ‘free’ water relative to that of ‘bound’ water also increases.44 Depending on w0 value the radius of the AOT reverse micellar nanocavity can vary over a wide range and it allows encapsulation of complex molecules within the water core. The size of reverse micelle is given by
i.e.w0 is the water/AOT mole ratio.
It is reported that the diameter of the AOT reverse micellar nanocavity is 8 nm at w0 = 11 and HSA is proposed to have side of 8 nm and depth of 3 nm.1 From this comparison of size, it can be said that the HSA molecule solubilized in water pool of reverse micelle must come in contact with the hydrophilic side chains of the surfactant and there is an electrostatic interaction between them which is of considerable importance to protein structure in reverse micelles. The property of the water pool in the reverse micelle is different from the water forming the outside layer of the pool. It is known that the charges of the surfactant tail groups play a predominant role at the outside layer.
The study of conformational change of BSA incorporated in AOT reverse micelle was carried out by Takeda et al.45 by monitoring the position of fluorescence emission maxima of tryptophan of BSA. It is reported that at low values of w0, λmax shifts to lower wavelength around 322 nm, but with increasing w0, λmax shifts to higher wavelength. Study of the spectral characteristics of Trp-214 in HSA fails to predict the actual polarity experienced by the protein molecule in the water pool of the reverse micellar cavity.19 In fact the peak position remains more or less unaffected even at high w0 values. This is because the tryptophan residue lies deeply buried within the hydrophobic pocket of the protein and experiences changes in microenvironment to a lesser extent.1 The actual picture can be obtained by monitoring the spectral characteristics of external probe EDMANA bound to HSA solubilized in the water pool of AOT reverse micelle. Water pool is created by injecting a solution of probe and protein into the surfactant dissolved in the non-polar phase. The pool size is increased by adding calculated amounts of water (in this case buffer) to the created pool. For our experiment, AOT concentration is maintained at 0.1 M and concentration of HSA is maintained at 100 μM. From initial value w0 = 1 the pool size is increased up to w0 = 48. Increasing pool size results in shifting of the position of emission maxima from 438 nm to 452 nm, i.e., 14 nm towards the red side with concomitant decrease in intensity at the emission maxima of the EDMANA-HSA complex (Fig. 9a and 9b). The variation of emission maxima and intensity with increasing water pool size (w0) is shown in Fig. 9c. These spectral variations with increasing pool size are indicative of greater polarity experienced by EDMANA bound to HSA. A similar observation was also reported by Davis et al.19 for Nile red bound to HSA in AOT reverse micelle and fluorescence of N-acetyl-tryptophanamide reported earlier which said that the microenvironment of the probe becomes more polar with increasing w0.46 Thus, the conformational change of HSA in a reverse micellar medium is efficiently monitored using extrinsic probe EDMANA.
 |
| | Fig. 9 (a) Steady state fluorescence spectra of EDMANA bound to HSA in 0.1 M AOT/n-heptane reverse micellar medium with increasing w0 values; (b) normalised spectra showing the shift in the position of emission maxima with increasing w0 values and (c) plot of variation of the position of emission maxima and emission intensity of EDMANA bound to HSA in 0.1 M AOT/n-heptane reverse micellar medium with increasing w0 values. | |
| | 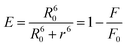 | (1) |
where E is the efficiency of energy transfer; F and F0 are the fluorescence intensities of donor in presence and absence of the acceptor respectively. R0 is expressed as| |  | (2) |
where k2 is the spatial orientation factor; n is the refractive index of the medium, ϕD is the fluorescence quantum yield of the donor and J is the overlap integral of emission spectrum of donor and absorption spectrum of acceptor (Fig. 10a). The term J is expressed as| |  | (3) |
where F(λ) is the normalized fluorescence intensity in the range λ and λ + Δλ; ε(λ) is the extinction coefficient of acceptor at λ. The overlap integral J is obtained by integrating the spectra in Fig. 10a. Using the values of k2 = 2/3, n = 1.333 and ϕD = 0.15,21 the data obtained are as follows: J = 3.54 × 10−13 cm2, R0 = 4.63 nm and r = 3.98 nm. The calculated distance between the probe and protein indicates that the probe is strongly bound to the protein molecules as is reflected in the binding constant of the probe-protein complex.
Fig. 10b shows the variation of fluorescence spectra of Trp-214 of HSA with increasing concentration of acceptor EDMANA. With increase in concentration of EDMANA, it is seen that the emission intensity of tryptophan decreases with the emergence of a new band at ∼480 nm. This implies that the radiation emitted by tryptophan of HSA is transferred to the probe EDMANA by the process of FRET leading to an increase in emission intensity of the probe molecule. The efficiency of energy transfer from tryptophan to the fluorescence probe is found to be ∼70% (Table 1), i.e., the energy transfer efficiency from tryptophan to the probe is quite high.
Conclusion
In the present work we have studied the binding of an external charge transfer fluorescence probe EDMANA with HSA protein by monitoring the characteristic fluorescence peak of the probe using steady state and time resolved fluorescence measurements. We have investigated structural and dynamical aspects of protein HSA in different micro-environments using the above extrinsic charge transfer fluorescence probe. The steady state anisotropy and REES values point towards the fact that the probe molecule remains bound within the hydrophobic cavity of the protein. Fluorescence quenching of EDMANA bound to HSA by acrylamide and FRET studies throw light on the probable location of the probe molecules within the hydrophobic cavity of HSA and the energy transfer efficiency from Trp-214 of HSA to the probe molecules. Increase in fluorescence lifetime with increase of protein concentration reinforces our conclusion that binding does occur between probe and protein. Urea denaturation of HSA has been monitored by the result of greater exposure of the probe molecules to the aqueous phase and the decrease in intensity with shifting of the emission maxima of the CT band towards the red end. The emission of EDMANA bound to HSA is also helpful in studying the conformational changes in HSA in AOT/n-heptane reverse micellar medium. Over all, the charge transfer fluorescence probe EDMANA can serve well as an extrinsic probe for the study of proteinous environment.
Acknowledgements
NG gratefully acknowledges the financial support received from Department of Science and Technology, India (Project No. SR/S1/PC-1/2003). RBS and SM thank CSIR, New Delhi for research fellowship. The authors are thankful to Dr Nilmoni Sarkar and Mr Debabrata Seth of Department of Chemistry, IITKGP for fluorescence lifetime measurements.
References
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef.
-
J. R. Brown, Albumin Structure, Function and Uses, ed. V. M. Rosenoer, M. Oratz, and M. A. Rothschild, Pergamon Press, Oxford, 1977, 27 Search PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CAS.
- D. C. Carter, X. M. He, S. H. Munson, P. D. Twigg, K. M. Gernert, M. B. Broom and T. Y. Miller, Science, 1989, 244, 1195–1198 CrossRef CAS.
- C. B. Berde, B. S. Hudson, R. D. Simoni and L. A. Sklar, J. Biol. Chem., 1979, 254, 391–400 CAS.
- E. L. Gelamo and M. Tabak, Spectrochim Acta A, 2000, 56, 2255–2271 CrossRef.
- E. L. Gelamo, C. H. Silva, H. Imasato and M. Tabak, Biochim. Biophys. Acta, 2002, 1594, 84–99 CrossRef CAS.
- H. Gharibi, S. Javadian and M. Hashemianzadeh, Colloids Surf. A Physicochem. Eng. Asp., 2004, 232, 77–86 CrossRef CAS.
- I Nowak and L. M. Shaw, Clin. Chem., 1995, 41, 1011–1017 CAS.
- R. Zini, D. Morin, P. Jouenne and J. P. Tillement, Life Sci., 1988, 43, 2103–2115 CrossRef CAS.
- M. Valle, M. Estaban, J. M. Rodriquez-Sasiain, R. Calvo and C. Aquirre, Res. Commun. Mol. Pathol. Pharmacol., 1996, 94, 73–88 Search PubMed.
- Y. Moriyama, D. Ohta, K. Hachiya, Y. Mitsui and K. Takeda, J. Protein Chem., 1996, 15, 265–271 CrossRef CAS.
- F. Moreno, M. Cortijo and Gonzalez-Jimenez, Photochem. Photobiol., 1999, 69, 8–15 CAS.
- U. Kragh-Hansen, Mol. Pharmacol., 1988, 34, 160–171 CAS.
- A. Barik, K. I. Priyadarsini and H. Mohan, J. Photochem., 2003, 77, 597–603 Search PubMed.
- A. Mallick and N. Chattopadhyay, Biophys. Chem., 2004, 109, 261–270 CrossRef CAS.
- G. N. Valsami, P. E. Macheras and M. A. Koupparis, Pharma. Res., 2004, 8, 888–892 Search PubMed.
- A. Mallick, B. Haldar and N. Chattopadhyay, J. Phys. Chem. B, 2005, 109, 14683–14690 CrossRef CAS.
- D. M. Davis and D. J. S. Birch, J. Fluoresc., 1996, 6, 23–32 CrossRef CAS.
- F.-Y. Wu, Z.-J. Ji, Y.-M. Wu and X.-F. Wan, Chem. Phys. Lett., 2006, 424, 387–393 CrossRef CAS.
- H. M. Rowel, K. Meidtner and J. Kroll, J. Agri. Food Chem., 2005, 53, 4228–4235 CrossRef.
- M. I. Kaldas, U. K. Walle, H. von der Woude, J. M. McMillan and T. Walle, J. Agri. Food Chem., 2005, 53, 4194–4197 CrossRef CAS.
- A. Papadopoulou, R. J. Green and R. A. Frazier, J. Agri. Food Chem., 2005, 53, 158–163 CrossRef CAS.
- A. Buzady, J. Savolainen, J. Erostyak, P. Myllyperkio, B. Somogyi and J. Korppi-Tommola, J. Phys. Chem. B, 2003, 107, 1208–1214 CrossRef CAS.
- J. K. Amisha Kamal, L. Zhao and A. H. Zewail, Proc. Natl. Acad. Sci., 2004, 101, 13411–13416 CrossRef CAS.
- H. Jun, S. Y. Hong, S. S. Yoon, C. Kang and M. Suh, Chem. Lett., 2004, 33, 690 CrossRef CAS.
- Y. Suzuki and K. Yokoyama, J. Am. Chem. Soc., 2005, 127, 17799–17802 CrossRef CAS.
- R. B. Singh, S. Mahanta, S. Kar and N. Gucchait, Chem. Phys., 2007, 342, 33–42 CrossRef CAS.
- S. Mahanta, R. B. Singh, S. Kar and N. Guchhait, J. Photochem. Photobiol., A, 2008, 194, 318–326 CrossRef CAS.
- A. Chakraborty, D. Seth, P. Setua and N. Sarkar, J. Phys. Chem. B, 2006, 110, 16607–16617 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- S. Nigam and G. Durocher, J. Phys. Chem., 1996, 100, 7135–7142 CrossRef CAS.
- S. Mukherjee and A. Chattopadhyay, J. Fluoresc., 1995, 5, 237–246 CrossRef CAS.
-
Topics in Fluorescence Spectroscopy, Vol. 2. Principles, ed. M. R. Eftink and J. R. LakowiczPlenum Press, New York, 1991, pp. 53–126 Search PubMed.
- A. Wallqvist, D. G. Covell and D. Thirumalai, J. Am. Chem. Soc., 1998, 120, 427–428 CrossRef CAS.
- B. Ahmed, M. K. A. Khan, S. K. Haq and R. H. Khan, Biophys. Biochem. Res. Commun., 2004, 314, 166–173 Search PubMed.
- J. Gonzalez-Jimenez and M. Cortijo, J. Protein Chem., 2002, 21, 75–79 CrossRef CAS.
- K. Wallevik, J. Biol. Chem., 1973, 248, 2650–2655 CAS.
- S. Muzammil, Y. Kumar and S. Tayyab, Proteins, 2000, 40, 29–38 CrossRef CAS.
- S. S. Krishnakumar and D. Panda, Biochemistry, 2002, 41, 7443–7452 CrossRef CAS.
- G.-G. Chang, T.-M. Huang and H.-C. Hung, Proc. Natl. Sci. Counc. ROC (B), 2000, 24, 89–100 Search PubMed.
- K. D. Tapas and A. Maitra, Adv. Colloid Surface Sci., 1995, 59, 95–193 Search PubMed.
- L. Magid, P. Walde, G. Zampieri, E. Battistel, Q. Peng, E. Trotta, M. Maestro and P. L. Luisi, Colloids Surf., 1988, 30, 193–207 CAS.
- M. Belletete, M. Lachapelle and G. Durocher, J. Phys. Chem., 1990, 94, 7642–7648 CrossRef CAS.
- K. Takeda and Y. Moriyama, Curr. Top. Colloid. Interface Sci., 1997, 1, 109–135 Search PubMed.
- S. Ferreira and E. Gratton, J. Mol. Liq., 1990, 45, 253–272 CrossRef CAS.
- P. R. Selvin, Nat. Struct. Biol., 2000, 7, 730–734 CrossRef CAS.
- B. Sengupta and P. K. Sengupta, Biochem. Biophys. Res. Comm., 2002, 299, 400–403 CrossRef CAS.
- Y. J. Hu, W. Li, Y. Liu, J. X. Dong and S. S. Qu, J. Pharm. Biomed. Anal., 2005, 39, 740–745 CrossRef CAS.
- K. Sahu, S. Mondal, K. Ghosh, D. Roy and K. Bhattacharyya, J. Chem. Phys., 2006, 124, 124909–124915 CrossRef.
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 


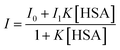

![Variation of fluorescence emission spectra of EDMANA-HSA complex with increasing acrylamide concentration in 100 μM HSA; Inset: Stern–Volmer plot of I0/Ivs. [acrylamide] in HSA.](/image/article/2009/PP/b814050b/b814050b-f7.gif)

