Bicarbonate-enhanced transformation of phenol upon irradiation of hematite, nitrate, and nitrite†
Serge Chirona, Stéphane Barbatia, Swapan Khanrabc, Binay K. Duttabd, Marco Minellac, Claudio Mineroc, Valter Maurinoc, Ezio Pelizzettic and Davide Vione*c
aLaboratoire Chimie Provence, Aix-Marseille Universités-CNRS (UMR 6264), 3 place Victor Hugo, 13331, Marseille cedex 3, France
bDepartment of Chemical Engineering, University of Calcutta, 92 Acharya Prafulla Chandra Road, 700009, Kolkata, India
cDipartimento di Chimica Analitica, Università di Torino, Via Pietro Giuria 5, 10125, Torino, Italy. E-mail: davide.vione@unito.it; Fax: +39-011-6707615; Tel: +39-011-6707633 Web: http://naturali.campusnet.unito.it/cgi.bin/docenti.pl/Show?_id=vione
dChemical Engineering Department, University of Technology Petronas, 31750, Tronoh, Perak Darul Ridzuan, Malaysia
First published on 29th October 2008
Abstract
Bicarbonate enhances the transformation of phenol upon irradiation of hematite, and phenol nitration upon irradiation of both nitrate and nitrite. Hematite under irradiation is able to oxidise the carbonate ion to the CO3−˙ radical, which in turn oxidises phenol to the phenoxyl radical faster compared to the direct photo-oxidation of phenol by hematite. The formation of CO3−˙ from hematite and carbonate under irradiation is supported by the detection of 3,3′-dityrosine from tyrosine, added as a probe for CO3−˙. It is shown that Fe(III) might be an important photochemical source of CO3−˙ in Fe-rich waters, e.g. waters that contain more than 1 mg L−1 Fe. The enhancement by bicarbonate of phenol nitration upon nitrate irradiation is probably accounted for by an increased photogeneration rate of nitrogen dioxide. The process could lead to enhanced phenol photonitration by nitrate in waters rich of inorganic carbon (>10 mM bicarbonate). Bicarbonate also increases the transformation and nitration rates of phenol upon nitrite photolysis. The effect is due to the combination of basification that enhances phenol nitrosation and nitration, and of peculiar bicarbonate chemistry. It is shown that bicarbonate-enhanced phenol nitration upon nitrite photolysis could be a significant photonitration pathway, leading to the generation of toxic nitrated compounds in natural waters in which the scavenging of hydroxyl radicals by nitrite is competitive with that of Dissolved Organic Matter (DOM).
Introduction
Photochemical processes in surface waters are important pathways for the transformation of dissolved compounds, including biorefractory pollutants1,2 and natural organic matter.3–5 They consist of direct photolysis of sunlight-absorbing molecules,6–8 transformation photosensitised by dissolved organic matter (DOM),9–11 and reaction with radical species that are produced by photochemical processes, such as ˙OH, CO3−˙, Cl2−˙, and ˙NO2.12–17Bicarbonate is the most abundant anion in freshwater. Its key role in acid–base equilibria has long been acknowledged, and more recent research has shown that it also interacts with surface-water photochemical processes. Bicarbonate and carbonate can scavenge reactive radical species such as ˙OH,18 producing the radical CO3−˙ that is involved in the degradation of electron-rich aromatics and of sulfur-containing compounds.13,14,19 Actually CO3−˙ is less reactive than ˙OH and other transients toward degradation processes,20,21 but its reactions with the radical scavengers that naturally occur in surface waters are also slower. The carbonate radical can therefore reach a higher steady-state concentration than ˙OH in the aquatic systems.19 Addition of bicarbonate has been shown to slightly inhibit the photodegradation of a series of xenobiotic compounds (atrazine, fluometuron, hexazinone, bensulfuron methyl, ciprofloxacin, clofibric acid10), to have no effect on that of the insecticide fipronil,9 and to significantly enhance the photoinduced transformation of dimethyl sulfide upon nitrate photolysis.22
The radical CO3−˙ could reach a higher steady-state concentration in surface waters compared to other reactive transients, not only because of slower consumption, but also due to further sources in addition to ˙OH + HCO3−/CO32−. The oxidation of carbonate by the excited triplet states of DOM could contribute some 10% to the overall generation rate of CO3−˙.19 To our knowledge, no data were available on the possible production of CO3−˙ from carbonate/bicarbonate and the Fe(III) (hydr)oxides under irradiation. It has been demonstrated that inorganic anions such as nitrite23 and chloride16 can enhance the degradation of organic substrates (phenol, carbamazepine) in the presence of irradiated Fe(III) oxide colloids. Substrate degradation upon irradiation of the oxide colloid alone could take place by charge transfer or via the ˙OH radicals produced by photolysis of the surface FeIII-OH groups of the oxide. However the efficiency of ˙OH photoproduction by Fe(III) oxides is rather low,24 almost negligible compared to the photoactive complex FeOH2+.25 Furthermore, charge-transfer reactions can be kinetically hampered with many organic compounds.24 In contrast the one-electron oxidation of nitrite to ˙NO2 and of chloride to Cl2−˙ by the irradiated oxides would be much faster, and the radicals thus formed could react with the organic substrates and considerably enhance their degradation.16,23 Bicarbonate and carbonate as major anions in freshwater could be involved in similar reactions. For this reason the effect of NaHCO3 on the kinetics of phenol degradation by irradiated hematite (α-Fe2O3) was studied in the present work. Hematite was adopted as a model of the Fe(III) (hydr)oxide colloids present in natural waters, which are photochemically active upon absorption of sunlight.26
Additionally, to our knowledge there is very limited information concerning the effect of bicarbonate on the photochemical production of harmful transformation intermediates from a given substrate. Nitrophenols from phenol nitration are an example of this kind of intermediates, and they can be formed in surface waters under irradiation.13 For this reason, the present work considers the effect of bicarbonate on the photogeneration of toxic nitrated phenols from phenol upon UV irradiation of nitrate and nitrite.
Experimental
Reagents and materials
Phenol (purity grade >98%), 2-nitrophenol (2-NP, 98%), 4-nitrophenol (4-NP, >99%), 4-nitrosophenol (4-NOP, 60%), tyrosine (99%), N,N-dimethyl-1-naphthylamine (99%), HCl (37%), NaH2PO4× H2O (>98%) and NaHCO3 (99.7%) were purchased from Aldrich; horseradish peroxidase from Sigma; NaNO2 (>99%), NaOH (99%), Na2HPO4× 2H2O (99.5%), and acetonitrile (Lichrosolv gradient grade) from VWR Int.; sulfanilic acid (>99%), 2,2′-dihydroxybiphenyl (>98%), 4,4′-dihydroxybiphenyl (>98%), and 4-phenoxyphenol (>95%) from Fluka. These reagents were used as received without further purification. The synthesis of α- Fe2O3 was carried out following the procedure of Leland and Bard.27 The radius distribution of the primary hematite particles was measured, after sonication of the suspension for 1 h, with an ALV-NIBS “High Performance Particle Sizer” Laser Light Scattering apparatus, connected to a ALV-5000/EPP Multiple Tau Digital Correlator. Particle radii were centred at around 170 nm, and the radius distribution plot is shown in ESI,† Fig. S1.Irradiation experiments
Irradiation was carried out in cylindrical Pyrex glass cells (4.0 cm diameter, 2.3 cm height), containing 5 mL of aqueous solution or suspension. The optical path length inside the cells was 0.4 cm. The solutions and the suspensions were magnetically stirred during irradiation. The stirring rate was limited not to perturb the surface layer via vortex formation. Anyway, it was sufficient to keep the suspension stable for the adopted irradiation time.Three different lamps were used for the irradiation, depending on the set of experiments. Hematite was irradiated under blue light using a 40 W Philips TL K03 lamp with emission maximum at 435 nm. Total photon flux in the cells was 2.2 × 10−7 einstein s−1, actinometrically measured with the ferrioxalate method.28 Such a photon flux is around 1.5 times higher than the flux of sunlight at mid-latitude during summertime at noon, in the relevant wavelength interval of 400–450 nm.29
Selective excitation of nitrate was obtained under a 100 W Philips TL 01 lamp with emission maximum at 313 nm. The photon flux in the cells measured by actinometry was 1.8 × 10−8 einstein s−1. Irradiance in the 290–400 nm wavelength interval was 5.6 W m−2, measured with a CO.FO.ME.GRA. (Milan, Italy) power meter.
Selective excitation of nitrite was carried out with an array of three 40 W Philips TL K05 lamps, with emission maximum at 365 nm. Photon flux in the cells was 1.2 × 10−7 einstein s−1, and irradiance between 290 and 400 nm was 32 W m−2. The emission spectra of the lamps and the absorption spectra of hematite, nitrate and nitrite are reported in ESI†, Fig. S2–S4. Emission spectra were measured with an Ocean Optics SD2000 CCD spectrophotometer, absorption spectra with a Varian Cary 100 Scan UV-Vis spectrophotometer.
Some runs were carried out under N2 atmosphere, obtained by purging the cells for 20 min prior to irradiation with a gentle flow of high-purity nitrogen (99.995%, purchased from SIAD, Bergamo, Italy). After irradiation the solutions were directly analysed, while the suspensions were first filtered through 0.45 μm Millipore Millex-LCR hydrophilic PTFE filter membranes. Solution pH was measured with a Metrohm 713 pH meter.
HPLC-DAD determinations
Sample analysis after irradiation was carried out with a VWR-Hitachi Elite liquid chromatograph coupled to a photodiode-array detector (HPLC-DAD). The instrument was equipped with L-2200 autosampler, L-2130 pump for low-pressure gradients, L-2300 column oven, Merck Hibar RT 250-4 column (25 cm length, 0.4 cm diameter) packed with LiChrospher 100 CH-18/2 (10 μm particle diameter), and L-2455 Diode Array Detector. Sample volume was set at 60 μL, column temperature at 40 °C, and isocratic elution was carried out at 1.0 mL min−1 flow rate with a 40:60 mixture of acetonitrile: aqueous H3PO4 (pH 2.8). Under these conditions the retention times were (min): phenol (4.59), 2-nitrophenol (8.75), and 4-nitrophenol (5.23). The DAD detector was set to acquire signal in the 200–300 nm interval, and 210 nm was used as the wavelength for quantification. For qualitative identification, the absorption spectra of the peaks corresponding to the two nitrophenols were compared with those of authentic standards.Some additional runs were carried out in the presence of nitrobenzene and nitrite. Nitrobenzene degradation was HPLC monitored by 1.0 mL min−1 elution with a 55:45 mixture of acetonitrile: aqueous H3PO4 (pH 2.8). The retention time under these conditions was 5.93 min and the detection wavelength was 264 nm.
HPLC-MS determinations
Whenever possible, the identification of phenol by-products in different irradiation experiments was carried out by comparing their full-scan MS spectra and their retention time with those of authentic standards. Negative atmospheric pressure chemical ionisation ((−)-APCI) and positive electrospray ((+)-ESP) mass spectrometry were performed on an Esquire 6000 ion trap system (Bruker, Bremen, Germany). By-product mass spectra were mainly characterised by their pseudo-molecular ions [M + H]+ or [M − H]− as base peak in positive and negative ionisation mode, respectively. The analytical separation was carried out with a Superspher 100 C-18 column (250 × 4 mm i.d., 5 μm) from Merck at a flow rate of 0.2 mL min−1. The mobile phase consisted of a binary mixture of solvents A (methanol) and B (water). The gradient was operated from 25% to 100% A for 25 min and then back to initial conditions in 5 min.Nitrosophenols were separated using a 100 × 2.1 mm i.d, 5 μm PGC Hypercarb column from Hypersil (Shandom, UK). Elution was carried out with methanol/acetonitrile/0.2 M formic acid (40/40/20 v:v) in the isocratic mode. In these conditions the retention times were 6.2 and 7.7 min for 4- and 2-nitrosophenol, respectively. MS detection was carried out as for the already-described analyses with the C-18 column, adopting (−)-APCI ionisation. The quantification of 4-nitrosophenol was carried out by comparison with an authentic standard. 2-Nitrosophenol was quantified under the hypothesis that its MS response was the same as for the 4-isomer.
Synthesis and determination of 3,3′-dityrosine
3,3′-Dityrosine was synthesised by incubating 0.5 mM L-tyrosine, 0.2 mg mL−1 (4.5 μM) horseradish peroxidase, and 500 μM H2O2 in 50 mM phosphate buffer (pH 7.4) for 20 min at 25 °C. The resulting mixture was centrifuged to remove the enzyme.30 The identification of tyrosine and 3,3′-dityrosine after the irradiation experiments was carried out using (+)-ESP mass spectrometry. The analytical separation was carried out with a MetaChem C-18 column (150 × 2 mm i.d., 3 μm) from Varian at a flow rate of 0.2 mL min−1. The mobile phase consisted of a binary mixture of solvents A (methanol) and B (0.1% formic acid in H2O). The gradient was operated from 0% to 50% A in 30 min and then back to the initial conditions in 5 min.Results and discussion
Irradiation of hematite with phenol
Blue light was adopted for the irradiation of hematite because it is absorbed by the oxide,27 and shows higher penetration in the aquatic systems compared to the UV.31 The initial degradation rates of 25 μM phenol, in the presence of 450 mg L−1 hematite (α-Fe2O3) and/or of 0.010 M NaHCO3, upon irradiation under blue light (λmax = 435 nm) are reported in Table 1, entries no. 1–3 (also see ESI,† Fig. S5, for the relevant time evolutions). In aerated solution the degradation rate of phenol was higher by at least one order of magnitude with hematite + bicarbonate than in the presence of the separate components. Moreover, the removal of dissolved oxygen under a gentle N2 flow decreased the degradation rate of phenol by around one order of magnitude (see Table 1, entry no. 4, and ESI,† Fig. S5).| No. | Conditions | pH | Phenol degr. rate/M s−1b |
|---|---|---|---|
| a Initial phenol concentration = 2.5 × 10−5 M, irradiation under blue light (lamp emission maximum at 435 nm).b Rates calculated as kC0 by fitting the phenol time evolution curves with the pseudo-first order exponential function Ct = C0 exp(−kt), where Ct is the concentration of phenol at the time t, C0 its initial concentration, and k the peudo-first order degradation rate constant. The error bounds (μ±σ) represent the goodness of the fit to the experimental data (intra-series variability). Reproducibility between repeated runs (3, 3′) was around 20%. | |||
| 1 | α-Fe2O3 450 mg L−1 | 6.0 | (4.3 ± 0.6) × 10−11 |
| 2 | NaHCO3 0.01M | 8.5 | (1.2 ± 0.4) × 10−10 |
| 3 | NaHCO3 0.01M + α-Fe2O3 450 mg L−1 | 8.4 | (1.6 ± 0.1) × 10−9 |
| 3′ | NaHCO3 0.01M + α-Fe2O3 450 mg L−1 | 8.4 | (1.3 ± 0.1) × 10−9 |
| 4 | NaHCO3 0.01M + α-Fe2O3 450 mg L−1, N2 atmosphere | 8.4 | (1.0 ± 0.2) × 10−10 |
| 5 | NaOH 0.01M + α-Fe2O3 450 mg L−1 | 12 | (1.4 ± 0.1) × 10−8 |
| 6 | NaH2PO4/Na2HPO4 buffer 0.01M + α-Fe2O3 450 mg L−1 | 5.7 | (3.8 ± 1.2) × 10−11 |
| 7 | NaH2PO4/Na2HPO4 buffer 0.01M + α-Fe2O3 450 mg L−1 | 7.7 | (9.4 ± 1.5) × 10−11 |
| 8 | NaH2PO4/Na2HPO4 buffer 0.01M + α-Fe2O3 450 mg L−1 | 8.4 | (7.3 ± 0.4) × 10−11 |
The addition of 0.01 M bicarbonate increased the pH of the phenol/hematite system from 6.0 to 8.4. Because of the acid–base equilibrium between phenol and the phenolate anion (pKa≅ 10),32 the ratio phenolate/phenol would pass from 1.0 × 10−4 at pH 6.0 to 0.025 at pH 8.4. Phenolate undergoes much faster degradation than phenol in the presence of irradiated hematite (see Table 1, entries 1 and 5), and the possible effect of pH was checked by addition of a phosphate buffer (NaH2PO4 + Na2HPO4, pH 8.4, total concentration 0.01 M). Phenol degradation rate with irradiated hematite and phosphate at pH 8.4 was at least 20 times lower than for hematite + NaHCO3 at the same pH (Table 1, entries 3, 3′ and 8). Phosphate is unlikely to influence significantly the photochemical behaviour of hematite because the photodegradation rate of phenol with phosphate buffer and hematite at pH 5.7 was comparable to that with hematite alone at pH 6.0 (Table 1, entries 1 and 6). These findings suggest that the enhancement of phenol photodegradation by bicarbonate cannot be accounted for by a mere basification of the solution.
Absorption of blue light by hematite (band gap around 2 eV) promotes electrons from the valence to the conduction band, leaving holes in the valence band.33 The valence-band holes of irradiated hematite are rather strong oxidants at neutral pH, with Eh+ = 2.3 V at pH 7,34 and would be able to oxidise the carbonate anion to the carbonate radical (E°(CO3−˙/CO32−) = 1.59 V35). The carbonate anion, which would be around 0.1 mM under the adopted experimental conditions based on the reported acidity constants,32 is known to form complexes on the hematite surface36 where it could undergo one-electron oxidation. Furthermore, the oxidation of the carbonate anion to CO3−˙ has been described in the presence of TiO2 under irradiation.37
The radical CO3−˙ is able to react at a significant rate with phenol (rate constant around 107 M−1 s−1,21), and the enhancement of phenol degradation by NaHCO3 could occur if the process of oxidation of CO32− to CO3−˙ and subsequent reaction with phenol were faster than the direct phenol transformation by irradiated α-Fe2O3. The latter reaction is rather slow (Table 1, entry no. 1), possibly because the monoelectronic oxidation of phenol is kinetically hampered as it requires a concerted proton-electron transfer to yield the phenoxyl radical.38
The oxidation of carbonate by the valence-band holes of α-Fe2O3 needs a parallel scavenging of conduction-band electrons. Electron scavenging could be carried out by surface-adsorbed oxygen species and/or by Fe(III) surface groups. The much higher degradation rate of phenol in aerated solution compared to the N2 atmosphere (Table 1, entries 3, 3′, 4) suggests that oxygen species would play a key role in the enhancement of phenol degradation in the presence of bicarbonate, probably by scavenging of conduction-band electrons.
The identification of the transformation intermediates of phenol by HPLC-MS in the presence of hematite + bicarbonate yielded three peaks with m/z 186 in APCI negative ionisation, and a peak with m/z 185 in ESI positive ionisation (also see ESI,† Fig. S6, S7). The peaks with m/z 185 could be attributed to deprotonated dihydroxybiphenyls and phenoxyphenols (MW 186 when undissociated). Injection of the available commercial standards indicated that 2-2′-dihydroxybiphenyl and 4-phenoxyphenol could possibly be present in the system. The ESI peak with m/z 185 could be a protonated biphenylquinone (MW 184), most likely arising on oxidative dehydrogenation of the corresponding dihydroxybiphenyl. Dihydroxybiphenyls and phenoxyphenols are typical products of the reaction between phenol (C6H5-OH) and the phenoxyl radical C6H5-O˙,39 and they are therefore compatible with a primary oxidation step of C6H5-OH to yield C6H5-O˙. This is the expected reaction in the presence of the radical CO3−˙, which can abstract an H atom from phenol to produce phenoxyl and bicarbonate:21
| CO3−˙ + C6H5-OH → HCO3− + C6H5-O˙ | (1) |
Additional evidence in favour of the oxidation of carbonate to CO3−˙ by irradiated hematite was obtained by use of tyrosine as a probe molecule for CO3−˙. It has been shown that tyrosine selectively yields 3,3′-dityrosine upon reaction with the carbonate radical, and the use of tyrosine as a probe is particularly useful because the carbonate radical is detected with difficulty by ESR spectroscopy.40 Formation of 3,3′-dityrosine was observed upon irradiation of α-Fe2O3 + NaHCO3 with tyrosine (see Fig. 1A). In contrast, the irradiation of α-Fe2O3 + tyrosine without bicarbonate yielded dihydroxyphenylalanine (DOPA) as a major intermediate, and no 3,3′-dityrosine was detected (see Fig. 1B).
 | ||
| Fig. 1 (a) HPLC-ESP-MS chromatogram of the tyrosine + hematite + bicarbonate system under irradiation. (b) HPLC-ESP-MS chromatogram of the tyrosine + hematite system under irradiation (DOPA = dihydroxyphenylalanine). | ||
It is possible to approximately assess the environmental significance of hematite as a source of CO3−˙ under the following hypotheses: (i) the reactivity of hematite is representative of that of Fe species in surface waters (at least among the Fe(III) (hydr)oxides it does not show a peculiarly high photoactivity27); and (ii) the enhancement by bicarbonate of the degradation rate of phenol in the presence of hematite under irradiation is equal to the generation rate of CO3−˙ by hematite. The rate enhancement by bicarbonate is around 1.4 × 10−9 M s−1 (see Table 1, entries 3,3′,8). The used radiation source, emitting 2.2 × 10−7 einstein s−1 over 12.6 cm2 was about 1.5 times more intense than summertime sunlight at noon in the 400–450 nm wavelength interval.41 The experimental pH value of 8.4 is reasonable for surface waters. The adopted hematite had an absorption coefficient of 9 × 10−3 L mg−1 cm−1 at the lamp emission maximum (435 nm; see ESI,† Fig. S2). At 450 mg L−1 loading (corresponding to 315 mg L−1 Fe) and 0.4 cm optical path length, hematite would absorb around 98% of the incident radiation. A 200-time dilution to 1.5 mg L−1 Fe would give 2% as the corresponding fraction of radiation absorption. A concentration of 1.5 mg L−1 Fe is reasonable for Fe-rich water systems,42 and is for instance typical of the Rhône river (southern France) before the delta.16 By scaling for lamp intensity and hematite absorption one would obtain 2 × 10−11 M s−1 as the generation rate of CO3−˙ in a surface water layer containing 1.5 mg L−1 Fe, under summertime noon conditions.
It is possible to make a comparison with the rate of CO3−˙ generation upon oxidation of CO32−/HCO3− by ˙OH, a major process for CO3−˙ production in surface waters.14,19,22 The experimental data of the present work have been obtained with [CO32−] ≈ 10−4 M. Table 2 reports the water composition data ([HCO3−], [CO32−], steady-state [˙OH] under mid-latitude summertime irradiation conditions, namely 22 W m−2 sunlight irradiance in the UV) of a number of surface-water samples with carbonate concentration near 0.1 mM.13,17,43 The use of [˙OH] allows the consideration of the consumption of the hydroxyl radicals by the natural scavengers, first of all DOM. For the relevant samples the rate of CO3−˙ generation upon oxidation of bicarbonate and carbonate by ˙OH was calculated as RCO3−˙ = {[˙OH] (k˙OH,HCO3− [HCO3−] + k˙OH,CO32− [CO32−])}, with k˙OH,HCO3− = 8.5 × 106 M−1 s−1 and k˙OH,CO32− = 3.9 × 108 M−1 s−1.20 The resulting values of RCO3−˙ are reported in Table 2 and are in the range of 0.6–4.1 × 10−11 M s−1, comparable to the 2 × 10−11 M s−1 rate obtained for hematite. Furthermore, 10−4 M carbonate would be present in the Greifensee lake (Switzerland) at pH 9 (upper limit for pH during summer19), with 2 × 10−16 M ˙OH in the surface layer,44 and 1 × 10−11 M s−1 as the corresponding CO3−˙ generation rate.19,44 Accordingly if hematite is representative of the photoreactivity of Fe species toward carbonate, in water bodies where total Fe exceeds 1 mg L−1 the iron-induced photooxidation of carbonate could be an important source of CO3−˙, comparable to the oxidation of CO32−/HCO3− by ˙OH.
| Lake Piccolo, Avigliana, NW Italy | Lake Grande, Avigliana, NW Italy | Rhône river before the delta, S Francea | Paddy fields, Rhône delta, S Francea | Ditch water, Rhône delta, S Francea | |
|---|---|---|---|---|---|
| a Total Fe in these samples has been determined as 1.5, 2.5 and 1.2 mg L−1 Fe, respectively. | |||||
| Literature reference | 13 | 13 | 17 | 43 | 43 |
| pH | 8.6 | 8.8 | 8.3 | 7.2 | 7.8 |
| HCO3−/M | 3.6 × 10−3 | 2.5 × 10−3 | 2.1 × 10−3 | 3.1 × 10−3 | 3.6 × 10−3 |
| CO32−/M | 9.6 × 10−5 | 1.0 × 10−4 | 2.7 × 10−5 | 2.5 × 10−5 | 7.5 × 10−5 |
| [˙OH]/M | 1.6 × 10−16 | 9.7 × 10−17 | 3.8 × 10−16 | 8.3 × 10−16 | 6.9 × 10−16 |
| RCO3−˙/M s−1 | 1.1 × 10−11 | 5.8 × 10−12 | 1.1 × 10−11 | 3.0 × 10−11 | 4.1 × 10−11 |
Irradiation of nitrate with phenol
Fig. 2 shows the initial degradation rate of 25 μM phenol (2A) and the initial formation rates of 2- and 4-nitrophenol (2B) upon UVB irradiation of 10 mM NaNO3, as a function of CNaHCO3. Phenol degradation rate decreased with increasing bicarbonate up to a minimum around 5 mM NaHCO3. Due to the subsequent increase, at 16 mM NaHCO3 the degradation rate of phenol was similar to that observed in the absence of bicarbonate. The initial formation rate of nitrophenols underwent little change up to 5 mM bicarbonate, then it started increasing. | ||
| Fig. 2 Initial rates of (a) phenol transformation and (b) nitrophenol formation, upon UVB irradiation of 10 mM NaNO3, as a function of the concentration of NaHCO3 or of the phosphate buffer (NaH2PO4 + Na2HPO4). Initial phenol was 2.5 × 10−5 M, pH varied from 6 to 8.5 depending on CNaHCO3. | ||
The addition of NaHCO3 to phenol and NaNO3 increased the pH from around 6 to 8.5, but very limited pH variation could be observed above 3–5 mM bicarbonate (see ESI,† Fig. S8). Indeed, pH was almost constant in the CNaHCO3 range where the enhancement effect of bicarbonate was operational. To assess the effect of pH, irradiation was also carried out in the presence of phosphate buffers with CNaH2PO4 + CNa2HPO4 = CNaHCO3, and pH within 0.1 units of that of the corresponding solution of NaHCO3. Fig. 2A shows that the rate of phenol degradation decreased monotonically with increasing phosphate (and pH). The trend of nitrophenols with phosphate (Fig. 2B) is less straightforward, but in all the cases the addition of the phosphate buffer decreased the rate of phenol nitration. All the data therefore suggest that the enhancement effect of bicarbonate above 5 mM concentration is not connected with pH.
The transformation of phenol upon nitrate photolysis is mainly due to reaction with ˙OH, photoproduced in reaction 2.44 The phosphate buffer is not able to alter significantly the steady-state [˙OH] in the system: from the literature rate constants20 one obtains that the percentage of ˙OH scavenging by phosphate would be <1%. The decrease of ˙OH photoproduction with increasing pH could be the consequence of the photoisomerisation of nitrate to peroxynitrous acid/peroxynitrite (pKa≈ 7).46,47 The acid HOONO can quickly be decomposed into the reactive species ˙OH and ˙NO2. Peroxynitrite, formed under basic conditions, is more stable and would not produce ˙OH upon decomposition.48–52 As a consequence, the rate of ˙OH generation would decrease with increasing pH.
| NO3− + hν + H+→˙OH + ˙NO2 | (2) |
| NO3− + hν→ ONOO− | (3) |
| HOONO ⇄ ONOO− + H+ | (4) |
| HOONO →˙OH + ˙NO2 | (5) |
| HOONO → NO3− + H+ | (6) |
A comparison of the rates of phenol photodegradation by nitrate in the presence of phosphate and of bicarbonate (Fig. 2A) shows that the initial decrease is slightly more marked with NaHCO3, possibly because of the significant scavenging of ˙OH by HCO3− and CO32−. In fact, based on the literature reaction rate20 and acidity32 constants, in the presence of 5 mM NaHCO3 10% of ˙OH would react with bicarbonate, 7% with carbonate, and the remaining 83% with phenol. Despite all the inhibitory effects carried out by bicarbonate, above 5 mM NaHCO3 there was an enhancement of both phenol transformation and nitrophenol formation.
In a previous study it has been reported that bicarbonate enhances the photodegradation of dimethyl sulfide upon nitrate photolysis.22 A possible explanation is that an ˙OH scavenger could increase the quantum yield of reaction 2, by consuming the hydroxyl radical when it is still inside the solvent cage. In this way the solvent-cage recombination between ˙OH and ˙NO2 would be inhibited, which would favour the diffusion of the oxidised scavenger (CO3−˙ in the present case) and of ˙NO2 out of the cage. Due to the inhibition of in-cage recombination, the overall generation rate of ˙OH + CO3−˙ + ˙NO2 in the presence of NaHCO3 could be significantly higher than that of ˙OH + ˙NO2 without NaHCO3, which could enhance the degradation and the nitration of phenol. This hypothesis implies that bicarbonate and carbonate would be able to effectively scavenge ˙OH in the solvent cage only for CNaHCO3 > 5 mM, below which they would only scavenge ˙OH in the solution bulk. Also note that bicarbonate concentration values above 5 mM are not uncommon in natural waters (see ESI,† Fig. S9).
An alternative explanation for the bicarbonate effect is the reaction between ONOO−, formed upon nitrate photoisomerisation (reaction 3), and CO2 to yield ˙NO2 and CO3−˙.53–57 Nitrogen dioxide and the carbonate radical would be able to enhance phenol nitration and total transformation, respectively, at elevated CNaHCO3.
The two described processes could well operate together. The minimum of phenol degradation rate as a function of CNaHCO3 would thus be a combination of two opposite trends: the decrease of ˙OH photoproduction by nitrate in neutral to basic pH, coupled with the scavenging of ˙OH by bicarbonate, and the strong enhancement of the generation rate of CO3−˙ at elevated bicarbonate. The parallel enhancement of the generation rate of ˙NO2 under the same conditions would also account for the significant increase of phenol photonitration above 5 mM NaHCO3.
Irradiation of nitrite with phenol
Fig. 3 reports the transformation rate of 25 μM phenol (3A) and the formation rate of nitrophenols (3B) as a function of the bicarbonate concentration, upon UVA irradiation of 10 mM NaNO2. A 27-time increase of phenol degradation rate could be observed between 0 and 3 mM NaHCO3, and in the same interval the formation rate of nitrophenols was increased by a factor of 9. Irradiation experiments in the presence of phosphate buffers with the same concentration of bicarbonate and the same pH (within ±0.1 units) were also carried out and are reported as well. The pH increase upon addition of phosphate increased the rates of phenol transformation and of nitrophenol formation at a considerable extent, but a bit less compared to bicarbonate, particularly in the case of phenol.![Initial rates of (a) phenol transformation and (b) nitrophenol formation, upon UVA irradiation of 10 mM NaNO2, as a function of the concentration of NaHCO3 or of the phosphate buffer (NaH2PO4 + Na2HPO4). The curves connecting the experimental data of phenol are just a guide for the eye. The nitrophenol curves in the presence of bicarbonate (△, 2-NP; ○, 4-NP) were fitted with equations of the form y = ax [(x + b) (x + c)]−1 (a, b, c: fitting parameters). The corresponding curves with phosphate (▲, 2-NP; ●, 4-NP) were fitted with y = dx (x + e)−1 (d, e: fitting parameters). Initial phenol was 2.5 × 10−5 M, pH varied from 6.5 to 8.5 depending on CNaHCO3.](/image/article/2009/PP/b807265p/b807265p-f3.gif) | ||
| Fig. 3 Initial rates of (a) phenol transformation and (b) nitrophenol formation, upon UVA irradiation of 10 mM NaNO2, as a function of the concentration of NaHCO3 or of the phosphate buffer (NaH2PO4 + Na2HPO4). The curves connecting the experimental data of phenol are just a guide for the eye. The nitrophenol curves in the presence of bicarbonate (△, 2-NP; ○, 4-NP) were fitted with equations of the form y = ax [(x + b) (x + c)]−1 (a, b, c: fitting parameters). The corresponding curves with phosphate (▲, 2-NP; ●, 4-NP) were fitted with y = dx (x + e)−1 (d, e: fitting parameters). Initial phenol was 2.5 × 10−5 M, pH varied from 6.5 to 8.5 depending on CNaHCO3. | ||
The increased transformation rate of phenol upon addition of the phosphate buffer is partially accounted for by increased nitration, which reached 35% yield with 2 mM phosphate. Formation of nitrosophenols was also very significant under these conditions and comparable to that of nitrophenols (see ESI,† Fig. S10). It has been shown that phenol nitrosation upon nitrite photolysis is favoured under basic conditions because of the efficient reaction of ˙NO2+˙NO with phenolate. Nitrosophenol oxidation could also contribute to the nitrophenol buildup.58 In contrast, very small variation of the ˙OH yield upon nitrite photolysis (reaction 7) could be observed under basic conditions. This is shown by the trend of the degradation of nitrobenzene, a known selective probe molecule for ˙OH,44 upon addition of phosphate (ESI,† Fig. S11).
Up to around 4–5 mM bicarbonate, both the degradation of phenol and the formation of nitrophenols were faster compared to the phosphate buffer (Fig. 3). The difference could be caused by the reaction between ˙NO and O2−˙ to give ONOO−,59 which could yield ˙NO2 and CO3−˙ in the presence of bicarbonate,53–57 enhancing the overall transformation and the nitration of phenol. The radical ˙NO is produced by nitrite photolysis, and HO2˙/O2−˙ (pKa = 4.8)45 can be formed in substantial amount in the presence of aromatic compounds, ˙OH radicals, and dissolved oxygen. Catechol (reaction 8) was identified by HPLC-MS among phenol transformation intermediates in the studied system (see ESI,† Fig. S12).
| NO2− + hν + H+→˙OH + ˙NO | (7) |
 | (8) |
| HO2˙⇄ O2−˙ + H+ | (9) |
| ˙NO + O2−˙→ ONOO− | (10) |
The decrease of phenol transformation and of nitrophenol formation rates at elevated bicarbonate could be accounted for by the depletion of reactive species in the reactions of O2−˙ and ˙NO2 with CO3−˙.21
| O2−˙ + CO3−˙→ O2 + CO32− | (11) |
| ˙NO2 + CO3−˙→ NO3− + CO2 | (12) |
A comparison with the nitrate system suggests that reaction 11 would play a more important role than reaction 12. The latter would take place as well upon nitrate irradiation, but no inhibition of phenol photonitration by nitrate was observed at high CNaHCO3 (Fig. 2B). Carbonate and bicarbonate might also be able to induce a solvent-cage effect on the photolysis of nitrite, but in such a case an enhancement of phenol phototransformation and photonitration would be expected at high CNaHCO3, which is not the case. Note that reaction 11 would not affect the formation of ONOO− from nitrate significantly, because the process takes place via photoisomerisation (reaction 3) and not through reaction 10.
The two effects of bicarbonate on phenol transformation upon nitrite photolysis, pH variation and peculiar chemistry, could hardly be separated in surface waters. Furthermore, the pH trend as a function of the bicarbonate concentration is very similar in the adopted experimental system and in natural waters (see ESI,† Fig. S8 and S9), which would enable a comparison between the present experimental results and the available field data on natural ecosystems. The assessment of the environmental importance of the bicarbonate-induced process in comparison with other known pathways of aromatic photonitration requires a description of the rate of phenol nitration as a function of phenol, nitrate and bicarbonate.
Unfortunately the complexity of the bicarbonate effect (pH variation, possible contribution of nitrosophenol oxidation, involvement of transformation intermediates in the generation of O2−˙, peroxynitrite chemistry) prevents the elaboration of a complete kinetic model, with soluble differential equations, which takes into account all the relevant phenomena. However, it is possible to adopt an empirical approach in which the two effects of bicarbonate (pH variation, formation of peroxynitrite) are both considered. In this way, the numerical fitting of the experimental data with reasonable functions would give lumped parameters that depend on the rate constants of the processes involved. First of all, at the environmental concentration levels of phenol and nitrite the formation rate of nitrophenols is linearly proportional to both [Phenol] and the photon flux absorbed by nitrite, IabsNO2− (see ESI,† Fig. S13–15, and related discussion). The effect of bicarbonate, as far as its mere impact on pH is considered, can be obtained from the phosphate buffer data, because the total concentration of salt (phosphate or bicarbonate) and the pH values were adjusted so as to be the same in the two cases. The pH-dependent effect shows a saturative behaviour with the concentration of salt, similar to the trend of pH vs. the concentration of bicarbonate or phosphate (see ESI,† Fig. S8). Upon fitting of the phosphate data (Fig. 3B) and from the results reported in ESI,† Fig. S13, S15, one can derive the following empirical equations for the formation rate of nitrophenols at environmental (i.e. low) concentrations of phenol and nitrite:
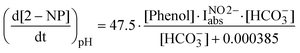 | (13) |
 | (14) |
Note that all concentrations are in molarity, and that IabsNO2− is the photon flux absorbed by nitrite, expressed in [einstein L−1 s−1]. IabsNO2− was chosen in the model instead of [NO2−] because the use of IabsNO2− can be extended to a wider range of irradiation conditions. Other details are reported as ESI.†
The overall effect of bicarbonate (which would depend on both pH and ONOO−) can be obtained by combining the functional forms derived from the fitting of the bicarbonate data of Fig. 3B, with the results reported in ESI† (Fig. S13, S15). The formation rate of nitrophenols can be expressed as follows:
 | (15) |
 | (16) |
Note that for 1 mM bicarbonate, namely the conditions of Fig. S13–S15 in ESI,†eqn (15) and eqn (16) would be transformed into d[2-NP]/dt = 50 [Phenol] IabsNO2− and d[4-NP]/dt = 30.5 [Phenol] IabsNO2−, as reported in ESI† for constant (1 mM) bicarbonate.
The role of the peroxynitrite chemistry would be obtained as the difference between the overall bicarbonate effect and that attributed to pH (eqn (15)−eqn (13) for 2-NP, eqn (16)−eqn (14) for 4-NP). In natural waters the presence of dissolved organic matter (DOM) and the associated scavenging of ˙OH radicals would have an impact, in particular on the pH-dependent effect that is linked with the production of ˙NO2 upon reaction between ˙OH and nitrite.58 In contrast DOM is unlikely to inhibit the nitration pathway that involves ONOO− and HCO3−, because it is not expected to influence the generation rate of ˙NO or to react with it, and could even enhance the generation of O2−˙ (see for instance reactions 8,9). Both ˙NO and O2−˙ are precursors of ONOO− (reaction 10). Accordingly, in the presence of variable interference by DOM, the formation of nitrophenols (2-NP + 4-NP) as enhanced by bicarbonate can be expected to vary from the full effect (ONOO− and pH, described by the sum of eqn (15) (2-NP) + eqn (16) (4-NP)), to that of peroxynitrite only (eqn (15) + eqn (16)−eqn (13)−eqn (14)). The plot of the resulting functions vs. bicarbonate (data not reported) shows that in the former case, the enhancement of nitration would be maximum for 1 mM bicarbonate, in the latter case for 0.4 mM bicarbonate (and no enhancement would be observed above 3.5 mM bicarbonate if the scavenging of ˙OH by DOM completely inhibits the pH-dependent effect).
The environmental significance of bicarbonate-enhanced phenol nitration can be assessed in comparison to nitration upon photolysis of nitrate and photooxidation of nitrite. The following calculations are referred to conditions observed in the water of the paddy fields in the Rhône delta (Southern France), containing 3 mM bicarbonate. It has been demonstrated that these conditions are particularly favourable to aromatic photonitration.42 In a water layer with a sunlight UV irradiance of 22 W m−2 (comparable to that observed at 9 a.m. on 15 July at mid latitude13), with 2.9 × 10−4 M nitrate, 2.4 × 10−5 M nitrite and a steady-state [˙OH] = 8.3 × 10−16 M, nitrate photolysis and nitrite photooxidation would produce [˙NO2] ≈ 1.5 × 10−9 M.42 Under these circumstances the initial formation rate of nitrophenols (NP = 2-NP + 4-NP) in the presence of 10−8 M phenol would be17 d[NP]/dt = 3.2 × 103 [Phenol] [˙NO2] = 4.8 × 10−14 M s−1, able to produce 1 nM nitrophenols in 5 to 6 hours.
Under the same irradiation conditions (22 W m−2 sunlight UV irradiance) the rate of photolysis of 2.4 × 10−5 M nitrite (reaction 7) would be:17 R˙OH,NO2− = 2.65 × 10−5 s−1 [NO2−] = 6.4 × 10−10 M s−1. From the photolysis quantum yield, Φ≈ 0.03 at λ = 340–365 nm51 one obtains IabsNO2− = R˙OH,NO2−Φ−1≅ 2 × 10−8 einstein L−1 s−1. Note that 365 nm is the emission maximum of the lamp adopted in the present work, and 340 nm is the wavelength where sunlight absorption by nitrite is most effective.29 In the presence of 3 mM bicarbonate and 10−8 M phenol, the formation rate of nitrophenols (d[NP]/dt)HCO3− would vary from 0.1 × 10−14 (eqn (15) + (16) − (13) −(14)) to 1.5 × 10−14 M s−1 (eqn (15) + (16)). Depending on the level of interference by DOM, bicarbonate could increase the formation rate of nitrophenols from 2 to 31%.
The case of the paddy fields of the Rhône delta would be intermediate because in the presence of 12.8 mg C L−1 NPOC (rate constant with ˙OH around 5 × 104 (mg C)−1 L s−1),13 3 mM HCO3− (rate constant with ˙OH 8.5 × 105 M−1 s−1)20 and 25 μM CO32− (3.9 × 108 M−1 s−1),20 24 μM nitrite (1 × 1010 M−1 s−1)20 would scavenge around 27% of photogenerated ˙OH. Under the reasonable hypothesis that the weight of the pH-dependent effect would be proportional to the fraction ξ of ˙OH that reacts with nitrite (that is around 1 under the conditions of Fig. 3B), the effect of bicarbonate on the formation rate of nitrophenols would be expressed as (d[NP]/dt)HCO3− = [eqn (15) + eqn (16) + (ξ− 1)(eqn (13) + eqn (14))]. With ξ = 0.27 as for the paddy fields of the Rhône delta one would get (d[NP]/dt)HCO3− = 4.8 × 10−15 M s−1, with an increase of about 10% with respect to the corresponding photonitration process without bicarbonate.
Conclusions
The present work shows that bicarbonate and carbonate are able to enhance the photochemical transformation, including the nitration where applicable, of phenol upon interaction with important photoactive species in surface waters such as Fe(III) oxide colloids, nitrate, and nitrite. Phenol transformation upon irradiation of hematite is considerably enhanced by bicarbonate, probably because the oxidation of CO32− to CO3−˙ and the reaction of CO3−˙ with phenol is faster than the direct oxidation of phenol by α-Fe2O3. If the photoreactivity of hematite is representative of that of Fe(III) species, the Fe(III)-induced oxidation of CO32− could be a major source of CO3−˙ in surface waters with more than 1 mg L−1 Fe.Bicarbonate enhances phenol nitration upon photolysis of nitrate, probably because it leads to increased production of nitrogen dioxide. Nitrate photolysis is an important photochemical source of nitrophenols in surface waters, comparable to the oxidation of nitrite.17,43 The importance of nitrate photolysis toward nitrophenol formation would be increased by a factor of at least two in waters rich of inorganic carbon (10 mM bicarbonate or higher). DOM is unlikely to influence the photonitration of phenol by nitrate; at most there could be an enhancement through a solvent-cage effect.59,60
Phenol transformation and nitration upon nitrite photolysis is considerably enhanced by bicarbonate, partially by basification and partially by peculiar chemistry. DOM could interfere with the pH-dependent process, but the pathway involving ONOO− could be important below 1 mM bicarbonate. However, bicarbonate is expected to significantly enhance the photonitration of phenol by nitrite if the interference of DOM (explicated through the scavenging of ˙OH) is limited.
Interestingly, the effects of bicarbonate on the photonitration processes induced by nitrate and by nitrite would be operational in different ranges of inorganic carbon. The nitrated intermediates that would be formed are pollutants of concern for their toxicity to aquatic organisms,61 and their ability to induce DNA damage.62
Acknowledgements
Financial support by PNRA-Progetto Antartide, INCA consortium (WG GLOB-CHEM) and Università di Torino - Ricerca Locale is gratefully acknowledged. SK acknowledges support from MIUR (Progetto India) and Compagnia di San Paolo (Torino, Italy) for his bursary.References
- B. L. Edhlund, W. A. Arnold and K. McNeil, Aquatic photochemistry of nitrofuran antibiotics, Environ. Sci. Technol., 2006, 40, 5422–5427 CrossRef CAS.
- K. Fenner, S. Canonica, B. I. Escher, L. Gasser, S. Spycher and H. C. Tülp, Developing methods to predict chemical fate and effect endpoints for use within REACH, Chimia, 2006, 60, 683–690 CrossRef CAS.
- K. H. Hefner, J. M. Fisher and J. L. Ferry, A multifactor exploration of the photobleaching of Suwannee river dissolved organic matter across the freshwater-saltwater interface, Environ. Sci. Technol., 2006, 40, 3717–3722 CrossRef CAS.
- A. V. Vahatalo and R. G. Zepp, Photochemical mineralization of dissolved organic nitrogen to ammonium in the Baltic Sea, Environ. Sci. Technol., 2005, 39, 6985–6992 CrossRef.
- J. Thomson, A. Parkinson and F. A. Roddick, Depolymerization of chromophoric natural organic matter, Environ. Sci. Technol., 2004, 38, 3360–3369 CrossRef CAS.
- D. L. Giokas and A. G. Vlessidis, Application of a novel chemometric approach to the determination of aqueous photolysis rates of organic compounds in natural waters, Talanta, 2007, 71, 288–295 CrossRef CAS.
- M. W. Lam and S. A. Mabury, Photodegradation of the pharmaceuticals atorvastatin, carbamazepine, levofloxacin, and sulfamethoxazole in natural waters, Aquat. Sci., 2005, 67, 177–188 CrossRef CAS.
- M. W. Lam, C. J. Young and S. A. Mabury, Aqueous photochemical reaction kinetics and transformations of fluoxetine, Environ. Sci. Technol., 2005, 39, 513–522 CrossRef CAS.
- S. S. Walse, S. L. Morgan, L. Kong and J. L. Ferry, Role of dissolved organic matter, nitrate, and bicarbonate in the photolysis of aqueous fipronil, Environ. Sci. Technol., 2004, 38, 3908–3915 CrossRef CAS.
- S. Canonica, Oxidation of aquatic organic contaminants induced by excited triplet states, Chimia, 2007, 61, 641–644 CrossRef CAS.
- A. Ter Halle and C. Richard, Simulated solar light irradiation of mesotrione in natural waters, Environ. Sci. Technol., 2006, 40, 3842–3847 CrossRef CAS.
- P. P. Vaughan and N. V. Blough, Photochemical formation of hydroxyl radical by constituents of natural waters, Environ. Sci. Technol., 1998, 32, 2947–2953 CrossRef.
- D. Vione, G. Falletti, V. Maurino, C. Minero, E. Pelizzetti, M. Malandrino, R. Ajassa, R. I. Olariu and C. Arsene, Sources and sinks of hydroxyl radicals upon irradiation of natural water samples, Environ. Sci. Technol., 2006, 40, 3775–3781 CrossRef CAS.
- J. Huang and S. A. Mabury, The role of carbonate radical in limiting the persistence of sulfur-containing compounds in sunlit natural waters, Chemosphere, 2000, 41, 1775–1782 CrossRef CAS.
- R. A. Larson and R. G. Zepp, Reactivity of the carbonate radical with aniline derivatives, Environ. Toxicol. Chem., 1988, 7, 265–274 CrossRef CAS.
- S. Chiron, C. Minero and D. Vione, Photodegradation processes of the antiepileptic drug carbamazepine, relevant to estuarine waters, Environ. Sci. Technol., 2006, 40, 5977–5983 CrossRef CAS.
- C. Minero, S. Chiron, G. Falletti, V. Maurino, E. Pelizzetti, R. Ajassa, M. E. Carlotti and D. Vione, Photochemical processes involving nitrite in surface water samples, Aquat. Sci., 2007, 69, 71–85 CrossRef CAS.
- P. L. Brezonik and J. Fulkerson-Brekken, Nitrate-induced photolysis in natural waters: Controls on the concentrations of hydroxyl radicals photo-intermediates by natural scavenging agents, Environ. Sci. Technol., 1998, 32, 3004–3010 CrossRef CAS.
- S. Canonica, T. Kohn, M. Mac, F. J. Real, J. Wirz and U. Von Gunten, Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (˙OH/˙O−) in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CAS.
- P. Neta, R. E. Huie and A. B. Ross, Rate constants for reactions of inorganic radicals in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1230 CAS.
- R. C. Bouillon and W. L. Miller, Photodegradation of dimethyl sulfide (DMS) in natural waters: Laboratory assessment of the nitrate-photolysis-induced DMS oxidation, Environ. Sci. Technol., 2005, 39, 9471–9477 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, D. Borghesi, M. Lucchiari and E. Pelizzetti, New processes in the environmental chemistry of nitrite. 2. The role of hydrogen peroxide, Environ. Sci. Technol., 2003, 37, 4635–4641 CrossRef CAS.
- P. Mazellier, G. Mailhot and M. Bolte, Photochemical behavior of the iron(III)/2,6-dimethylphenol system, New J. Chem., 1997, 21, 389–397 Search PubMed.
- D. W. King, R. A. Aldrich and S. E. Charnecki, Photochemical redox cycling of iron in NaCl solutions, Mar. Chem., 1993, 44, 105–120 CAS.
- P. M. Bohrer, B. Sulzberger, P. Reichard and S. M. Kraemer, Effect of siderophores on the light-induced dissolution of colloidal iron(III) (hydr)oxides, Mar. Chem., 2005, 93, 179–193 CrossRef.
- J. K. Leland and A. J. Bard, Photochemistry of colloidal semiconducting iron oxide polymorphs, J. Phys. Chem., 1987, 91, 5076–5083 CrossRef CAS.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Chemical actinometry, Pure Appl. Chem., 2004, 76, 2105–2146 CrossRef CAS.
- D. Vione, C. Minero, V. Maurino and E. Pelizzetti, Seasonal and water column trends of the relative role of nitrate and nitrite as ˙OH sources in surface waters, Ann. Chim. (Rome), 2007, 97, 699–711 CrossRef CAS.
- M. N. Alvarez, G. Peluffo, P. Wardman and R. Radi, Reaction of the carbonate radical with spin-trap 5,5′-dimethyl-1-pyrrolidone-N-oxide in chemical and cellular systems: Pulse radiolysis, electron paramagnetic resonance, and kinetic-competition studies, Free Radical Biol. Med., 2007, 43, 1523–1533 CrossRef CAS.
- S. Markager and W. F. Vincent, Spectral light attenuation and the absorption of UV and blue light in natural waters, Limnol. Oceanogr., 2000, 45, 642–650 CAS.
- A. E. Martell, R. M. Smith, R. J. Motekaitis, Critically selected stability constants of metal complexes database, version 4.0, released November 1997.
- B. C. Faust and M. R. Hoffmann, Photoinduced reactive dissolution of α-Fe2O3 by bisulfite, Environ. Sci. Technol., 1986, 20, 943–948 CAS.
- B. C. Faust, M. R. Hoffmann and D. W. Bahnemann, Photocatalytic oxidation of sulfur dioxide in aqueous suspensions of α-Fe2O3, J. Phys. Chem., 1989, 93, 6371–6381 CrossRef CAS.
- R. E. Huie, C. L. Clifton and P. Neta, Electron-transfer reaction rates and equilibria of the carbonate and sulfate radical anions, Radiat. Phys. Chem., 1991, 38, 477–481 CrossRef CAS.
- J. R. Bargar, J. D. Kubicki, R. Reitmeyer and J. A. Davis, ATR-FTIR spectroscopic characterization of coexisting carbonate surface complexes on hematite, Geochim. Cosmochim. Acta, 2005, 69, 1527–1542 CrossRef CAS.
- A. J. Feitz and T. D. Waite, Kinetic modeling of TiO2-catalyzed photodegradation of trace levels of microcystin-LR, Environ. Sci. Technol., 2003, 37, 561–568 CrossRef CAS.
- I. J. Rhile, T. F. Markle, H. Nagao, A. G. Di Pasquale, O. P. Lam, M. A. Lockwood, K. Rotter and J. M. Mayer, Concerted proton-electron transfer in the oxidation of hydrogen-bonded phenols, J. Am. Chem. Soc., 2006, 128, 6075–6088 CrossRef CAS.
- J. Platz, O. J. Nielsen, T. J. Wallington, J. C. Ball, M. D. Hurley, A. M. Straccia, W. F. Schneider and J. Sehested, Atmospheric chemistry of the phenoxy radical, C6H5O(˙): UV spectrum and kinetics of its reaction with NO, NO2 and O2, J. Phys. Chem. A, 1998, 102, 7964–7974 CrossRef CAS.
- M. N. Alvarez, G. Peluffo, L. Folkes, P. Wardman and R. Radi, Reaction of the carbonate radical with the spin-trap 5,5-dimethyl-1-pyrroline-N-oxide in chemical and cellular systems: Pulse radiolysis, electron paramagnetic resonance, and kinetic-competition studies, Free Radical Biol. Med., 2007, 43, 1523–1533 CrossRef CAS.
- R. Frank and W. Klöpffer, Spectral solar photon irradiance in Central Europe and the adjacent North Sea, Chemosphere, 1988, 17, 985–994 CrossRef.
- E. M. White, P. P. Vaughan and R. G. Zepp, Role of the photo-Fenton reaction in the production of hydroxyl radicals and photobleaching of dissolved organic matter in a coastal river of the southeastern United States, Aquat. Sci., 2003, 65, 402–414 CrossRef CAS.
- S. Chiron, C. Minero and D. Vione, Occurrence of 2,4-dichlorophenol and of 2,4-dichloro-6-nitrophenol in the Rhône river delta (Southern France), Environ. Sci. Technol., 2007, 41, 3127–3133 CrossRef CAS.
- J. Hoigné, Formulation and calibration of environmental reaction kinetics: Oxidations by aqueous photooxidants as an example, in Aquatic Chemical Kinetics, ed. W. Stumm, Wiley, New York, 1990, pp. 43–70 Search PubMed.
- P. Warneck and C. Wurzinger, Product quantum yields for the 305-nm photodecomposition of NO3− in aqueous solution, J. Phys. Chem., 1988, 92, 6278–6283 CrossRef CAS.
- G. Mark, H.-G. Korth, H.-P. Schuchmann and C. von Sonntag, The photochemistry of aqueous nitrate ion revisited, J. Photochem. Photobiol., A., 1996, 101, 89–103 CrossRef CAS.
- S. Goldstein and J. Rabani, Mechanism of nitrite formation by nitrate photolysis in aqueous solution: The role of peroxynitrite, nitrogen dioxide, and hydroxyl radical, J. Am. Chem. Soc., 2007, 129, 10597–10601 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. J. Harrison, R. I. Olariu and C. Arsene, Photochemical reactions in the tropospheric aqueous phase and on particulate matter, Chem. Soc. Rev., 2006, 35, 441–463 RSC.
- J. W. Coddington, J. K. Hurst and S. V. Lymar, Hydroxyl radical formation during peroxynitrous acid decomposition, J. Am. Chem. Soc., 1999, 121, 2438–2443 CrossRef CAS.
- G. Merenyi, J. Lind, G. Czapski and S. Goldstein, The decomposition of peroxynitrite does not yield nitroxyl anion and singlet oxygen, Proc. Nat. Acad. Sci. USA, 2000, 97, 8216–8218 CrossRef CAS.
- G. Merenyi, J. Lind, S. Goldstein and G. Czapski, Mechanism and thermochemistry of peroxynitrite decomposition in water, J. Phys. Chem. A, 1999, 103, 5685–5691 CrossRef CAS.
- R. M. Uppu, J. N. Lemercier, G. L. Squadrito, H. W. Zhang, R. M. Bolzan and W. A. Pryor, Nitrosation by peroxynitrite: Use of phenol as a probe, Arch. Biochem. Biophys., 1998, 358, 1–16 CrossRef CAS.
- S. Goldstein, G. Czapski, J. Lind and G. Merenyi, Carbonate radical ion is the only observable intermediate in the reaction of peroxynitrite with CO2, Chem. Res. Toxicol., 2001, 14, 1273–1276 CrossRef CAS.
- G. L. Squadrito and W. A. Pryor, Mapping the reaction of peroxynitrite with CO2: Energetics, reactive species, and biological implications, Chem. Res. Toxicol., 2002, 15, 885–895 CrossRef CAS.
- S. Goldstein, G. Czapski, J. Lind and G. Merenyi, Carbonate radical is the only observable intermediate in the reaction of peroxynitrite with CO2, Chem. Res. Toxicol., 2001, 14, 1273–1276 CrossRef CAS.
- S. Goldstein, D. Meyerstein, R. van Eldik and G. Czapski, Evidence for adduct formation between ONOO− and CO2 from high-pressure pulse radiolysis, J. Phys. Chem. A, 2000, 104, 9712–9714 CrossRef CAS.
- R. M. Uppu, G. L. Squadrito, R. M. Bolzan and W. A. Pryor, Nitration and nitrosation by peroxynitrite: Role of CO2 and evidence for common intermediates, J. Am. Chem. Soc., 2000, 122, 6911–6916 CrossRef CAS.
- D. Vione, V. Maurino, E. Pelizzetti and C. Minero, Phenol photonitration and photonitrosation upon nitrite photolysis in basic solution, Int. J. Environ. Anal. Chem., 2004, 84, 493–504 CrossRef CAS.
- M. Fischer and P. Warneck, Photodecomposition of nitrite and undissociated nitrous acid in aqueous solution, J. Phys. Chem., 1996, 100, 18749–18756 CrossRef CAS.
- J. Dzengel, J. Theurich and D. W. Bahnemann, Formation of nitroaromatic compounds in advanced oxidation processes: photolysis versus photocatalysis, Environ. Sci. Technol., 1999, 33, 294–300 CrossRef CAS.
- G. E. Howe, L. L. Marking, T. D. Bills, J. J. Rach and F. L. Mayer, Effects of water temperature and pH on toxicity of terbufos, trichlorfon, 4-nitrophenol and 2,4-dinitrophenol to the amphipod Gammarus-pseudolimnaeus and rainbow trout (Oncorhyncus mykiss), Environ. Toxicol. Chem., 1994, 13, 51–66 CrossRef CAS.
- S. Chiron, S. Barbati, M. De Meo and A. Botta, In vitro synthesis of 1,N-6-etheno-2′-deoxyadenosine and 1,N-2-etheno-2′-deoxyguanosine by 2,4-dinitrophenol and 1,3-dinitropyrene in presence of a bacterial nitroreductase, Environ. Toxicol., 2007, 22, 222–227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S15. See DOI: 10.1039/b807265p |
| This journal is © The Royal Society of Chemistry and Owner Societies 2009 |