Dose dependence of DNA repair in rainbow trout (Oncorhynchus mykiss) larvae exposed to UV-B radiation
David L.
Mitchell
*a,
Tristan
Adams-Deutsch
b and
Mark H.
Olson
b
aDepartment of Carcinogenesis, The University of Texas MD Andersen Cancer Center, 1808 Park Road 1C, Smithville, TX 78957. E-mail: dmitchel@mdanderson.org
bDepartment of Biology, Franklin & Marshall College, Lancaster, PA 17604
First published on 27th October 2008
Abstract
We examined survival and DNA damage in rainbow trout larvae (Oncorhynchus mykiss) resulting from a range of sublethal to lethal ultraviolet-B exposures in the presence and absence of photoreactivating radiation. We found that after low, sublethal UV-B exposures trout larvae are quite proficient at photoenzymatic repair but that this capacity decreases exponentially with the total incident UV-B dose. The relationship between the dose dependence of PER and trout development and vulnerability in fisheries and in the natural environment are discussed.
Introduction
Because the ultraviolet-B (UV-B) (285–315 nm) spectrum overlaps with the absorption spectrum of DNA, prolonged exposure to UV-B often results in DNA damage that can pose a significant threat to the survival of aquatic and terrestrial organisms.1,2 Two major mechanisms that repair the major types of DNA damage are nucleotide excision repair (NER) and photoenzymatic repair (PER). PER is ubiquitous in the plant and animal kingdoms, but is absent in placental mammals.3,4 The mechanism of PER is simple, and comprises the activity of a small number of proteins and the energy associated with UV-A/blue light absorption.4 NER involves multiple proteins and requires metabolic energy in the form of ATP. Unlike PER which is specific to photodamage, NER is a generalized repair mechanism for DNA damage (for a review see reference 5).Fish are generally thought to use both NER and PER to repair DNA photodamage. This idea is based on a number of studies on either tropical (e.g. Poecilia formosa, Carassius auratus)6–9 or marine (e.g. Engraulis mordax, Gadus morhua)10–13 fishes. In a previous study, we examined interspecific variation in PER and NER in larval stages of five temperate freshwater fish species representing four families and three orders.14 We focused on larvae because fish are most vulnerable to UVR during early life history stages when high surface area to volume ratios and limited avoidance capabilities can result in high exposure levels to a large proportion of cells. Previous work on a variety of systems has shown that it is very probable that the DNA repair capacity in young fish (larvae) is fully operative and, in fact, proceeds at a faster rate than older fish, declining with development.15 Our earlier results show some degree of NER in the larvae of all species tested. However, we found evidence of PER in only two of the five species. In the current study we revisited PER in rainbow trout (Oncorhynchus mykiss, Salmoniformes, Salmonidae), one of the species lacking this pathway. Using reduced levels of incident UV-B, we found that trout larvae were well equipped to remove DNA damage using PER after low doses of UV-B, but that with increasing doses the larvae removed correspondingly fewer CPDs in the presence of photorepair radiation (PRR). The underlying mechanism for this change in PER is not known, but may result from PER saturation or damage to the repair machinery itself.
Materials and methods
Fish
The rainbow trout used in our experiment were obtained from the Trout Lodge Hatchery in Sumner, Washington, as were the experimental animals in our previous study.14 Eggs and sperm were collected from brood stock that had been in captivity for multiple generations. Fertilization was conducted indoors and eggs were kept indoors at the hatchery until they reached the eyed egg stage, when they were shipped overnight in a Styrofoam cooler to Franklin and Marshall College in Lancaster, Pennsylvania. Upon arrival, eggs were transferred to 40 L aquarium filled with spring water and housed in a 12 °C incubator on a 12 h light:12 h dark cycle using full-spectrum fluorescent aquarium lights. Hatching was highly synchronous and began five days after shipment. Post-hatch larvae were left in the aquarium under the same incubation conditions as the eggs. Our experiment was conducted on 3 d post-hatch larvae (mean total length = 16.2 mm). Larvae were allocated into groups of 7 in 39 × 30 mL quartz dishes filled with 20 mL spring water and then exposed to the various light conditions.Irradiation chamber
Exposures to UVR and PRR were conducted at 12 °C in a controlled environment chamber (Thermal Product Solutions, New Columbia, PA). The lamp system consisted of a 30 cm diameter opaque wheel with forty 5.1 cm holes into which quartz vials are placed. The wheel rotated slowly (2 rpm) at 24 cm beneath the UV-B lamps and 32 cm above the PRR source. two 40 W Q-panel 340 bulbs (Q-Panel, Cleveland OH) intercalated with two 40 W visible light fluorescent bulbs (CoolWhite, General Electric). The UV-B source consisted of three Spectroline XX15B lamps each containing two 7.5 W BLE 1T158 bulbs (Spectronics, Westbury, NY, USA) yielding a spectral output extending from the UV-C (∼260 nm) through the UV-A spectra with peak emission at 300–329 nm (Fig. 1A).16 These lamps were covered with cellulose acetate (cutoff < 290 nm) to remove the low levels of UVC emitted by the lamps. UV-B doses were measured at 301 nm using an IL-1400A Radiometer/Photometer attached to an IL-1403 UV-B Phototherapy Radiometer with a SEL 240/UV-B-1/TD detector. The emission spectrum of the PRR source included a UV-A and visible component with no UV-B (Fig. 1B) emitted by two 40 W Q-panel 340 bulbs (Q-Panel, Cleveland OH) intercalated with two 40 W visible light fluorescent bulbs (CoolWhite, General Electric). | ||
| Fig. 1 Emission spectra. The emission spectra for the lamp systems used to induce DNA damage (Panel A) and to photoreactivate damage after induction (Panel B). | ||
Experimental design
Two approaches were used to evaluate DNA damage and repair in rainbow trout larvae and included Protocol 1, a chronic combined irradiation procedure designed to mimic what the organisms would actually experience in a natural sunlight environment (i.e., UV-B + PRR) and Protocol 2, an acute dose repair experiment in which the UV-B was delivered in a short period of time and then followed by incubation in the dark or under PRR conditions for different time periods up to 48 h. In Protocol 1, we used a two factor experimental design with UV-B exposure level as one factor and the presence or absence of PRR as the second factor. Larvae were exposed from above to 0, 8.4, 24.0, 41.8, 59.6, 81.5 and 104.5 kJ m−2 UV-B and from below to a constant dose of PRR during the 12 h exposure period. The different UV-B dose rates were controlled by placing stainless-steel mesh screens on top of the quartz dishes resulting in 23, 40, 57, 78, and 100% transmittance. The presence or absence of PRR was controlled using opaque metal discs to block PRR from below. Most treatments had 3 replicates with an additional 5 replicates kept in the dark as controls to estimate and correct for any non UVR-induced mortality. For each dose, 5 larvae per replicate were returned to a dark incubator and checked daily for survival for 144 h. For Protocol 2 (the acute experiments) larvae were initially exposed from above to 0, 6.8, 13.5 and 20.3 kJ m−2 UV-B in the absence of any PRR from below. Immediately after UV-B irradiation, the larvae were placed in the dark or exposed to a constant dose of PRR from below for 3, 6, 24, and 48 h. In the absence of PRR, NER alone was operative; in the presence of PRR both NER and PER functioned to repair CPDs.DNA extraction and damage analysis
Subsequent to UV exposure and repair, two larvae from each replicate were immediately frozen and stored at −20 °C. Frozen samples were later thawed and DNA extracted from one fish per replicate using standard techniques. The DNA concentration and purity were determined by absorbance at 260/280 nm using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). CPDs were quantified using radioimmunoassay (RIA). Briefly, the RIA is a competitive binding assay between very small amounts of radiolabeled UV-irradiated DNA and 1–2 ug sample DNA for an antiserum raised against UV-irradiated DNA. Under these conditions, antibody binding to an unlabeled competitor inhibits antibody binding to the radiolabeled ligand. These details, as well as those concerning the specificities of the RIAs and standards used for quantification, are described in detail by Mitchell.17,18 Survival and CPD frequencies from protocol 1 were analyzed with two-way Analysis of Variance (ANOVA) with UV-B exposure level and PRR presence/absence as main effects. Because survival was 100% in all dark controls, adjustments to survival in experiment units were not necessary. CPD frequencies were ln-transformed to normalize residuals. CPD frequencies from protocol 2 were analyzed with three-way ANOVA with UV-B exposure level, PRR presence/absence, and recovery time as main effects. This analysis was conducted on CPD frequencies that were standardized by dark control frequencies to account for the fact that samples were analyzed in two batches.Results
The first part of this study (Protocol 1) was designed to examine the effects of simulated environmental solar exposures on the efficiency of NER and PER (i.e., UV-B + PRR). Survival of rainbow trout larvae was significantly affected by the UV-B exposure level and the presence or absence of PRR (Fig. 2). Two-way ANOVA was highly significant and included main effects of exposure level and PRR (Overall Model: F11,22 = 30.8, p < 0.0001; Main effect of UV-B Level: F5,22 = 63.1, p < 0.0001; Main effect of PRR: F1,22 = 4.5, p < 0.05). In addition, there was a significant interaction of the two main effects (F5,22= 3.5, p < 0.05), indicating that the effect of PRR on survival was dependent on the level of UV-B exposure. At UV-B exposures of 8.4 kJ m−2, a large proportion of the larvae showed complete survival after 144 h with or without PRR. The prominent shoulder on the survival curve suggests that both NER and PER are functional in rainbow trout larvae. Between 60 and 13% survival was observed after 24 kJ m−2 with and without PRR, respectively. Although there was some survival after 41.8 kJ m−2, there was none observed at the higher doses, either in the presence or absence of PRR. | ||
| Fig. 2 Survival. The proportion of trout larvae alive at 144 h after the 12 h UV-B treatment with (○) and without (●) PRR are shown. The means and standard errors are shown. | ||
The dose dependence of PER is shown in Fig. 3. The frequencies of CPDs were quantified in the larvae immediately after the 12 h treatment with and without PRR exposure. All larvae were alive at the time of sacrifice at the end of the experiment (i.e., 12 h). Background CPD levels were very low in the absence of UV-B (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) = 3 CPDs/mb DNA) but accumulated in response to increasing amounts of exposure. CPD frequencies after 12 h exposure increased as a function of UV-B and were affected by the presence/absence of PRR, as well as the interaction between PRR and UV-B exposure (ANOVA: Overall Model: F11,22 = 130.6, p < 0.0001, Main effect of UV-B exposure: F5,22 = 273.9, p < 0.0001, Main effect of PRR: F1,22 = 18.4, p < 0.0001, Interaction: F5,22 = 9.1, p < 0.0001). At lower exposure levels the number of CPDs remaining in the larvae immediately after the 12 h co-exposure regimen was lower in the presence of PRR. However, as the dose increased beyond 41.8 kJ m−2, CPD frequencies with or without PRR showed no further accumulation and no significant differences between larvae exposed to PRR versus those that were not.
= 3 CPDs/mb DNA) but accumulated in response to increasing amounts of exposure. CPD frequencies after 12 h exposure increased as a function of UV-B and were affected by the presence/absence of PRR, as well as the interaction between PRR and UV-B exposure (ANOVA: Overall Model: F11,22 = 130.6, p < 0.0001, Main effect of UV-B exposure: F5,22 = 273.9, p < 0.0001, Main effect of PRR: F1,22 = 18.4, p < 0.0001, Interaction: F5,22 = 9.1, p < 0.0001). At lower exposure levels the number of CPDs remaining in the larvae immediately after the 12 h co-exposure regimen was lower in the presence of PRR. However, as the dose increased beyond 41.8 kJ m−2, CPD frequencies with or without PRR showed no further accumulation and no significant differences between larvae exposed to PRR versus those that were not.
 | ||
| Fig. 3 Cyclobutane pyrimidine dimer frequencies in fish larvae after combined induction and repair radiation. CPDs were measured in larvae immediately after increasing exposures to UV-B with (○) and without (●) PRR. Mean and standard errors are shown (n = 3). | ||
The data in Fig. 3 were re-plotted in Fig. 4 to better show the relationship between the UV-B dose and the normalized amount of damage remaining after 12 h PRR. In Fig. 4 we excluded the two highest doses since the system had saturated above 59.6 kJ m−2. At relatively low UV-B exposures, rainbow trout are quite efficient at removing CPDs using PER. We observed an exponential relationship between UV-B dose and PER up to 59.6 kJ m−2. At higher doses this capability is severely attenuated.
![Dose-dependence of photoenzymatic repair in larvae exposed to combined chronic radiation. The proportion of cyclobutane pyrimidine dimers removed from the DNA of rainbow trout larvae [1 − (+PRR/−PR)] exposed for 12 h to increasing doses of UV-B in the presence and absence of photoenzymatic radiation is shown. Standard errors were determined by propagating the errors from Fig. 2.](/image/article/2009/PP/b807469k/b807469k-f4.gif) | ||
| Fig. 4 Dose-dependence of photoenzymatic repair in larvae exposed to combined chronic radiation. The proportion of cyclobutane pyrimidine dimers removed from the DNA of rainbow trout larvae [1 − (+PRR/−PR)] exposed for 12 h to increasing doses of UV-B in the presence and absence of photoenzymatic radiation is shown. Standard errors were determined by propagating the errors from Fig. 2. | ||
The second part of this study (Protocol 2) was designed to complement the first part and to separate DNA damage induction from light- and dark-dependent repair responses. Using this approach, specific UV-B dose effects (i.e., CPD yields) on NER and PER could be determined separately. In Fig. 5 the amounts of CPDs induced by the three different doses used in the acute study are shown. The relationship between the induction of CPDs and dose is clearly linear (r2 = 0.9952). A slight background level of CPDs was observed (i.e., 1.1 ± 0.2 CPDs/mb) and was subtracted from the data shown in Fig. 6. From these results we concluded that the time differences used for the different doses (i.e., 30, 60 and 90 min) did not affect the yield. In other words, there was no significant CPD repair (by NER or PER) during the duration of the exposures up to 90 min. From Fig. 1 it can be seen that a significant portion of the radiation generated by the UV-B bulbs extends into the UV-AII (315–340) and UV-AI (340–400 nm) regions of the UV spectrum. The fact that the T0 dose response does not deviate from linearity indicates that no PER occurred. Hence, either the amount of PRR delivered during this procedure was too small to affect CPD frequencies or the action spectrum for PER in rainbow trout does not have a significant UV-A component.
 | ||
| Fig. 5 Dose response curve in fish larvae exposed to single acute doses of UV-B radiation. The frequency of CPDs is shown after each of the 3 UV-B exposures used in the acute experiments and include 0, 6.8 (30 min), 13.5 (60 min) and 20.3 (90 min) kJ m−2 UV-B radiation measured at 301 nm. Mean and standard errors are shown (n = 3). | ||
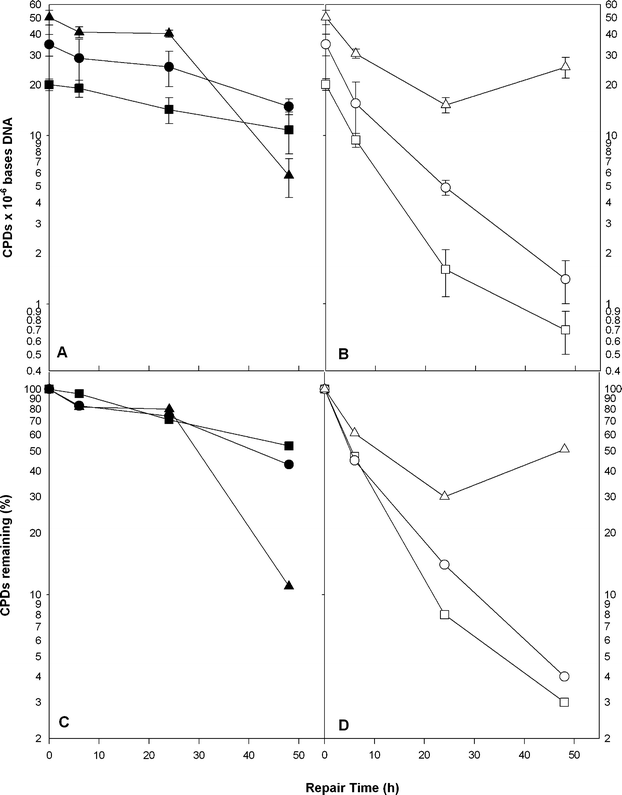 | ||
| Fig. 6 Cyclobutane dimer repair in fish larvae exposed to increasing doses of UV-B radiation. CPD frequencies are shown immediately after UV-B irradiation (T0) and at 6, 24 and 48 h (Panels A and B). Panel A shows dark repair (closed symbols) and Panel B shows repair in samples exposed to PRR during repair (open symbols). Doses include 6.8 (■,□), 13.5 (●,○) and 20.3 (▲,△) kJ m−2 UV-B measured at 301 nm. Mean and standard errors are shown (n = 5–6). Panels C and D represent the means of the data in A and B normalized to T0 at 100%. | ||
In Fig. 6 the CPD repair kinetics is shown for the three different dose/damage levels. The number of CPDs/mb DNA is shown at T0 (immediately after irradiation) and at 6, 24 and 48 h post-irradiation. The left panel (A) shows repair in the absence of PRR (i.e., NER alone) and the right panel (B) shows the effects in the presence of PRR (i.e., combined NER + PER). Three-way ANOVA was highly significant (F23,72= 15.25, p < 0.0001) and indicated significant effects by all three factors; two-way interactions between exposure and PRR, between exposure level and recovery time, and a three-way interaction (Table 1). The interaction of exposure level and ±PRR occurs because PER is severely inhibited at the high dose (Panel B). In addition, the three-way interaction suggests that PER function over time varies among the three exposure levels. However, under PRR conditions we are observing the combined repair activity of both PER and NER. If we assume that PRR does not affect the rate of NER, then we can subtract the amount of NER determined under dark conditions from the total amount of damage repaired under PRR to yield the net PER at each time and dose level. In Fig. 7 the proportion of lesions repaired at the three different time points by NER (black bar) and PER (white bar) can be seen at 6.8 (Panel A), 13.6 (Panel B) and 20.3 (Panel C) kJ m−2. It is evident that, for the most part, at increasing time points the proportion of damage repaired by PER decreases while the proportion of damage repaired by NER increases. Indeed, after 48 h incubation in the dark all of the damage can be repaired by NER.
| Source | Numerator degrees of freedom | F-statistic | p-value |
|---|---|---|---|
| Exposure duration | 2 | 61.09 | 0.0001 |
| ±PRR | 1 | 77.25 | 0.0001 |
| Recovery time | 3 | 29.09 | 0.0001 |
| Exposure duration X ±PRR | 2 | 7.30 | 0.001 |
| Exposure duration X recovery time | 6 | 2.66 | 0.022 |
| ±PRR X recovery time | 3 | 2.08 | 0.11 |
| Exposure duration X ±PRR X recovery time | 6 | 4.53 | 0.001 |
 | ||
| Fig. 7 The dose-dependence of dark and light-dependent DNA repair in trout larvae. This histogram uses the data from Fig. 5 to compare the extent of CPD removal at 6, 24 and 48 h post UV-B irradiation with increasing doses of UV-B radiation. NER is represented by dark bars and PER by white bars. | ||
Discussion
Using a simulated environmental exposure that included simultaneous DNA damage induction and light-dependent repair and an acute study that separated damage induction from the light and dark repair processes we show that both NER and PER are important repair process in rainbow trout. We observed a partnership between these two repair systems in fish where a reduction in PER in response to high, environmentally relevant UV-B doses (e.g., ∼20 kJ m−2) was compensated for by a less labile and more persistent NER system. This interplay of NER and PER has previously been reported in marine fishes.1,12,13A comparison of the D37 values of the survival curves indicates that rainbow trout larvae are able to withstand 1.5- to 2-fold greater UV-B in the presence of PRR, but that the benefit of PRR is lost at high UVB doses. These data are consistent with a comparative survival study in rainbow trout and other freshwater species in which we examined NER and PER in larvae exposed to ∼138 kJ m−2 UV-B and found no survival 72 h after the 12 h exposure period, regardless of the PRR conditions.14 The current data re-examined one aspect of this earlier work in finer detail and show that rainbow trout are very sensitive to the biological consequences of UV-B (i.e., survival and CPD repair) and that this sensitivity is dose-dependent.
We observed a threshold of CPD accumulation at UV-B doses > 41.8 kJ m−2. The saturation of DNA damage levels at high doses suggests that a steady-state level is reached whereby the rates of induction and repair are equivalent and result in no further damage accumulation. It is doubtful that photochemical saturation of CPDs is reached in these organisms since the maximum induction (i.e., ∼200 CPDs/megabase) is at least an order of magnitude below the photochemical saturation level of CPDs.19 In addition, all values were within the linear range of the semi-log standard curve generated by the RIA. Hence, the saturation of CPDs at UV-B doses > 41.8 kJ m−2 is not easily explained but may result from the robust NER observed in this species exposed to high UV-B doses.14 Although none of the larvae exposed to UV-B doses > 41.8 kJ m−2 survived to the 144 h endpoint they were alive at the time they were sacrificed for DNA damage analysis at 12 h and most likely retained functional NER for at least 12 h.
Hence, we believe that the primary mechanism involved in the maintenance of a DNA damage equilibrium at the higher doses is NER. It is interesting to note that this equilibrium occurs at ∼ 200 CPDs/mb, suggesting that, in the absence of PER, NER is able to maintain a balance between the dose rate and the repair rate. Several other factors may also come into play:1 We and others have shown that DNA repair is inducible in fish with a significant increase in DNA repair capacity observed 8 h after prior exposure to visible light.20,21 Hence, the induction of PER or NER by the visible light portion of the PRR treatment and the resulting overall up-regulation of DNA repair efficiency may contribute to the observed DNA damage equilibrium; and2 It is also possible that photoprotective mechanisms (e.g., melanization) or UV-induced hyperplasia may have occurred during the 12 h treatment and effectively mitigated the amount of UV-B absorbed later in the treatment time course.
In our earlier research into the UV response in common freshwater fishes14 we found that NER was robust in all species tested, including rainbow trout. These studies were performed at a single fairly high UV-B dose (up to 138 kJ m−2) and under simulated environmental conditions (i.e., UV-B + PRR). By separating damage induction from repair in the current study we find greater NER efficiencies at high UV-B doses compared to lower doses where PER is the dominant repair system. The co-dependence of PER by NER in fish is not a new finding.12,13 However, our current study extends this finding to freshwater species and, more importantly, shows that the extent of this co-dependence is variable and dependent on the level of DNA damage induced.
The mechanisms underlying the UV-B dose effects on PER and NER are intriguing and merit some discussion. Because PER is a multi-step process, the exponential decrease in PER as a function of dose (Fig. 4) suggests that there is more than one target affected by high UV-B doses. It is very doubtful that the DNA damage substrate (i.e., CPDs) is limiting at the high dose. When the acute DNA repair data shown in Fig. 6 are normalized to 100% at the T0 dose (see Panels C and D) the rates of PER after the 6.8 and 13.6 kJ m−2 doses are indistinguishable, suggesting that the substrate levels at these doses are non-limiting and that PER is working at maximum efficiency throughout the entire experiment. However, at the high 20.3 kJ m−2 dose the initial rate of PER appears comparable to the lower doses but deteriorates after 6 h when the CPD substrate is at or below the T0 levels observed for the lower dose, which repairs normally. These data suggest that the level of CPDs is not limiting and not responsible for the observed decrease in PER.
The loss of PER at high doses may result from damage to the repair machinery itself (i.e., to the proteins involved). Since both PER and NER are complex reactions requiring more than a single substrate this is not an unreasonable hypothesis. PER is a fairly simple reaction in which a photolyase-cofactor complex binds to a CPD forming an enzyme-substrate complex that is stable in the absence of light. In the presence of sufficient levels of light, photon energy is absorbed and the CPD is photolyzed. The presence of multiple substrates (e.g., photolyases, cofactors, photons) and multiple functions (e.g., tracking, binding, absorption, and photolysis) lends itself to this multi-target mechanism. However, the fact that we only see this deterioration in PER and not in NER contradicts this idea. Indeed, the fact that the NER complex is built from a large number of essential proteins and yet is less sensitive to the effects of high UV-B argues against any direct UV effects on the structure or function of DNA repair enzymes.
Finally, another more mundane physico-chemical mechanism may lay at the heart of this observation. It is evident from Fig. 7 that as the UV-B dose (i.e., exposure time) increases, the amount of PER progressively decreases up to 48 h. The increased UV-B dose may induce hyperproliferation or melanization in the epidermal cells of the larvae, thus shielding more and more of the damaged cells from UV-B as well as PRR. Given this scenario, the longer exposure times may allow a corresponding increase in photoprotection against further damage while at the same time reducing the amount of PRR required for efficient PER. NER would be unaffected by these factors. In fact, photolyase binding to CPDs would also be unaffected by increased photoprotection and could, possibly, enhance NER by mimicking the DNA repair proteins that initially recognize and bind the DNA damage site (e.g., XPC, DDB2; ref. 5). Such “molecular hijacking” has been observed for different transcription factors in mammalian systems.22
The dose dependence of PER that we observe in rainbow trout is consistent with early studies on PER in bacteria.23 and suggests that the results of our previous studies14 regarding the absence of PER in brook trout (Salvelinus fontinalis) and northern pike (Esox lucius) should be reconsidered in light of these new findings. Northern pike would be of particular interest because their eggs and larvae adhere to aquatic macrophytes24 and thus are exposed to solar UV-B. In contrast, rainbow trout and brook trout larvae spend their first few weeks post-hatching in interstitial spaces of gravel substrates.25,26 Such “safe” environmental conditions may or may not exist in commercial hatcheries. The fact that we did see evidence of PER in two other species, bluegill (Lepomis macrochirus) and yellow perch (Perca flavescens),14 suggests that the dose dependence of PER may vary between species. In the future, examination of DNA repair in fish larvae should be conducted over a broad range of UV doses, since the biological response may be qualitatively different depending upon dose. Research into the biological responses of fish species to UV-B stress is an important focus for future studies in environmental photobiology.
Abbreviations
| CPD | Cyclobutane pyrimidine dimer |
| mb | Megabase |
| NER | Nucleotide excision repair |
| PER | Photoenzymatic repair |
| PRR | Photorepair radiation |
Acknowledgements
This research was supported by NSF grant DEC-IRCEB-0210972 to M. H. Olson and D. L. Mitchell and by NIEHS Center grant ES07784.References
- D. Mitchell and D. Karentz, The induction and repair of DNA photodamage in the environment, in Environmental UV Photobiology, ed. A. R. Young, J. Moan, L. O. Bjorn and W. Nultsch, 1993, Plenum Press, New York, pp. 345–377 Search PubMed.
- D. L. Mitchell, DNA Damage Induced by Ultraviolet Radiation, in Encyclopedia of Molecular Biology: and Molecular Medicine, ed. R. A. Meyers, Chernow Publishers, New York, 2nd edn, 2004 Search PubMed.
- G. B. Sancar, Enzymatic photoreactivation: 50 years and counting, Mutat. Res., 2000, 451(1–2), 25–37 CrossRef CAS.
- A. Sancar and G. B. Sancar, DNA repair enzymes, Ann, Rev. Biochem., 1988, 57, 29–68 CrossRef CAS.
- J. E. Cleaver and D. L. Mitchell, Ultraviolet radiation carcinogenesis, Cancer Med., ed. D. W. Kufe, R. C. Bast Jr, W. N. Hait, W. K. Kong, R. E. Pollock, R. R. Weichselbaum, J. F. Holland, E. Frei III, 7th edn, BC Decker, Inc., Hamilton-London, 2006, pp. 283–291 Search PubMed.
- P. M. Achey, A. D. Woodhead and R. B. Setlow, Photoreactivation of pyrimidine dimers in DNA from thyroid cells of the teleost, Poecilia formosa, Photochem. Photobiol., 1979, 29, 305–10 CrossRef CAS.
- S. Yasuhira and A. Yasui, Visible light-inducible photolyase gene from the goldfish Carassius auratus, J. Biol. Chem., 1992, 267, 25644–25647 CAS.
- E.-H. Park and A.-K. Yi, Photoreactivation rescue and dark repair demonstrated in UV-irradiated embryos of the self-fertilizing fish Rivulus ocellatus marmoratus (Teleostei; Aplocheilidae), Mutat. Res., 1989, 217, 19–24 CAS.
- D. L. Mitchell, J. A. Meador, M. Byrom and R. B. Walter, The Resolution of DNA Damage in Xiphophorus fishes, Marine Biotech., 2001, 3, S61–S71 Search PubMed.
- S. E. Kaupp and J. R. Hunter, Photorepair in larval anchovy, Engraulis mordax, Photochem Photobiol., 1981, 33, 253–6 CrossRef CAS.
- H. I. Browman, R. D. Vetter, C. A. Rodriguez, J. J. Cullen, R. F. Davis, E. Lynn and J. F. St Pierre, Ultraviolet (280–400 nm)-induced DNA damage in the eggs and larvae of Calanus finmarchicus G. (Copepoda) and Atlantic cod (Gadus morhua), Photochem. Photobiol., 2003, 77, 397–404 CrossRef CAS.
- J. D. Regan, R. D. Snyder, A. A. Francis and B. L. Olla, Excision repair of ultraviolet- and chemically-induced damage in the DNA of fibroblasts derived from two closely-related species of marine fishes, Aquatic Toxicol., 1983, 4, 181–188 CrossRef CAS.
- J. D. Regan, W. L. Carrier, C. Samet and B. L. Olla, Photoreactivation in two closely related marine fishes having different longevities, Mech. Ageing Develop., 1982, 18, 59–66 Search PubMed.
- M. H. Olson and D. L. Mitchell, Interspecific variation in UV defense mechanisms among temperate freshwater fishes, Photochem. Photobiol., 2006, 82, 606–610 CrossRef CAS.
- D. L. Mitchell and P. S. Hartman, The regulation of DNA repair during development, BioEssays, 1990, 12, 74–79 CrossRef CAS.
- C. E. Williamson, G. Grad, H. J. De Lange and S. Gilroy, Temperature-dependent ultraviolet responses in zooplankton: implications of climate change, Limnol Oceanogr., 2002, 47, 1844–1848.
- D. L. Mitchell, Radioimmunoassay of DNA damaged by ultraviolet light, in Technologies for Detection of DNA Damage and Mutations, ed. G. Pfeifer, Plenum Publishing Corp., New York, 1996, pp. 73–85 Search PubMed.
- D. L. Mitchell, Quantification of DNA photoproducts in mammalian cell DNA using radioimmunoassay, in Methods in Molecular Biology, DNA Repair Protocols, 2nd edn, Dr. Daryl S. Henderson, The Humana Press Inc., Totowa, NJ, 2006, pp. 239–249 Search PubMed.
- S. Wang, Photochemistry and photobiology of nucleic acids, Academic press, New York, San Francisco, London, 1976, vol. 1 Search PubMed.
- S. Yasuhira and A. Yasui, Visible light-inducible photolyase gene from the goldfish Carassius auratus, J. Biol. Chem., 1992, 267, 25644–25647 CAS.
- D. L. Mitchell, J. T. Scoggins and D. C. Morizot, DNA-repair in the variable platyfish (Xiphophorus variatus) irradiated in vivo with ultraviolet-B light, Photochem. Photobiol., 1993, 58, 455–459 CrossRef CAS.
- D. G. Johnson, A. Coleman, K. L. Powell and M. C. MacLeod, High-affinity binding of the cell cycle-regulated transcription factors E2F1 and E2F4 to benzo[a]pyrene diol epoxide-DNA adducts, Mol. Carcinog., 1997, 20, 216–23 CrossRef CAS.
- J. Jagger, Photoreactivation, Bacteriol. Rev., 1958, 22, 99–142 CAS.
- W. B. Scott and E. J. Crossman, Freshwater fishes of Canada, Bull. Fish. Res. Board Can., 1979, 184 Search PubMed.
- G. L. Chandler and T. C. Bjornn, Abundance, growth and interactions of juvenile steelhead relative to time of emergence, Trans. Amer. Fish. Soc., 1988, 117, 432–443 Search PubMed.
- Snucins, E. J. R. A. Curry and J. M. Gunn, Brook trout (Salvelinus fontinalis) embryo habitat and timing of alevin emergence in a lake and a stream, Can. J. Zool., 1992, 70, 423–427 CrossRef.
| This journal is © The Royal Society of Chemistry and Owner Societies 2009 |