Fluorescence quenching of CdSe quantum dots by tertiary amines and their surface binding effect†
Raquel E.
Galian
a and
Juan C.
Scaiano
*b
aDepartamento de Química Analítica, Instituto de Ciencia Molecular, Universidad de Valencia, 50 Dr Moliner, 46100, Burjassot, Valencia, Spain
bDepartment of Chemistry, University of Ottawa, Ottawa, Ontario, Canada K1N 6N5
First published on 18th October 2008
Abstract
The effect of different amines on the fluorescence of quantum dots has been discussed in the literature but the mechanism involved is not fully understood. We have studied the interaction of core CdSe quantum dots of different sizes and core-shell CdSe quantum dots with tertiary amines, such as aliphatic amines (triethylamine) and bicyclic amines (DABCO and quinuclidine), using fluorescence techniques. Fluorescence quantum yield decreases with increasing concentration of the amine following a linear Stern–Volmer behaviour (Kq = 80–330 M−1), but relatively small changes were observed in the fluorescence lifetimes. In order to get insights into the mechanism involved, small nanoparticles (2.1 nm) were synthesized and the interaction QD surface–amine was analysed by NMR and FTIR spectroscopy.
Introduction
The use of semiconductor quantum dots (QD) as fluorescence probes has increased in recent years along with their application in different areas such as medicine, chemistry and engineering. This interest is related to their broad absorption spectra and strong and tunable fluorescence.1–4The many applications of QD in biology,5 include the tumor targeting and imaging in animals and the development of functional nanoparticles able to interact with biological molecules such as peptides, proteins and nucleic acids.6 It is important to point out the presence of the amine moieties in some of these structures. We are interested in understanding the interaction between simple amines and nanoparticles in order to correlate possible behaviors in more complex structures.
The interaction of QD with some aliphatic amines has been previously reported, however different effects on the QD luminescence were observed. Addition of n-butylamine to CdSe QD caused a decrease in the fluorescence quantum yield with no significant effect on the fluorescence lifetime. It was proposed that the amine occupied hole sites giving a decrease in the density of luminescent centers. This binding can be described by a similar model as enzyme/ligand systems.7,8 Fluorescence quenching processes may involve dynamic quenching or static quenching. The latter involves pre-association between fluorophore and quencher.
The effect of ethylenediamine and 1,6-hexanediamine or aromatic compounds such as o-phenylenediamine on the QD surface support a binding complexation model for the latter and passivation of the surface at low concentration or aggregation at high concentration for aliphatic amines.9
We have recently reported the binding of 4-aminoTEMPO to CdSe QD with an association constant of 8 × 106 M−1, determined by EPR spectroscopy.10,11 The much higher fluorescence quenching by 4-amino-TEMPO compared to TEMPO was attributed to the strong binding of the amine to the QD surface. The mechanism proposed for the fluorescence quenching by both nitroxides involves reversible charge or electron transfer or paramagnetic-assisted spin relaxation.12 The size of QD, the presence of a ZnS shell and the binding properties of the nitroxides play an important role in the quenching process, as was demonstrated in a comparative study using core and core-shell QD.13 The presence of n-alkylamines in the synthesis of CdSe QD has been studied.14 The chain length, stereochemical effect and electron donating activity were discussed, leading to the proposal that amines formed a Cd–amine complex in the synthesis solution and act as caping ligand passivating the QD surface.
The use of tertiary amines as capping material for QD, has been reported by Winnik et. al.15,16 They used polydimethylaminoethyl methacrylate that can act as multidentate ligand and make the nanoparticles soluble in methanol, where they are usually insoluble.15–17 On the other hand, a dramatic fluorescence enhancement has been reported for microcrystalline semiconductor particles of CdS or Cd3As2 when they interact with simple amines such as triethylamine at concentrations lower than 1 mM; the authors suggested that the amine binds to lower energy trap sites increasing the quantum yield of the emission, but the binding site involved in the interaction was not described.18 Given the diverse observations in the QD-amine studies reported, we decided to explore the interaction of simple tertiary amines (Chart 1) such as triethylamine (TEA), 1,4-diazabicyclo[2.2.2]octane (DABCO) and quinuclidine (QNU) with QD. The effect of the size of the core nanoparticle and the presence of ZnS shell has also been explored.
 | ||
| Chart 1 Structure of the tertiary amine, 1 = triethylamine, 2 = DABCO and 3 = quinuclidine. | ||
Experimental
Amines, cadmium oxide (CdO, 99.99%), trioctylphosphine (TOP, 90%), trioctylphosphine oxide (TOPO, 99%) and Se (100 ME) were purchased from Sigma-Aldrich and used as received except for triethylamine, it was distilled twice before use. Tetradecylphosphonic acid was purchased from Alfa Aesar.The green QD (λmaxabs = 475 nm and 516 nm) and orange QD (λmaxabs = 573 nm) were synthesized following the methodology described by Peng et. al.19 Briefly CdO (51 mg), TOPO (3.7768 g) and TDPA (0.2232 g) were heated in a flask under argon until the solution become clear at around 300 °C. The Se powder (41 mg) was dissolved in TOP (2.4 mL) and this solution was quickly added to the mixture at 270 °C. The reaction was stopped at different times in order to get the desired QD size. A commercial core-shell QD (λmaxabs = 506 nm, Adirondack green has been purchased from Evidenttech,20 was the same type of material employed in earlier studies.13 In all cases the trioctylphosphine and trioctylphosphine oxide (TOP/TOPO) have been used as ligands that make the nanoparticles soluble in toluene.
Fluorescence spectra were obtained in a PTI instrument. The excitation wavelength was 380 nm and the spectral resolution was set at 2 nm. All experiment were done under air (aerobic conditions) since there was no significant difference compared to experiments under nitrogen (anaerobic conditions, see electronic supplementary information, ESI, Fig. SI-8†). In all cases the solvent used for the fluorescence determination was toluene.
The quenching studies have been performed under air and the concentration of the QD was adjusted to give an absorbance of 0.2 at the excitation wavelength.
Infrared spectroscopy (FTIR, Nexus 670 spectrometer, OMNIC software) was used to check the TOPO binding to the QD surface on the home-made nanoparticles and to analyse the solid complex performed between small green QD (2.1 nm) and TEA.
Results and discussion
It is well known that the size of the nanoparticles is related to their absorption maximum and the surface quality is related to their fluorescence emission.1 The maximum in absorbance, corresponding to the first exciton peak, was used to determine the size of the home-made QD according to a literature method.21Table 1 summarizes the photophysical properties of the nanoparticles used in this work. The small full width at half maximum (FWHM) in the fluorescence spectra and the absence of the surface trap emission band at longer wavelength indicate that the nanoparticles used were of good quality. For the green QD the fluorescence quantum yield was around 0.05.| QD | λ max abs/nm | Diameter/nma | λmaxem/nm | FWHM/nmb |
|---|---|---|---|---|
| a Calculated according to standard methods.21 b Full width at half maximum. c Synthesized following the literature.19 d Value given by Evidenttech. | ||||
| Small greenc | 475 | 2.1 | 490 | 27 |
| Greenc | 516 | 2.5 | 527 | 25 |
| Orangec | 573 | 3.6 | 586 | 30 |
| Green core-shell | 506 | 2.4d | 526 | 27 |
The addition of triethylamine (18 mM) to a green QD solution (2.5 nm) showed a blue shift in the absorbance spectra from 516 nm to 512 nm (Fig. 1). The fluorescence intensity of the QD decreases with the increasing concentration of TEA. Interestingly we have observed, at each particular addition, the fluorescence decreases with the time (Fig. 2) and reaches a plateau around 60 minutes after the addition.
 | ||
| Fig. 1 Absorption spectra of 3.7 μM of green CdSe QD in the absence (○) and in the presence (■) of 18 mM of TEA, which was recorded 1 hour after the addition. | ||
 | ||
| Fig. 2 Fluorescence spectra (λmaxexc 380 nm) of 3.7 μM of green CdSe QD (filled circle) obtained at different times after the addition of 18 mM of TEA. The times are before TEA addition, immediately after the addition (filled square), 5 min (open circle), 30 min (open triangle), 60 min (open square) and 90 min (filled triangle); note that the last two overlap. | ||
This general behavior is independent of the TEA or QD concentration (Fig. 3 and 4) and the media (aerobic or anaerobic conditions, see Fig. SI-1.†)
In order to obtain the Stern–Volmer quenching constant (Kq) for the process, five additions of different concentrations of the amine were made. The fluorescence spectra were recorded 60 minutes after the addition of the amine to ensure the stabilization of the intensity reading. Much to our surprise, linear Stern–Volmer plots were observed with all the amines and the quenching constants values obtained from the slopes are reported on Table 2 (eqn (1), Fig. 5).
| F0/F = 1 + Kq[Q] = 1 + kqτ0[Q] | (1) |
| QD | Stern–Volmer constant, Kq/M−1 | |
|---|---|---|
| TEA a,b | DABCO c,b | |
| a Data obtained 1 hour after the quencher addition. b Errors based on Stern–Volmer analysis for several quencher concentrations, as shown in Fig. 5. For quinuclidine, the effect was similar to DABCO but of smaller magnitude. c Data obtained immediately after the addition. d Small fluorescence quenching. | ||
| Green, 2.5 nm | 329 ± 13 | 21.2 ± 0.9 |
| Orange, 3.6 nm | 105 ± 1 | 6.5 ± 0.4 |
| Core-shell, 2.4 nm | 79 ± 5 | —d |
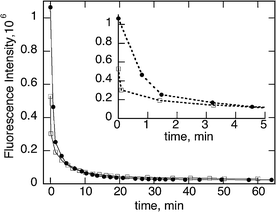 | ||
| Fig. 3 Fluorescence intensity vs. time of green CdSe QD at 5.2 μM (□) and 7.2 μM (●) after the addition of 18 mM of TEA. The inset is an expansion of the first 5 minutes, also illustrating better the importance of concentration on luminescence immediately after amine addition; note that the higher concentration also absorbs more of the excitation light. | ||
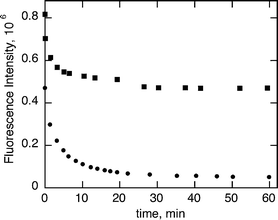 | ||
| Fig. 4 Fluorescence intensity vs. time of 5.2 μM of green CdSe QD with 3.6 mM (■) and 18 mM (●) of TEA. | ||
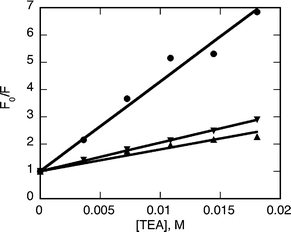 | ||
| Fig. 5 Stern–Volmer plots for green (●), orange (▼) and green core-shell QD (▲) in the presence of TEA, obtained 1 hour after each addition of quencher. | ||
The smaller the QD diameter the higher the quenching efficiency observed, as noted in related work.10–12 The presence of a ZnS shell on the QD surface reduces about four times this efficiency for the same approximate size of QD, due to the reduction in surface defect sites present in the core-shell QD and/or the additional protection offered by the shell towards the quencher, as was observed for nitroxide quenching of core-shell QD.13
When DABCO was used as a quencher no absorbance shift was observed. Further, a different kinetic behavior was obtained after the addition of the each quencher concentration, the fluorescence decreased immediately after the addition followed by a 10% increase for green (see Fig. SI-2†) or orange QD and 20% for the core-shell QD. In the last case the fluorescence intensity was higher than the original value (see Fig. SI-3†). In the presence of quinuclidine the effect was similar but lower than for DABCO.
In the presence of TEA we observed a small decrease in fluorescence lifetime for green QD with the increasing amine concentration (see Fig. SI-4†); thus, some dynamic fluorescence quenching seems to be involved in the quenching mechanism. In the case of core-shell QD the fluorescence lifetime did not change significantly in the presence of different TEA concentrations. With a Kq of 329 M−1 (for green core only CdSe QD) and a lifetime 50 ns the kq would be around 6.6 × 109 M−1 s−1, i.e., approaching the diffusion controlled rate, for dynamic quenching, although in this case (see ESI†) static and dynamic quenching are probably involved. The former requires pre-association of the fluorophore and quencher.
It is well known that after photon absorption by a semiconductor QD, an exciton (an electron–hole pair, equation (a) Chart 2) is generated which can recombine to emit a photon. The presence of electron donor compounds close to the surface can influence this emission pathway.
 | ||
| Chart 2 Mechanism proposed for the interaction of CdSe QD with TEA. | ||
The oxidation of TEA by the valence-band holes and the consequent steady-state photocurrents have been observed in CdS nanoparticle assemblies.22,23 In addition, TEA has been proposed to generate a stable monomer free radical after deprotonation in the presence of CdS semiconductor. This system is able to photocatalyze the polymerization of methyl methacrylate.24
According to the examples mentioned above electron transfer from the amine to the photogenerated hole would be anticipated. Given the electron donor ability of the amines examined here, triethylamine should be the best. The lower the availability of the free electron pair of the amine (DABCO and quinuclidine) the lower the quenching efficiency. We have observed a clear fluorescence quenching for TEA in contrast to the luminescence enhancement reported by Dannhouser et. al.18 for CdS. This is one of the few examples of fluorescence enhancement caused by an aliphatic amine. In another example, cyclohexylamine reversed the quenching caused by 4-aminoTEMPO;10 it is unclear if a similar mechanism (quenching reversal) may provide an interpretation of the results reported by Dannhouser et. al.18
After the excitation of the nanoparticle, the photogenerated hole (h+) will accept an electron from the amine giving rise to the corresponding radical cation. Our results seem to suggest that an initial approach of the amine to the QD surface (equation (b), Chart 2) will favor the electron transfer process from the amine to the hole of the nanoparticle following excitation (equation (c), Chart 2). In all cases, the decrease observed immediately after the addition of the amine is tentatively attributed to a favorable electron transfer process (see Chart 2).
The addition of an organic acid such as p-toluensulfonic acid to a QD–TEA solution causes a recovery of the fluorescence after 1 h of the acid addition (Fig. 6). The lone electron pair of the amine moiety is key for the fluorescence quenching, and probably mediates the electron transfer process. This fluorescence recovery may indicate reversible binding of the amine to the QD surface, with the amine being effectively removed from the surface by protonation in the presence of a strong acid. In order to check the binding of TEA to the QD surface and the possibility to exchange the ligand TOPO by the amine 31P NMR experiments were carried out.
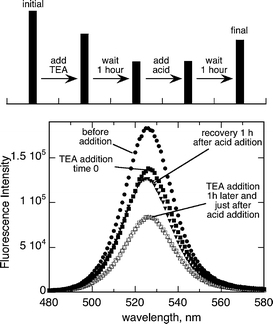 | ||
| Fig. 6 Steady state fluorescence spectra (λmax 380 nm) of 1.2 μM of green CdSe Q-dots in toluene after the addition of TEA (time 0 and 1 h) and the effect of p-toluensulfonic acid (time 0 and 1 h). The bar graph at the top shows the evolution of the experiment and the intensity at the corresponding maxima. | ||
It is known that when the size of the nanoparticles is reduced the particle has proportionally more surface, and interaction with other molecules can be enhanced. The effect of high concentration of butylamine on CdSe QD of 1–2 nm in diameter has been described; the relative weak absorption band at 445 nm changes to a broad strong band at 414 nm. The mechanism proposed relies on the binding of the amine to the QD surface.8 As a proof of concept, small green QD (2.1 nm in diameter) were synthesized and studied by FTIR and NMR techniques. In agreement with the blue shift observed for the other QD, a 7 nm blue shift was observed for this batch of nanoparticles (data not shown). Since the UV maximum is related to the size of the nanoparticle these changes suggest a reorganization of the CdSe QD surface structure or reduction of the nanoparticle diameter.
The P–O stretching band is shifted by >20 cm−1 relative to bulk TOPO25 on the QD surface (Fig. 7). When a high concentration of TEA was added to a QD solution and allowed to exchange for a couple of days (1 day under reflux and 1 day at room temperature with stirring) a solid complex was obtained. It was practically insoluble in all polar and no polar solvents but we were able to obtain an FTIR spectrum of this mixture. It was compared with the original QD spectrum (Fig. 8). Detailed FTIR spectra are shown in Fig. SI-5 and SI-6.† The absence of TOPO band (1260 cm−1) and the presence of amine band at (3450 cm−1) is consistent with passivation of the QD by the amine through surface substitution. This hypothesis was confirmed by 31P NMR.
 | ||
| Fig. 7 FTIR spectra of TOPO (black) and small green CdSe QD (gray) dispersed in KBr. | ||
 | ||
| Fig. 8 FTIR spectra of QD–TEA solid complex (black) and small green CdSe QD (gray) dispersed in KBr. | ||
The 31PNMR spectrum of the QD showed a typical broad band (20–40 ppm) due to the TOPO and TOP bound to the QD surface. After the addition of TEA to the QD NMR tube a sharp pick at 43.5 ppm was observed, which corresponds to TOPO (see Fig. SI-7†) release to the solution (Fig. 9).This experiment confirms the ligand exchange of TOPO by TEA. A similar behaviour has been observed for the addition of polydimethylaminoethyl methacrylate to CdSe/ZnS quantum dots used to passivate the surface.15
 | ||
| Fig. 9 The 31P NMR spectra of QD in toluene d8 before (A) and after (B) the addition of TEA at room temperature. Long time of accumulation was used in order to obtain a good (S/N) ratio. | ||
The presence of organic compounds on the surface of the nanoparticles can influence their luminescence properties, giving rise to the fluorescence enhancement or decrease depending on their nature. They can occupy surface traps or interact with the electron–hole pair generated upon nanoparticle excitation. The fluorescence quenching of QD by tertiary amines is size-dependent and is affected by the efficiency in the surface passivation and presumably the ‘quality’ of the QD. Core-shell QD showed relatively inefficient quenching.
Binding of TEA to hole-trapping emitting sites could explain the quenching effect observed. On the other hand, the partial recovery of the fluorescence of QD by cyclic tertiary amines can be understood if they bind to surface defects, that otherwise would trap excited electron and prevent fluorescence, or the creation of new fluorescent centers (QDs-amine). The access of the quencher to the QD core through the carbon chain of the ligands TOPO/TOP and the electron donor property of the amines play an important role in fluorescence quenching and could be the responsible for the intensity decrease with the time after the addition of each quencher concentration. This study is key to the understanding of the interaction between more complex amines and QD.
Conclusions
Our results show that for both core and core-shell QD the dominant effect of amine addition is a reduction of the fluorescence quantum yield. In earlier work with 4-amino TEMPO, we concluded that binding involved two distinct processes.10 A rapid one in which the quencher binds to available surface sites, followed by a second, more demanding one in which quencher binding occurs at the expense of TOPO displacement. Stepwise binding also occurs in the case of disulfides.26 The involvement of fast and slow processes, as shown in Fig. 2 and 3 is consistent with this two-stage mechanism for TEA; further FTIR and NMR data also support TOPO substitution as a requirement for binding. The fact that some of the luminescence is recovered by acid addition is consistent with TEA binding through the nitrogen lone pair (essentially as a Lewis base), but without structural changes in the amine, since these would be unlikely to be reversible.Acknowledgements
We acknowledge the generous financial support of the Natural Sciences and Engineering Research Council of Canada and of the Government of Ontario. REG wants to acknowledge the MEC for the award of a Ramón y Cajal contract.Notes and references
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- A. P. Alivisatos, J. Phys. Chem., 1996, 100, 13226–13239 CrossRef CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- A. M. Smith and S. Nie, Analyst, 2004, 129, 672–677 RSC.
- R. F. Heuff, J. L. Swift and D. T. Cramb, Phys. Chem. Chem. Phys., 2007, 9, 1870–1880 RSC.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- C. Landes, C. Burda, M. Braun and M. A. El-Sayed, J. Phys. Chem. B, 2001, 105, 2981–2986 CrossRef CAS.
- C. Landes and M. A. El-Sayed, J. Phys. Chem. A, 2002, 106, 7621–7627 CrossRef CAS.
- J.-G. Liang, S.-S. Zhang, X.-P. Ai, X.-H. Ji and Z.-K. He, Spectrochim. Acta, Part A, 2005, 61, 2974 CrossRef.
- V. Maurel, M. Laferriere, P. Billone, R. Godin and J. C. Scaiano, J. Phys. Chem. B, 2006, 110, 16353–16358 CrossRef CAS.
- J. C. Scaiano, M. Laferriere, R. E. Galian, V. Maurel and P. Billone, Phys. Status Solidi A, 2006, 203, 1337–1343 CrossRef CAS.
- M. Laferriere, R. E. Galian, V. Maurel and J. C. Scaiano, Chem. Commun., 2006, 257–259 RSC.
- E. Heafey, M. Laferrière and J. C. Scaiano, Photochem. Photobiol. Sci., 2007, 6, 580–584 RSC.
- K. Nose, H. Fujita, T. Omata, S. Otsuka-Yao-Matsuo, H. Nakamura and H. Maeda, J. Luminesc., 2007, 126, 21 CrossRef CAS.
- X. S. Wang, T. E. Dykstra, M. R. Salvador, I. Manners, G. D. Scholes and M. A. Winnik, J. Am. Chem. Soc., 2004, 126, 7784–7785 CrossRef CAS.
- M. Wang, J. K. Oh, T. E. Dykstra, X. Lou, G. D. Scholes and M. A. Winnik, Macromolecules, 2006, 39, 3664–3672 CrossRef CAS.
- M. Wang, T. D. Dykstra, X. Lou, M. R. Salvador, G. D. Scholes and M. A. Winnik, Angew. Chem., Int. Ed., 2006, 45, 2221–2224 CrossRef CAS.
- T. Dannhauser, M. O'Neil, K. Johansson, D. Whitten and G. McLendon, J. Phys. Chem., 1986, 90, 6074–6076 CrossRef CAS.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 183–184 CrossRef CAS.
- For further information visit web page www.evidenttech.com.
- W. W. Yu, L. Qu, Q. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- R. Baron, C.-H. Huang, D. M. Bassani, A. Onopriyenko, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2005, 44, 4010–4015 CrossRef CAS.
- V. Pardo-Yissar, T. Bourenko, J. Wasserman and I. Willner, Adv. Mater., 2002, 14, 670–673 CrossRef CAS.
- R. Ojah and S. K. Dolui, J. Photochem. Photobiol., A, 2005, 172, 121 CrossRef CAS.
- J. E. B. Katari, V. L. Colvin and A. P. Alivisatos, J. Phys. Chem., 1994, 98, 4109–4117 CrossRef.
- P. S. Billone, L. Maretti, V. Maurel and J. C. Scaiano, J. Am. Chem. Soc., 2007, 129, 14150–14151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. SI-1–SI-8. See DOI: 10.1039/b807580h |
| This journal is © The Royal Society of Chemistry and Owner Societies 2009 |