A new entry into cis-3-amino-2-methylpyrrolidines viaring expansion of 2-(2-hydroxyethyl)-3-methylaziridines†
Matthias
D'hooghe
,
Wim
Aelterman
and
Norbert
De Kimpe
*
Department of Organic Chemistry, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, B-9000, Ghent, Belgium
First published on 6th November 2008
Abstract
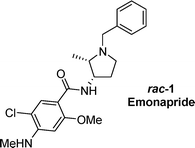
3-Amino-2-methylpyrrolidines were prepared via a novel protocol, involving the reductive ring closure and O-deprotection of γ-acetoxy-α-chloroketimines towards 2-(2-hydroxyethyl)-3-methylaziridines, followed by ring expansion of the latter into 3-bromopyrrolidines via intermediate bicyclic aziridinium salts and consecutive nucleophilic displacement of the bromo atom by azide towards 3-azidopyrrolidines. A final reduction of the azide moiety furnished 3-amino-2-methylpyrrolidines in high yields. Thus, a new formal synthesis of the antipsychotic emonapride was developed through preparation and further aroylation of cis-3-amino-1-benzyl-2-methylpyrrolidine.
Introduction
3-Aminopyrrolidines have received considerable interest from a medicinal point of view due to their broad applicability. For example, 7-(3-aminopyrrolidin-1-yl)-substituted naphthyridones and quinolones (such as clinafloxacine and tosufloxacine) have been described as broad-spectrum antibacterial agents,1 and different types of 3-aminopyrrolidines have been reported in the framework of anti-cancer research.2 Furthermore, the antipsychotic emonapride 1, a benzamide derived from the 3-amino-2-methylpyrrolidine scaffold, is of significant pharmacological interest.3 Consequently, a variety of 3-amino-2-methylpyrrolidine derived benzamides has been prepared and evaluated in terms of potential bioactivity.4 Despite the pharmaceutical importance of the 3-amino-2-methylpyrrolidine motif, few general and convenient approaches towards this class of compounds are known. 3-Amino-1-benzyl-2-methylpyrrolidine has been prepared from the corresponding pyrrolidin-3-oneviaimination with hydroxylamine and subsequent reduction,3 and different chiral 3-amino-1-(2-hydroxy-1-phenylethyl)-2-methylpyrrolidines have been synthesized via asymmetric conjugate additions to chiral bicyclic lactams followed by reductive cleavage.5 The first asymmetric synthesis of (2R,3R)-3-amino-1-benzyl-2-methylpyrrolidinevia a diastereoselective reductive alkylation has been reported starting from (S)-malic acid in 11 steps6 and, more recently, a one-pot synthesis of 2-substituted 3-nitropyrrolidines has been published based on a multicomponent domino reaction between imines and 3-nitro-1-(methanesulfonyloxy)propane.7In the present paper, a short and straightforward synthetic approach towards cis-3-amino-2-methylpyrrolidines viaring expansion of 2-(2-hydroxyethyl)-3-methylaziridines is disclosed. Moreover, the high yielding preparation of cis-3-amino-1-benzyl-2-methylpyrrolidine implies a new formal synthesis of the antipsychotic emonapride 1.
Results and discussion
As a model substrate for ring expansions of aziridines into pyrrolidines, the synthesis of 2-(2-hydroxyethyl)-2,3-dimethylaziridine 4 was envisaged. Thus, regiospecific alkylation of α-chloroketimine 28 was performed by treatment of the corresponding 3-chloro-1-azaallyl anion, obtained viaα-deprotonation by means of 1.2 equivalents of lithium diisopropylamide (LDA) in ice-cooled THF for one hour, with 1.05 equivalents of 1-bromo-2-[(trimethylsilyl)oxy]ethane in THF, affording the functionalized ketimine 3 in 70% yield (Scheme 1). Further reaction of the latter imine 3 with 2 molar equivalents of sodium borohydride in methanol under reflux furnished the corresponding 2-(2-hydroxyethyl)aziridine 4 as a 1/1 mixture of cis- and trans-isomers through hydride addition across the imino bond and subsequent cyclization upon expulsion of chloride (Scheme 1). During the reductive cyclization, hydride-induced deprotection of the silyl ether occurred, resulting in a free hydroxyl terminus. Attempted purification of aziridine 4 by distillation led only to a moderate yield of 44% and a purity of 85%. Treatment of distilled 2-(2-hydroxyethyl)aziridine 4 with 1.2 equivalents of triphenylphosphine and 1.2 equivalents of N-bromosuccinimide (NBS) in THF at room temperature for 20 hours furnished 3-bromopyrrolidine 6 as a mixture of cis- and trans-isomers, which could be separated and isolated by means of column chromatography on silica gel, albeit in low yield due to the small scale experiment. The formation of pyrrolidine 6 can be rationalized considering the initial replacement of the hydroxyl group by a bromo atom, followed by intramolecular nucleophilic substitution by nitrogen upon expulsion of bromide. The thus formed bicyclic aziridinium salt 5 is prone to ring opening by bromide towards pyrrolidine 6 (Scheme 1). Alternatively, the reaction could proceed via an oxyphosphorane intermediate instead of via the bromide, leading to the same bicyclic intermediate 5. The ring expansion of 2-(2-hydroxyethyl)aziridines into pyrrolidines via intermediate 1-azoniabicyclo[2.1.0]pentanes comprises an unexplored field of research, as only one literature example is known to date.9 Previously, the alcoholysis of different α-chloro-γ-[(trimethylsily)oxy]ketimines such as 3 has been described, affording a stereospecific entry into cis-2-alkoxy-3-aminooxolanes,10 which underlines the synthetic versatility of this class of imines.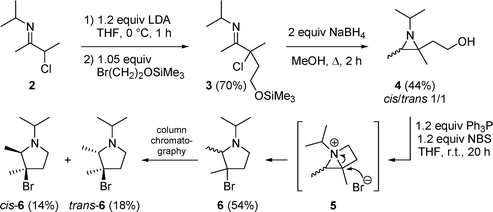 | ||
| Scheme 1 | ||
Despite the synthetic relevance of the transformations depicted in Scheme 1, the low yield and purity in which aziridine 4 was isolated called for a different approach. As the hydride-induced in situ formation of a free hydroxyl group might be responsible for side reactions in the synthesis of 2-(2-hydroxyethyl)aziridine 4, a different type of O-protecting group was required which is less reactive towards sodium borohydride. Thus, γ-acetoxyketimines 8a–c were prepared viaimination of 5-acetoxy-3-chloropentan-2-one 711 utilizing 4 equivalents of a primary amine in diethyl ether in the presence of 0.6 equivalents of titanium(IV) chloride at room temperature (Scheme 2). Subsequent reduction of the imino moiety and ring closure by means of 1.5 equivalents of sodium borohydride in methanol at room temperature furnished 2-(2-acetoxyethyl)aziridines 9a–c as mixtures of cis- and trans-isomers (1/1–1/3) in excellent yields (Scheme 2). The O-protecting group in the latter aziridines 9 was easily removed upon reaction with a methanolic solution of 1.5 equivalents of potassium carbonate at room temperature for 24 hours, yielding 2-(2-hydroxyethyl)aziridines 10a–c in high yields and purity (Scheme 2). The ratio of cis/trans-isomers for aziridines 9 and 10 could be assigned by comparison of 13C NMR values for both isomers with literature data of analogous 2,3-dimethylaziridines.12
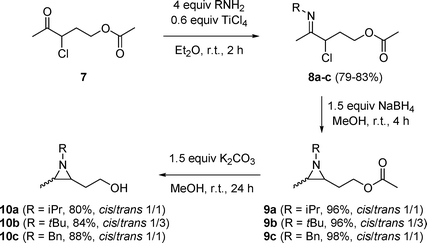 | ||
| Scheme 2 | ||
In the next step, 2-(2-hydroxyethyl)-3-methylaziridines 10 were converted into 3-bromo-2-methylpyrrolidines 11a–cvia intermediate bicyclic aziridinium salts 12 by the action of 1.2 equivalents of triphenylphosphine and 1.2 equivalents of NBS in THF at room temperature (Scheme 3). Although cis- and trans-pyrrolidines 11 could be separated by means of column chromatography, a convenient alternative for this laborious method comprised heating of the mixtures of cis/trans-11 in DMSO at 70–80 °C for 5–20 hours, followed by aqueous workup and extraction with diethyl ether. In this way, only trans-pyrrolidines trans-11 were obtained in high yields (49–52% after purification by column chromatography).
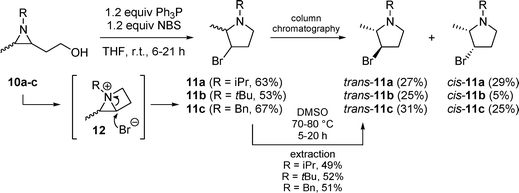 | ||
| Scheme 3 | ||
At first instance, an equilibrium shift from the bridged bicyclic ammonium ions 12–formed by heating of the mixture of cis/trans-11 in DMSO–to the corresponding carbenium ions by spontaneous ring opening, followed by neutralization of the latter carbenium ions by bromide was assumed to be responsible for the preferential formation of the more stable trans-pyrrolidines trans-11. However, this assumption could not be supported by experimental observations, as no isomerisation of cis-11 into trans-11 occurred by simply heating pure cis-pyrrolidines cis-11 under the same conditions (DMSO, 70 °C, 20 h). At present, no conclusive explanation can be provided for the experimental observation that only trans-pyrrolidines trans-11 were obtained upon heating of the mixtures of cis/trans-11 in DMSO at 70–80 °C for 5–20 hours, followed by aqueous workup and extraction with diethyl ether. The fact that heating of cis/trans-pyrrolidines cis/trans-11 was performed on the crude reaction mixture, in which presumably some bromine or another weak Lewis acid activator was present, might be of importance in that respect.
3-Bromopyrrolidines 11 constitute suitable substrates for further functionalization upon nucleophilic substitution of the halo atom. Treatment of trans-11a with 2 equivalents of potassium cyanide in DMSO for 24 hours at 90 °C afforded the corresponding cis-3-cyanopyrrolidinecis-13via a clean SN2 protocol (Scheme 4). Recently, a variety of 2-substituted pyrrolidine-3-carbonitriles has been prepared as potential therapeutics for the treatment of glaucoma.13 When azide was used instead of cyanide, 3-azidopyrrolidines 14a–c were formed utilizing 3–5 equivalents of sodium azide in DMSO at 80–90 °C for 8–18 hours (Scheme 4). Remarkably, 3-azido-1-isopropylpyrrolidine 14a was obtained as a mixture of cis/trans-isomers (cis/trans 3/2), whereas 3-azido-substituted 1-tert-butyl- and 1-benzylpyrrolidines 14b and 14c were formed predominantly as cis-isomers (cis/trans > 20/1). The formation of trans-3-azidopyrrolidines 14 can be explained considering the formation of an intermediate bicyclic aziridinium salt 15, followed by ring opening by azide at the more hindered position. In this case, double Walden inversion –i.e. two times SN2 reaction – results in retention of configuration, affording trans-pyrrolidines as the substitution products. Obviously, the competition between direct SN2 substitution and formation of a constrained bicyclic intermediate in these reactions is dependant on the type of nucleophile (cyanidevs.azide) and, to a lesser extent, the N-substituent in the substrate (isopropylvs. tert-butyl and benzyl). In the literature, nucleophilic substitutions of 3-halo- or 3-(sulfonyloxy)pyrrolidines usually proceed via a direct SN2 process, although also double substitution reactionsvia intermediate bicyclic aziridinium salts are known.14 The preference for one of these routes has been described to be dependant on different factors such as the solvent,15 the type of nucleophile16 and the relative stereochemistry of the substrate.17
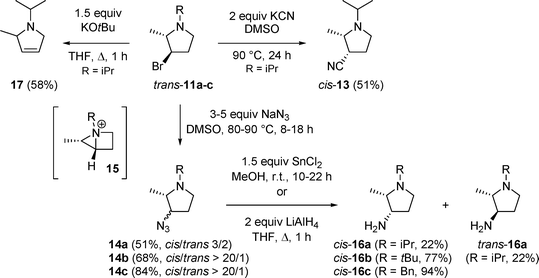 | ||
| Scheme 4 | ||
The final goal of this work comprised the synthesis of biologically relevant 3-amino-2-methylpyrrolidines 16, which were obtained by azide reduction upon treatment of pyrrolidines 14 with 1.5 equivalents of tin(II) chloride in methanol at room temperature (for R = iPr and tBu) or with 2 equivalents of lithium aluminium hydride in THF under reflux (for R = Bn) (Scheme 4). The relative stereochemistry of pyrrolidines 11, 13, 14 and 16 could be assigned based on NOE effects between the 2-methyl group and the hydrogen atoms at the 2- and the 3-position (for example, a NOE of 1.8% and 2.5% was observed between 2-H and 3-H for cis-1-isopropylpyrrolidines 13 and 14a, respectively). Besides substitution reactions, dehydrobromination of 3-bromopyrrolidine trans-11a was evaluated using 1.5 equivalents of potassium tert-butoxide in THF under reflux, affording 2-methyl-3-pyrroline 17 in 58% yield (Scheme 4).
The stereochemical assignment of cis-/trans-pyrrolidines 11a–c, 14a and 16a was further supported by careful analysis of spectroscopical data. In the literature, 2,3-disubstituted pyrrolidines are characterized by a so called “γ-gauche effect” in 13C NMR for the carbon atom present at the 2-position. This effect results in a significant difference in chemical shift Δ(δtrans−δcis) of 3–5 ppm between cis and trans isomers.18 Also for pyrrolidines 11a–b, 14a and 16a, a Δ(δtrans−δcis) ranging from 2.2 to 3.8 was observed, which confirmed the postulated stereochemical assignment.
The above-described methodology for the preparation of cis-3-amino-1-benzyl-2-methylpyrrolidinecis-16c allows the formal synthesis of the antipsychotic (±)-emonapride 1 in high overall yield, as the coupling of pyrrolidinecis-16c with 5-chloro-2-methoxy-4-(methylamino)benzoic acid in the presence of ethyl chloroformiate and triethylamine has been described in the literature to afford emonapride 1 (Scheme 5).3 In the light of the pharmaceutical importance of emonapride 1 and its derivatives, the present methodology offers a useful and efficient alternative for the limited number of known synthetic methodologies towards this class of compounds.
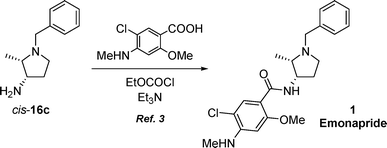 | ||
| Scheme 5 | ||
In conclusion, a novel and efficient approach towards biologically relevant 3-amino-2-methylpyrrolidines has been developed starting from γ-acetoxy-α-chloroketimines. This methodology involves the reductive ring closure and O-deprotection of γ-acetoxy-α-chloroketimines towards 2-(2-hydroxyethyl)-3-methylaziridines, followed by ring expansion of the latter into 3-bromopyrrolidines via intermediate bicyclic aziridinium salts and consecutive nucleophilic displacement of the bromo atom by azide. A final reduction of the azide moiety afforded the title compounds in good yields. Furthermore, the straightforward preparation of cis-3-amino-1-benzyl-2-methylpyrrolidine implies a new formal synthesis of the antipsychotic emonapride.
Experimental section
Synthesis of N-[2-chloro-1-methyl-2-(2-trimethylsilyloxyethyl)propylidene] isopropylamine 3
To an ice-cooled solution of diisopropylamine (12 mmol) in dry THF (10 mL) under nitrogen atmosphere was added n-BuLi (12 mmol, 2.5 M in hexane) via a syringe. After 10 minutes, a solution of α-chloroketimine 28 (10 mmol) in THF (5 mL) was added via a syringe, after which the resulting solution was stirred for 1 hour at 0 °C. Subsequently, a solution of 1-bromo-2-[(trimethylsilyl)oxy]ethane (10.5 mmol) in THF (5 mL) was added via a syringe at 0 °C, and the resulting solution was stirred for 15 hours at room temperature. Afterwards, the reaction mixture was poured into an aqueous solution of NaOH (15 mL, 0.5 M) and extracted with Et2O (3 × 15 mL). Drying (K2CO3), removal of the drying agent and evaporation of the solvent afforded the crude imine 3, which was purified by distillation (Bp. 60–62 °C/0.05 mmHg).
1H NMR (270 MHz, CDCl3): δ 0.10 (9H, s); 1.10 (6H, d, J = 6 Hz); 1.70 (3H, s); 1.99 (3H, s); 2.0-2.4 (2H, m); 3.6-3.8 (1H, m); 3.76 (2H, t, J = 7 Hz). 13C NMR (68 MHz, CDCl3): δ−0.5, 12.9, 23.2, 28.4, 44.5, 50.7, 59.3, 74.9, 164.5. IR (NaCl): νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N = 1651 cm−1. MS (70 eV): m/z (%): no M+; 248/50 (7); 228 (5); 147/9 (92); 132 (17); 112 (23); 96 (20); 84 (100); 73 (21).
N = 1651 cm−1. MS (70 eV): m/z (%): no M+; 248/50 (7); 228 (5); 147/9 (92); 132 (17); 112 (23); 96 (20); 84 (100); 73 (21).
Synthesis of 2-(2-hydroxyethyl)-1-isopropyl-2,3-dimethylaziridine 4
To an ice-cooled solution of imine 3 (5 mmol) in methanol (10 mL) was added NaBH4 (10 mmol) in small portions, and the resulting mixture was heated under reflux for 2 hours. Afterwards, the reaction mixture was poured into water (15 mL) and extracted with CH2Cl2 (3 × 15 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded the crude aziridine 4, which was purified by distillation (Bp. 50–54 °C/0.3 mmHg).1H NMR (270 MHz, CDCl3): δ 1.05 (3H, d, J = 6.6 Hz); 1.09 and 1.10 (6H, 2 × d, J = 6.3 Hz); 1.00-1.24 (11H, m); 1.25 and 1.26 (6H, 2 × s); 1.61 (4H, ∼t, J = 6.3 Hz); 2.19 (2H, ∼septet, J = 6.3 Hz); 3.56–3.92 (4H, m). 13C NMR (68 MHz, CDCl3): δ 14.9, 17.2, 20.6, 23.1, 23.2, 23.3, 23.4, 35.9, 36.5, 41.8, 43.2, 43.5, 44.4, 52.8, 52.9, 60.77, 60.81. IR (NaCl): νOH = 3570–3050 cm−1. MS (70 eV): m/z (%): 157 (M+, 1); 142 (5); 128 (3); 126 (6); 124 (5); 114 (16); 112 (13); 98 (10); 96 (4); 94 (5); 86 (32); 84 (29); 82 (8); 70 (79); 67 (6); 58 (10); 55 (12); 53 (6).
Synthesis of 3-bromo-1-isopropyl-2,3-dimethylpyrrolidine 6
To a solution of aziridine 4 (5 mmol) in THF (30 mL) was added N-bromosuccinimide (6 mmol) and triphenylphosphine (6 mmol) at room temperature, after which the mixture was stirred for 20 hours at room temperature. Afterwards, the reaction mixture was poured into water (30 mL) and extracted with Et2O (3 × 25 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded a residue, to which Et2O (25 mL) was added. After a second filtration, Et2O (25 mL) was added and the suspension was stored at −20 °C for 15 hours. A final filtration and evaporation of the solvent afforded the crude pyrrolidinecis/trans-6, which was purified by column chromatography on silica gel (hexane/EtOAc/MeOH 90/7/3) in order to separate both isomers.cis-3-Bromo-1-isopropyl-2,3-dimethylpyrrolidine cis-6
Rf 0.11 (hexane/EtOAc/MeOH 90/7/3). 1H NMR (270 MHz, CDCl3): δ 0.91 and 1.15 (6H, 2 × d, J = 6.6 Hz); 1.14 (3H, d, J = 5.9 Hz); 1.79 (3H, s); 1.97 (1H, d × d × d, J = 14.1, 9.9, 7.7 Hz); 2.27 (1H, q, J = 5.9 Hz); 2.46 (1H, d × d × d, J = 14.1, 7.9, 3.6 Hz); 2.68 (1H, t × d, J = 9.9, 3.6 Hz); 3.01-3.12 (1H, m); 3.09 (1H, septet, J = 6.6 Hz). 13C NMR (68 MHz, CDCl3): δ 13.3, 16.8, 22.1, 30.0, 42.3, 43.1, 47.4, 65.8, 75.5. IR (NaCl): νmax = 2958, 1448, 1375, 1358, 1100 cm−1. MS (70 eV): m/z (%): 219/21 (M+, 11); 204/6 (49); 140 (50); 137 (11); 125 (21); 124 (61); 122 (12); 110 (18); 98 (91); 96 (21); 94 (31); 84 (22); 83 (28); 82 (25); 81 (19); 69 (17); 56 (100); 53 (19); 49 (11). Anal. Calcd for C9H18BrN: C 49.10, H 8.24, N 6.36. Found: C 49.73, H 8.56, N 6.52.trans-3-Bromo-1-isopropyl-2,3-dimethylpyrrolidine trans-6
Rf 0.29 (hexane/EtOAc/MeOH 90/7/3). 1H NMR (270 MHz, CDCl3): δ 1.00 and 1.11 (6H, 2 × d, J = 6.6 Hz); 1.02 (3H, d, J = 6.6 Hz); 1.77 (3H, s); 2.04 (1H, d × d × d, J = 13.6, 9.0, 7.6 Hz); 2.27 (1H, d × d×d, J = 13.6, 7.1, 2.6 Hz); 2.82-3.00 (2H, m); 3.00 (1H, septet, J = 6.6 Hz); 3.36 (1H, q, J = 6.6 Hz). 13C NMR (68 MHz, CDCl3): δ 16.9, 22.3, 17.9, 28.5, 42.2, 45.6, 50.4, 68.0, 72.2. IR (NaCl): νmax = 2960, 1500, 1378, 1210, 1170, 1074 cm−1. MS (70 eV): m/z (%): 219/21 (M+, 16); 204/6 (77); 140 (58); 137 (16); 125 (45); 124 (87); 122 (14); 110 (33); 98 (99); 96 (22); 94 (36); 83 (30); 82 (35); 81 (20); 70 (16); 56 (100). Anal. Calcd for C9H18BrN: C 49.10, H 8.24, N 6.36. Found: C 49.39, H 8.38, N 6.27.Synthesis of N-(4-acetoxy-2-chloro-1-methylbutylidene)amines 8
General procedure: To a solution of 5-acetoxy-3-chloropentan-2-one 711 (10 mmol) in dry Et2O (25 mL) was added amine (40 mmol), followed by the gentle addition of a solution of TiCl4 (6 mmol) in pentane (10 mL) at 0 °C, after which the resulting mixture was stirred for 2 hours at room temperature. Afterwards, the reaction mixture was poured into an aqueous solution of NaOH (15 mL, 0.5 M) and extracted with Et2O (3 × 25 mL). Drying (K2CO3), removal of the drying agent and evaporation of the solvent afforded the crude imine 8, which was purified by distillation. For the synthesis of N-benzylimine 8c, a mixture of 1 equiv of benzylamine and 3 equiv of Et3N was used instead of 4 equiv of benzylamine.N-(4-Acetoxy-2-chloro-1-methylbutylidene)isopropylamine 8a
Bp. 53–56 °C/0.03 mmHg. 1H NMR (270 MHz, CDCl3): δ 1.10 and 1.13 (6H, 2 × d, J = 6.3 Hz); 1.94 (3H, s); 2.05-2.35 (2H, m); 3.69 (1H, septet, J = 6.3 Hz); 4.15–4.30 (2H, m); 4.49 (1H, d × d, J=8.9, 6.0 Hz). 13C NMR (68 MHz, CDCl3): δ 12.9, 20.9, 23.0, 34.1, 50.8, 61.2, 64.0, 163.2, 170.9. IR (NaCl): νCO = 1740 cm−1, νCN = 1658 cm−1. MS (70 eV): m/z (%): 218/220 (M+, 0.4); 184/6 (3); 144/6 (5); 134/6 (3); 133/5 (21); 120 (3); 119 (2); 118 (5); 112 (2); 108 (2); 104 (2); 98 (7); 85 (4); 84 (37); 82 (4); 80 (2); 76 (4); 70 (2); 68 (2); 67 (3); 61 (3); 60 (2); 55 (5); 54 (3); 53 (2); 49 (2); 42 (100).
Synthesis of 2-(2-acetoxyethyl)-3-methylaziridines 9
General procedure: To an ice-cooled solution of imine 8 (5 mmol) in methanol (10 mL) was added NaBH4 (7.5 mmol) in small portions, and the resulting mixture was stirred at room temperature for 4 hours. Afterwards, the reaction mixture was poured into water (15 mL) and extracted with CH2Cl2 (3 × 15 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded aziridine 9. The isomers cis/trans-9a and cis/trans-9b were separated by preparative gas chromatography.cis-2-(2-Acetoxyethyl)-1-isopropyl-3-methylaziridine cis-9a
1H NMR (270 MHz, CDCl3): δ 1.09 and 1.11 (6H, 2 × d, J = 6.3 Hz); 1.15 (3H, d, J = 5.6 Hz); 1.36-1.58 (3H, m); 1.60-1.88 (2H, m); 2.07 (3H, s); 4.20 (2H, t, J = 6.5 Hz). 13C NMR (68 MHz, CDCl3): δ 13.7, 21.0, 21.9, 27.5, 37.8, 40.1, 61.1, 63.2, 171.1. IR (NaCl): νCO = 1738 cm−1. MS (70 eV): m/z (%): 185 (M+, 4); 170 (5); 142 (14); 126 (24); 125 (6); 112 (45); 110 (16); 100 (9); 85 (7); 84 (16); 83 (13); 82 (100); 72 (5); 71 (5); 70 (64); 69 (8); 68 (12); 67 (6); 58 (5); 57 (6); 56 (23); 55 (25); 54 (13); 42 (100). Anal. Calcd for C10H19NO2: C 64.83, H 10.34, N 7.56. Found: C 64.62, H 10.07, N 7.44.
trans-2-(2-Acetoxyethyl)-1-isopropyl-3-methylaziridine trans-9a
1H NMR (270 MHz, CDCl3): δ 1.10 (6H, d, J = 6.1 Hz); 1.28 (3H, d, J = 5.9 Hz); 1.10-1.25 (1H, m); 1.45-1.92 (3H, m); 2.06 (3H, s); 2.21 (1H, septet, J = 6.1 Hz); 4.18 (2H, d × d, J = 7.4, 5.8 Hz). 13C NMR (68 MHz, CDCl3): δ 11.1, 21.0, 22.9, 23.0, 32.5, 38.2, 41.5, 50.8, 63.0, 171.1. IR (NaCl): νCO = 1739 cm−1. MS (70 eV): m/z (%): 185 (M+, 4); 170 (5); 142 (14); 126 (24); 125 (6); 112 (45); 110 (16); 100 (9); 85 (7); 84 (16); 83 (13); 82 (100); 72 (5); 71 (5); 70 (64); 69 (8); 68 (12); 67 (6); 58 (5); 57 (6); 56 (23); 55 (25); 54 (13); 42 (100). Anal. Calcd for C10H19NO2: C 64.83, H 10.34, N 7.56. Found: C 64.59, H 10.10, N 7.54.
Synthesis of 2-(2-hydroxyethyl)-3-methylaziridines 10
General procedure: To a solution of aziridine 9 (10 mmol) in dry methanol (20 mL) was added K2CO3 (15 mmol), and the resulting mixture was stirred at room temperature for 24 hours. Afterwards, the reaction mixture was poured into water (25 mL) and extracted with CH2Cl2 (3 × 15 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded the crude aziridine 10, which was purified by distillation.2-(2-Hydroxyethyl)-1-isopropyl-3-methylaziridine 10a
Bp. 85–92 °C/8 mmHg. 1H NMR (270 MHz, CDCl3): δ 1.11, 1.13, 1.15, 1.16 and 1.28 (2 × 9H, 5 × d, J = 6.2 Hz); 1.39-2.25 (2 × 5H, m); 3.57-3.92 (2 × 2H, m). 13C NMR (68 MHz, CDCl3): δcis-10a 13.6, 21.8, 21.9, 29.9, 37.6, 40.8, 61.1, 61.3. δtrans-10a 10.8, 22.7, 23.0, 32.8, 36.2, 42.6, 50.9, 60.3. IR (NaCl): νOH = 3540–3020 cm−1. MS (70 eV): m/zcis-10a (%): 143 (M+, 4); 128 (17); 112 (57); 100 (27); 98 (11); 84 (11); 70 (100); 57 (17); 56 (82); 55 (14). m/ztrans-10a (%): 143 (M+, 8); 128 (19); 112 (55); 100 (28); 99 (11); 98 (13); 84 (25); 70 (100); 56 (84). Anal. Calcd for C8H17NO: C 67.09, H 11.96, N 9.78. Found: C 67.27, H 12.19, N 7.66.Synthesis of 3-bromo-2-methylpyrrolidines 11
General procedure: To a solution of aziridine 10 (5 mmol) in THF (30 mL) was added N-bromosuccinimide (6 mmol) and triphenylphosphine (6 mmol) at room temperature, after which the mixture was stirred for 6–21 hours at room temperature. Afterwards, the reaction mixture was poured into water (30 mL) and extracted with Et2O (3 × 25 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded a residue, to which Et2O (25 mL) was added. After a second filtration, Et2O (25 mL) was added and the suspension was stored at −20 °C for 15 hours. A final filtration and evaporation of the solvent afforded the crude pyrrolidinecis/trans-11, which was purified by column chromatography on silica gel in order to separate both isomers.cis-3-Bromo-1-isopropyl-2-methylpyrrolidine cis-11a
Rf 0.15 (hexane/EtOAc/MeOH 90/8/2). 1H NMR (270 MHz, CDCl3): δ 0.95 and 1.12 (6H, 2 × d, J = 6.4 Hz); 1.14 (3H, d, J = 6.3 Hz); 2.15-2.47 (2H, m); 2.65 (1H, d × t, J = 8.9, 6.5 Hz); 2.84–3.05 (3H, m); 4.40 (1H, d × t, J = 6.6, 5.3 Hz). 13C NMR (68 MHz, CDCl3): δ 15.6, 16.7, 21.9, 34.5, 44.6, 48.5, 55.5, 58.2. IR (NaCl): νmax = 2960, 1450, 1380, 1188 cm−1. MS (70 eV): m/z (%): 205/7 (M+, 24); 190/2 (100); 148/50 (11); 126 (43); 111 (26); 110 (26); 96 (20); 85 (13); 84 (77); 83 (15); 82 (22); 81 (10); 80 (16); 69 (38); 68 (22); 67 (29); 57 (34); 56 (69); 55 (28); 54 (12); 53 (11). Anal. Calcd for C8H16BrN: C 46.62, H 7.82, N 6.80. Found: C 46.49, H 7.73, N 6.85.trans-3-Bromo-1-isopropyl-2-methylpyrrolidine trans-11a
Rf 0.11 (hexane/EtOAc/MeOH 90/8/2). 1H NMR (270 MHz, CDCl3): δ 0.97 and 1.12 (6H, 2 × d, J = 6.6 Hz); 1.14 (3H, d, J = 6.3 Hz); 1.98-2.11 and 2.32-2.50 (2H, 2 × m); 2.74–3.10 (3H, m); 3.02 (1H, septet, J = 6.6 Hz); 3.89 (1H, d × t, J = 7.6, 5.0 Hz). 13C NMR (68 MHz, CDCl3): δ 15.5, 18.9, 21.9, 33.8, 45.2, 49.3, 54.5, 65.1. IR (NaCl): νmax = 2960, 1455, 1381, 1363, 1208, 1171 cm−1. MS (70 eV): m/z (%): 205/7 (M+, 20); 190/2 (100); 148/50 (15); 126 (20); 111 (33); 110 (25); 96 (21); 84 (45); 83 (11); 82 (16); 80 (12); 69 (37); 68 (22); 67 (21); 57 (23); 56 (52); 55 (23); 54 (10). Anal. Calcd for C8H16BrN: C 46.62, H 7.82, N 6.80. Found: C 46.81, H 8.06, N 6.93.Synthesis of cis-3-cyano-1-isopropyl-2-methylpyrrolidinecis-13
To a solution of pyrrolidinetrans-11a (2 mmol) in DMSO (10 mL) was added KCN (4 mmol), and the resulting mixture was heated at 90 °C for 24 hours. Afterwards, the reaction mixture was poured into water (15 mL) and extracted with Et2O (3 × 10 mL). The combined organic extracts were then washed with brine (2 × 20 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded 3-cyanopyrrolidine cis-13, which was purified by column chromatography on silica gel (hexane/EtOAc/MeOH 76/19/5).Rf 0.20 (hexane/EtOAc/MeOH 76/19/5). 1H NMR (270 MHz, CDCl3): δ 0.93 and 1.13 (6H, 2 × d, J = 6.6 Hz); 1.20 (3H, d, J = 5.9 Hz); 1.97 and 2.18 (2H, 2 × d, J = 12.9 Hz); 2.53 (1H, d × d×d, J = 9.6, 7.9, 6.5 Hz); 2.60-2.72 and 2.85-3.01 (2H, m); 2.81-2.95 (1H, m); 3.07 (1H, septet, J = 6.6 Hz). 13C NMR (68 MHz, CDCl3): δ 13.8, 22.1, 18.1, 26.9, 35.0, 44.3, 47.4, 59.7, 121.9. IR (NaCl): νCN = 2213 cm−1. MS (70 eV): m/z (%): 152 (M+, 13); 137 (100); 109 (6); 95 (23); 84 (7); 71 (5); 70 (8); 68 (10); 57 (15); 56 (18). Anal. Calcd for C9H16N2: C 71.01, H 10.59, N 18.40. Found: C 71.16, H 10.70, N 18.32.
Synthesis of 3-azido-2-methylpyrrolidines 14
General procedure: To a solution of pyrrolidinetrans-11 (6 mmol) in DMSO (30 mL) was added NaN3 (18-30 mmol), and the resulting mixture was heated at 80–90 °C for 8–18 hours. Afterwards, the reaction mixture was poured into water (45 mL) and extracted with Et2O (3 × 30 mL). The combined organic extracts were then washed with brine (2 × 50 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded 3-azidopyrrolidine 14, which was purified by column chromatography on silica gel.cis-3-Azido-1-isopropyl-2-methylpyrrolidine cis-14a
Rf 0.18 (hexane/EtOAc/MeOH 76/19/5). 1H NMR (270 MHz, CDCl3): δ 0.90 and 1.12 (6H, 2 × d, J = 6.6 Hz); 1.09 (3H, d, J = 6.3 Hz); 1.89 and 2.10 (2H, d × d×d × d, J = 13.4, 9.0, 7.3, 4.5 Hz); 2.55 (1H, t × d, J = 9.0, 7.3 Hz); 2.81-2.98 (2H, m); 3.00 (1H, septet, J = 6.6 Hz); 3.78 (1H, d × t, J = 7.3, 4.5 Hz). 13C NMR (68 MHz, CDCl3): δ 13.5, 14.2, 22.0, 28.7, 43.6, 47.5, 58.4, 64.3. IR (NaCl): νN3 = 2100 cm−1. MS (70 eV): m/z (%): 168 (M+, 14); 153 (13); 125 (12); 113 (15); 98 (8); 85 (18); 84 (28); 83 (21); 82 (14); 70 (100); 68 (14); 68 (14); 56 (86); 55 (17). Anal. Calcd for C8H16N4: C 57.11, H 9.59, N 33.30. Found: C 57.37, H 9.95, N 33.54.trans-3-Azido-1-isopropyl-2-methylpyrrolidine trans-14a
Rf 0.20 (hexane/EtOAc/MeOH 76/19/5). 1H NMR (270 MHz, CDCl3): δ 0.96, 1.11 and 1.13 (9H, 3 × d, J = 6.3 Hz); 1.68-1.81 and 2.08-2.21 (2H, m); 2.61-2.73 and 2.82-3.05 (4H, m); 3.51 (1H, t × d, J = 8.1, 4.0 Hz). 13C NMR (68 MHz, CDCl3): δ 15.8, 18.3, 21.9, 28.7, 45.0, 48.8, 61.0, 67.3. IR (NaCl): νN3 = 2085 cm−1. MS (70 eV): m/z (%): 168 (M+, 7); 153 (7); 125 (11); 113 (15); 98 (9); 85 (16); 84 (25); 83 (12); 70 (100); 68 (11); 56 (69); 55 (13). Anal. Calcd for C8H16N4: C 57.11, H 9.59, N 33.30. Found: C 57.43, H 9.82, N 33.46.Synthesis of 3-amino-2-methylpyrrolidines 16a,b
General procedure: To a solution of pyrrolidine 14 (1 mmol) in dry methanol (3 mL) under nitrogen atmosphere was added SnCl2 (1.5 mmol), and the resulting mixture was stirred for 10–22 hours at room temperature. Afterwards, the reaction mixture was poured into an aqueous solution of NaOH (30 mL, 0.5 M) and extracted with CH2Cl2 (3 × 20 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded the crude pyrrolidine 16, which was purified by column chromatography on silica gel.cis-3-Amino-1-isopropyl-2-methylpyrrolidine cis-16a
Rf 0.11 (CH2Cl2/MeOH/NH4OH(25%) 43.7/10.9/1). 1H NMR (270 MHz, CDCl3): δ 0.98 and 1.15 (6H, 2 × d, J = 6.6 Hz); 1.04 (3H, d, J = 6.3 Hz); 1.44-1.55 and 1.96-2.15 (2H, 2 × m); 2.56 (1H, ∼q, J = 8.6 Hz); 2.79 (1H, pent, J = 6.3 Hz); 2.92 (1H, t × d, J = 8.6, 4.0 Hz); 2.99 (1H, septet, J = 6.6 Hz); 3.30 (1H, q, J = 6.3 Hz). 13C NMR (68 MHz, CDCl3): δ 12.6, 15.4, 21.6, 32.5, 44.4, 48.8, 54.3, 58.7. IR (NaCl): νNH2 = 3560–3000 cm−1. MS (70 eV): m/z (%): 142 (M+, 26); 127 (31); 99 (38); 86 (25); 85 (25); 84 (50); 82 (23); 80 (24); 72 (26); 71 (32); 70 (26); 57 (88); 56 (100). Anal. Calcd for C8H18N2: C 67.55, H 12.75, N 19.69. Found: C 67.72, H 12.91, N 19.51.trans-3-Amino-1-isopropyl-2-methylpyrrolidine trans-16a
Rf 0.14 (CH2Cl2/MeOH/NH4OH(25%) 43.7/10.9/1). 1H NMR (270 MHz, CDCl3): δ 0.96 and 1.14 (6H, 2 × d, J = 6.5 Hz); 1.11 (3H, d, J = 6.2 Hz); 1.33-1.46 (1H, m); 1.45-1.90 (2H, m); 2.15 (1H, d × q, J = 12.7, 7.9 Hz); 2.34 (1H, pent, J = 6.0 Hz); 2.70 (1H, d × t, J = 8.9, 8.2 Hz); 2.88 (1H, t × d, J = 8.9, 4.0 Hz); 2.90–3.02 (1H, m); 3.03 (1H, septet, J = 6.5 Hz). 13C NMR (68 MHz, CDCl3): δ 14.7, 17.1, 21.8, 32.5, 44.0, 48.4, 58.4, 64.5. IR (NaCl): νNH2 = 3590–3000 cm−1. MS (70 eV): m/z (%): 142 (M+, 40); 127 (47); 99 (54); 86 (39); 85 (42); 84 (64); 80 (36); 72 (41); 71 (46); 70 (41); 58 (40); 57 (97); 56 (100). Anal. Calcd for C8H18N2: C 67.55, H 12.75, N 19.69. Found: C 67.80, H 12.97, N 19.81.Synthesis of cis-3-amino-1-benzyl-2-methylpyrrolidinecis-16c
To an ice-cooled solution of pyrrolidine 14c (4 mmol) in dry THF (10 mL) was added LiAlH4 (8 mmol) in small portions, and the resulting suspension was heated under reflux for 1 hour. Afterwards, water (3 mL) was added at 0 °C in order to neutralize the excess of LiAlH4. The mixture was stirred for 10 minutes, after which the grey suspension was filtered over K2CO3 and celite. The filter cake was then washed thoroughly with dry ether (3 × 20 mL). Removal of the solventin vacuo afforded pyrrolidinecis-16c (purity > 97% based on GC).1H NMR (270 MHz, CDCl3): δ 1.12 (3H, d, J = 6.4 Hz); 1.35-1.51 and 1.96-2.19 (5H, 2 × m); 2.34 (1H, quint, J = 6.4 Hz); 2.89 (1H, t × d, J = 8.7, 2.3 Hz); 3.14 (1H, d, J = 13.1 Hz); 3.21-3.29 (1H, m); 3.97 (1H, d, J = 13.1 Hz); 7.19-7.38 (5H, m). 13C NMR (68 MHz, CDCl3): δ 13.6, 33.0, 51.8, 54.6, 57.9, 63.0, 126.8, 128.1, 128.8, 139.5. IR (NaCl): νNH2 = 3380–3200 cm−1. MS (70 eV): m/z (%): 190 (M+, 1); 173 (12); 147 (19); 91 (30); 65 (6); 56 (100). Anal. Calcd for C12H18N2: C 75.74, H 9.53, N 14.72. Found: C 75.96, H 9.70, N 14.87.
Synthesis of 1-isopropyl-2-methyl-3-pyrroline 17
To a solution of pyrrolidine 11a (2.5 mmol) in THF (5 mL) was added KOtBu (3.75 mmol), and the resulting mixture was heated under reflux for 1 hour. Afterwards, the reaction mixture was poured into water (10 mL) and extracted with Et2O (3 × 10 mL). Drying (MgSO4), removal of the drying agent and evaporation of the solvent afforded pyrroline 17 (purity > 95% based on GC).1H NMR (270 MHz, CDCl3): δ 1.03 and 1.12 (6H, 2 × d, J = 6.4 Hz); 1.15 (3H, d, J = 6.3 Hz); 2.92 (1H, septet, J = 6.4 Hz); 3.46 and 3.67 (2H, 2 × d, J = 13.9 Hz); 3.68-3.83 (1H, m); 5.62 and 5.71 (2H, 2 × d, J = 6.1 Hz). 13C NMR (68 MHz, CDCl3): δ 18.9, 22.0, 21.5, 50.9, 55.8, 62.0, 125.9, 133.4. IR (NaCl): νmax = 2960, 1382, 1365, 1180 cm−1. MS (70 eV): m/z (%): 125 (M+, 19); 110 (67); 82 (19); 80 (7); 68 (100); 55 (21). Anal. Calcd for C8H15N: C 76.74, H 12.07, N 11.19. Found: C 76.97, H 12.26, N 11.04.
Acknowledgements
The authors are indebted to the “Fund for Scientific Research – Flanders (Belgium)” (FWO–Vlaanderen) and to Ghent University (GOA) for financial support.References
- D. Bouzard, P. Di Cesare, M. Essiz, J. P. Jacquet, B. Ledoussal, P. Remuzon, R. E. Kessler and J. Fung-Tomc, J. Med. Chem., 1992, 35, 518 CrossRef CAS.
- (a) M. Takamatsu, M. Matsui and Y. Ikeda, Eur. Pat. Appl., 1989 Search PubMed , EP 327709 A2; M. Takamatsu, M. Matsui and Y. Ikeda, Chem. Abstr., 1990, 113, 71314 Search PubMed; (b) Y. Ito, H. Kato, S. Yasuda, N. Kato, T. Yoshida, and Y. Yamamoto, Jpn. Kokai Tokkyo Koho 1994, JP 06016624 A; Chem. Abstr., 1994, 121, 9155 Search PubMed; (c) K. Nakada, S. Kashimoto, M. Tajima, T. Ooe, K. Chiba, and K. Shibamori, Jpn. Kokai Tokkyo Koho 1996, JP 08073460 A; Chem. Abstr., 1996, 125, 58488 Search PubMed; (d) Y. Tsuzuki, K. Tomita, K. Shibamori, Y. Sato, S. Kashimoto and K. Chiba, J. Med. Chem., 2004, 47, 2097 CrossRef; (e) M. Atanasova, S. Ilieva and B. Galabov, Eur. J. Med. Chem., 2007, 42, 1184 CrossRef CAS.
- S. Iwanami, M. Takashima, Y. Hirata, O. Hasegawa and S. Usada, J. Med. Chem., 1981, 24, 1224 CrossRef CAS.
- J. Ohmori, K. Maeno, K. Hidaka, K. Nakato, S. Sakamoto and S.-i. Tsukamoto, PCT Int. Appl., 1995 Search PubMed , WO 9508533 A1; J. Ohmori, K. Maeno, K. Hidaka, K. Nakato, S. Sakamoto and S.-i. Tsukamoto, Chem. Abstr., 1995, 123, 285754 Search PubMed.
- C. J. Andres, P. H. Lee, T. H. Nguyen and A. I. Meyers, J. Org. Chem., 1995, 60, 3189 CrossRef CAS.
- (a) P. Q. Huang, S. L. Wang, H. Zheng and X. S. Fei, Tetrahedron Lett., 1997, 38, 271 CrossRef CAS; (b) P. Q. Huang, S. L. Wang, J. L. Ye, Y. P. Ruan, Y. Q. Huang, H. Zheng and J. X. Gao, Tetrahedron, 1998, 54, 12547 CrossRef CAS.
- N. Baricordi, S. Benetti, G. Biondini, C. De Risi and G. P. Pollini, Tetrahedron Lett., 2004, 45, 1373 CrossRef CAS.
- (a) N. De Kimpe, R. Verhé, L. De Buyck, L. Moens and N. Schamp, Synthesis, 1982, 43 CrossRef CAS; (b) N. De Kimpe, P. Sulmon and N. Schamp, Angew. Chem. Int. Ed., 1985, 24, 88.
- W. Van Brabandt, R. Van Landeghem and N. De Kimpe, Org. Lett., 2006, 8, 1105 CrossRef CAS.
- N. De Kimpe, W. Aelterman, K. De Geyter and J.-P. Declercq, J. Org. Chem., 1997, 62, 5138 CrossRef CAS.
- E. R. Buchman, J. Am. Chem. Soc., 1936, 58, 1803 CrossRef CAS.
- N. De Kimpe and L. Moens, Tetrahedron, 1990, 46, 2965 CrossRef CAS.
- (a) D. W. Old and D. T. Dinh, PCT Int. Appl., 2007 Search PubMed , WO 2007140197 A1; D. W. Old and D. T. Dinh, Chem. Abstr., 2007, 148, 33531 Search PubMed; (b) D. W. Old and D. T. Dinh, U.S. Pat. Appl. Publ. 2008, US 2008033023 A1; D. W. Old, and D. T. Dinh, Chem. Abstr., 2008, 148, 222012 Search PubMed.
- (a) T. Rosen, S. W. Fesik, D. T. W. Chu and A. G. Pernet, Synthesis, 1988, 40 Search PubMed; (b) M. A. Williams and H. Rappoport, J. Org. Chem., 1994, 59, 3616 CrossRef; (c) G. Giardina, G. Dondio and M. Grugni, Synlett, 1995, 55 Search PubMed.
- P. Di Cesare, D. Bouzard, M. Essiz, J. P. Jacquet, B. Ledoussal, J. R. Kiechel, P. Remuzon, R. E. Kessler, J. Fung-Tomc and J. Desiderio, J. Med. Chem., 1992, 35, 4205 CrossRef.
- (a) J. K. Thottathil and J. L. Moniot, Tetrahedron Lett., 1986, 27, 151 CrossRef CAS; (b) K. Hashimoto and H. Shirahama, Tetrahedron Lett., 1991, 32, 2625 CrossRef CAS; (c) T. R. Webb and C. Eigenbrot, J. Org. Chem., 1991, 56, 3009 CrossRef CAS; (d) P. Gmeiner, F. Orecher, C. Thomas and K. Weber, Tetrahedron Lett., 1995, 36, 381 CrossRef CAS.
- C. Gallina, C. Marta, C. Colombo and A. Romeo, Tetrahedron, 1971, 27, 4681 CrossRef CAS.
- (a) H. Beierbeck and J. K. Saunders, Can. J. Chem., 1980, 58, 1258 CAS; (b) J. K. Whitesell, T. Lacour, R. Lovell, J. Pojman, P. Ryan and A. Yamada-Nosaka, J. Am. Chem. Soc., 1988, 110, 991 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: 10.1039/b816617j |
| This journal is © The Royal Society of Chemistry 2009 |