Aminoacid-derived mercaptoimidazoles†
Alain
Crépin
a,
Nicolas
Wattier
b,
Sylvain
Petit
b,
Laurent
Bischoff
*c,
Corinne
Fruit
c and
Francis
Marsais
d
aCurrent adress: Val-de-Pharm, Parc industriel d'Incarville, BP 606, 27106, Val-de-Reuil Cedex, France. Fax: +33 02 32 25 79 15
bCNRS UMR 6014 COBRA B.P., 08 76131, Mont-Saint-Aignan Cedex, France. Fax: +33 02 35 52 29 62
cUniversité de Rouen, place Emile Blondel, 76130, Mont-Saint-Aignan, France. E-mail: laurent.bischoff@univ-rouen.fr
dLCOFH, IRCOF-INSA Rouen, B.P., 08 76131, Mont-Saint-Aignan Cedex, France
First published on 31st October 2008
Abstract
Starting from suitably protected aminoacids, mercaptoimidazoles were synthesized either from the acid or including the amine nitrogen itself. A preliminary optimisation study led to efficient conditions for the obtention of the imidazole ring. These conditions are compatible with the presence of aminoacid or dipeptide scaffolds.
Introduction
Peptidomimetics1 have been thoroughly studied for decades as aminoacid or peptide surrogates. Their main requirements in terms of chemical properties consist of a structural mimicry of natural peptides, though avoiding their shortcomings. Thus, the amide bond surrogate should be stable towards enzymatic hydrolysis, display a low toxicity and/or good bioavailability. Most peptidomimetics either feature a replacement of the scissile amide bond itself for instance with azapeptides2 or fluoroolefins,3 or incorporate aminoacid side-chains into constrained structures such as azabicycloalkanes of various sizes.4 In this work, we focused our attention towards the incorporation of the N-terminus amine or C-terminus acid of aminoacids into a rigid, aromatic heterocyclic scaffold (Fig. 1). In view to explore new series of potential zinc exopeptidases inhibitors, the 4-mercaptoimidazole scaffold was chosen as a binding motif for the metal.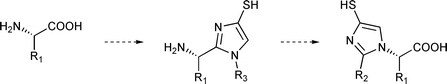 | ||
| Fig. 1 N - or C-terminus aminoacid-derived mercaptoimidazoles. | ||
In the past thirty years,5thiol inhibitors have been widely developed. The imidazole ring which is found in histidine often accounts for the chelation of the zinc cation in the binding site of the enzyme.6 In addition, mercaptoimidazoles are appealing targets for their antioxidant properties. This moiety is present in natural compounds such as ergothioneine or ovothiol.7 In this paper, we wish to report a validation of a general method for the conversion of aminoacids into their mercaptoimidazole analogs.
Results and discussion
As far as the synthesis of 4-mercaptoimidazoles is concerned, few procedures exist. Condensation can be achieved either from an alkylsulfanyl enamine using propane phosphonic anhydride8 or from N-acyl, N-alkylaminothioacetamide, readily available from aminoacetonitrile precursors (Fig. 2). The cyclization step requires a selective electrophilic activation of the carboxamide, over the thioamide. This can be achieved if the dehydrating reagent has a better affinity for oxygen than nitrogen or sulfur. Previously, Hopkins et al.9 used trimethylsilyl triflate. This procedure, which was successfully employed later,10 and is amenable to selenoimidazoles,11 is very efficient and allows smooth formation of 4-mercaptoimidazoles. Further workup of the reaction requires the use of NaBH4 to avoid obtaining disulfides.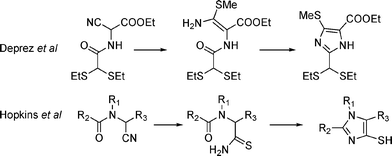 | ||
| Fig. 2 Strategies described in the literature. | ||
In the case of aminoacids and peptides, we chose (Fig. 3) the method described in ref. 9, believing that those mild conditions would be suitable with highly-functionalized aminoacid derivatives. Preliminary results, however, called for further optimisation of the cyclisation step and the introduction of a subsequent protection step.
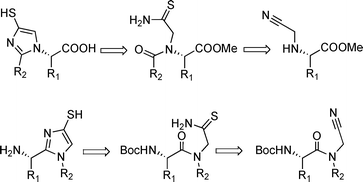 | ||
| Fig. 3 Retrosynthetic analysis. | ||
Unraveling the reactivity of this system was initially conducted with N-acetyl-N-benzylaminothioacetamide 1 as a model substrate before being applied to aminoacids. As shown in Scheme 1, several parameters were examined to reach an efficient one-pot process. Preparation of the reagent was accomplished by treating N-benzyl aminoacetonitrile with excess thioacetic acid in pyridine12 followed by acetylation. Dehydration of this compound with TMSOTf/Et3N following the literature conditions led to a modest yield of 4-mercaptoimidazole.
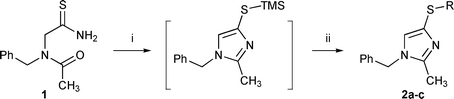 | ||
| Scheme 1 Reagents and conditions: i, TMSOTf, Et3N, CH2Cl2, −78 °C to RT, 6.5 h; ii, see conditions in Table 1. | ||
In addition, the imidazole thiol may be isolated as a thiol or disulfide but characterisation of this compound is facilitated by protecting the thiol moiety. On the one hand, pKa values13 indicate that 4-mercaptoimidazoles predominantly exist under a zwitterionic form, since 1,5-dimethyl-4-mercaptoimidazole exhibited a pKa of 2.3 for the thiol and 10.3 for the imidazole itself. On the other hand, as free thiols, mercaptoimidazoles in particular are very potent anti-oxidants, and they have a high propensity to undergo oxidative dimerisation. Their corresponding disulfides were more commonly isolated. Thus, in situ electrophilic trapping was used as a mean to facilitate the isolation of the products. As listed in Table 1, in situ protection of the thiol was performed with benzylic halides or di-tert-butyl dicarbonate Boc2O, though the reaction seemed rather slow in this case. We also noticed that treatment with NaBH4 prior to S-protection was unnecessary if the reaction is conducted under inert atmosphere (entries 3 and 4). The optimal conditions consisted in treating the crude reaction medium with methanol before protection of the thiol moiety with Boc2O (Table 1, entry 6). We suggest that S-desilylation occurs with methanol, the free thiol being further protected. Having this result in hand, we examined whether the method was applicable to aminoacid substrates. Aminoacids in which the carboxyl group was replaced with the heterocycle were obtained according to Scheme 2.
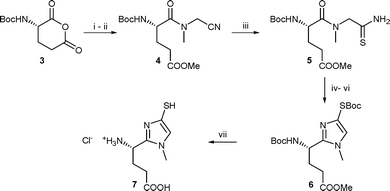 | ||
| Scheme 2 Reagents and conditions: i, CH3–NH–CH2CN, dioxane; ii, EDCI, DMAP in dry MeOH, 56% overall; iii, 8 equiv CH3COSH, pyridine, 65%; iv, TMSOTf, Et3N, CH2Cl2, −78 °C to RT; v, MeOH, 15 min; vi, Boc2O, Et3N, DMAP, CH2Cl2, 53%; vii, 1.5 M HCl, 50% aq dioxane, 40 °C, 100% | ||
For instance, with glutamic acid, opening of the anhydride with a N-cyanomethyl amine such as sarcosine nitrile afforded the amide 4 with a good regioselectivity in favour of the α-carbonyl.14 To facilitate the purification step, the crude reaction mixture was treated with EDCI/DMAP in anhydrous methanol, leading to the amidoester 4. Thioacetic acid-mediated addition of H2S on the nitrile afforded thioamide 5 in a 65% yield. NMR spectra of most cyanomethyl amides and aminothiocarbonyl amides in CDCl3 revealed two rotamers (see experimental section).
It is worth noticing that the use of TMSOTf under our optimized conditions successfully led to compound 6 (53% yield) from a more sensitive functionalized substrate such as 5. Final deprotection under standard acidic hydrolysis conditions of the thiol, side-chain acid and amine afforded the pure compound 7 as its hydrochloride salt. 1H NMR in D2O exhibited a rapid exchange of the proton at C5 of the imidazole ring, due to an easy thiol/thione tautomerism.
As far as the “N-terminus” mimetic was concerned, the cyanomethylamine was prepared directly from the amino group of the aminoacid, as depicted in Scheme 3.
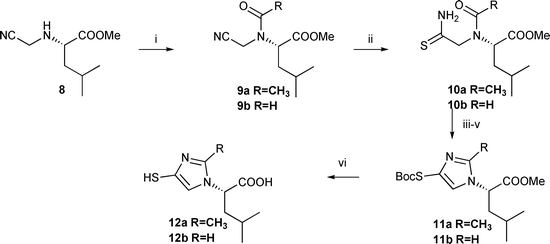 | ||
| Scheme 3 Reagents and conditions: i, RCOOAc, 50% (9a), 95% (9b); ii, 8 equiv CH3COSH, pyridine, 78% (10a), 71% (10b); iii, TMSOTf, Et3N, CH2Cl2, −78 °C to RT; iv, MeOH, 15 min; v, Boc2O, Et3N, DMAP, CH2Cl2, 60% (11a), 66% (11b); vi, 1.5 M HCl, 50% aq dioxane, 40 °C, 100% | ||
Cyanomethylation of aminoacid methyl esters is a known reaction that can be performed smoothly either by direct alkylation with chloroacetonitrile,15 or via aminomethylation in presence of benzotriazole derivatives.16 The free cyanomethylamine 8 can be readily purified by flash chromatography on silica gel without noticeable degradation. Subsequent acetylation or formylation was carried out with acetic anhydride or mixed formic acetic anhydride. Compounds 10 were cyclized in good yields with TMSOTf and S-Boc protected as described above, to produce the 4-mercaptoimidazole 12 after final acidic hydrolysis. This new isoleucine derivative 12 bears a potential zinc ligand as a surrogate of the amine moiety.
Further applications of this work were devoted to the evaluation of a dipeptidic substrate. We were interested in knowing whether the formation of the imidazole ring was compatible with the presence of the secondary amide bond. Examination of the reactivity of the dipeptide-derived compound 13 proved that our conditions are amenable to starting materials which contain peptidic linkages. In addition, quenching with methyl iodide instead of Boc2O at the end of the cyclisation process afforded the S-methyl derivative 15b. This allowed an efficient synthesis of both dipeptides 15b and 16a (Scheme 4).
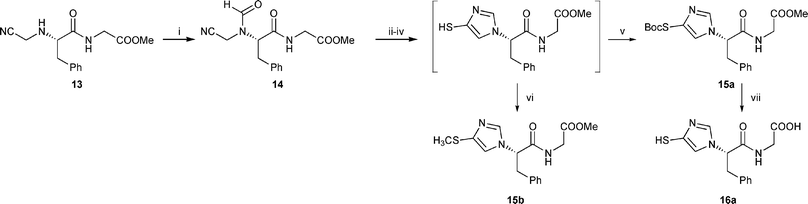 | ||
| Scheme 4 Reagents and conditions: i, HCOOH, Ac2O, 57%; ii, 8 equiv CH3COSH, pyridine; iii, TMSOTf, Et3N, CH2Cl2, −78 °C to RT; iv, MeOH, 15 min; v, Boc2O, Et3N, DMAP, CH2Cl2, 42%; vi, CH3I, Et3N, 26%; vii, 1.5 M HCl, 50% aq dioxane, 40 °C, 100%. | ||
Recently, 2-substituted 1-benzyl-4-methyl imidazoles were described as acid mimics in aminoacid series. Their preparations used palladium-catalyzed cyclisation of N-allyl oxime aminoacid derivatives17 or electrocyclization of azomethine ylides.18 We were interested in examining whether our mercaptoimidazoles could serve as precursors of similar, unsubstituted imidazoles. Initial attempts at desulfurisation of S-Boc derivatives with Raney nickel were unsuccessful. We could, however, overcome this poor reactivity by using the methylsulfanyl imidazole 17. The latter was readily converted to the unsubstituted imidazole by treatment with Raney nickel19 in refluxing ethanol overnight as shown in Scheme 5.
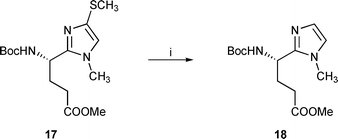 | ||
| Scheme 5 Reagents and conditions: i, Raney nickel, EtOH, reflux, 95% | ||
Summary and conclusion
This work allowed an efficient and concise synthesis of optically active aminoacid-derived mercaptoimidazoles and imidazoles. These compounds constitute a new series of heterocyclic mimics of aminoacid and peptides, which opens the way towards new enzyme inhibitors or antioxidants. In addition, as the conditions used for the formation of the heterocycle are compatible with peptides, wide screening of compounds with different aminoacid scaffolds is now envisageable.Experimental
Unless otherwise stated, reactions were performed under a nitrogen atmosphere using freshly distilled solvents. All reactions were monitored by thin-layer chromatography with Merck silica gel 60 F254 pre-coated aluminum plates (0.25 mm). Flash chromatography was performed with indicated solvents using silica gel (particle size 30–63 μm) purchased from Merck. 1H and 13C NMR spectra were recorded on a Bruker Avance at 300 MHz for 1H and 75 MHz for 13C. Chemical shifts are reported relative to TMS, calibrated with chloroform or deuterium oxide. Coupling constants J are given in Hz.N-(2-Amino-2-thioxoethyl)-N-benzylacetamide 1
To a solution of N-benzyl-N-cyanomethyl acetamide (2.34 g, 12.4 mmol) in CH2Cl2 (23 cm3) was added pyridine (23 cm3), followed by thioacetic acid (7.57 g, 99.6 mmol). Stirring was kept 16 hr at RT. After concentration in vacuo, the residue was purified by flash chromatography through silica gel, using [EtOAc:cyclohexane = 3:2], (Rf 0.28) as an eluent.Compound 1 (2.14 g, 77%) was obtained as a stench, pale yellow oil. (Found: C, 57.29; H 8.02; N 12.38. C11H14N2OS requires C, 57.39; H, 8.21; N, 12.41); 1H NMR ratio of rotamers 4:1, major compound : δH(300 MHz; CDCl3) 2.15 (3H, s), 4.29 (2H, s), 4.64 (2H, s), 7.11–7.33 (5H, m), 7.77 (1H, br s), 8.27 (1H, br s); δC(75 MHz; CDCl3) 21.9, 53.4, 57.4, 127.0, 128.4, 129.5, 135.8, 173.2, 204.2
1-Benzyl-2-methyl-4-(Bocsulfanyl)-1H-imidazole 2c
General procedure for the preparation of protected mercaptoimidazoles, e.g.2c:A solution of N-(2-amino-2-thioxoethyl)-N-benzylacetamide 1 (489 mg, 2.2 mmol) and Et3N (1.26 cm3, 9.0 mmol) in anhydrous CH2Cl2 (15 cm3) was cooled to −78 °C and trimethylsilyl triflate (1.27 cm3, 6.6 mmol) was added dropwise. After stirring 15 min at −78 °C and 6.5 h at RT, methanol was added and stirring continued during 15 min. The solvents were evaporated and a solution of DMAP (27 mg, 0.22 mmol) and anhydrous Et3N (2 cm3, 14.4 mmol) in CH2Cl2 (15 cm3) was added to the residue. To this solution was added dropwise a solution of Boc2O (1.9 g, 8.8 mmol) in anhydrous CH2Cl2 (5 cm3). After stirring overnight and concentration in vacuo, the residue was purified by flash chromatography through silica gel, eluent [EtOAc:cyclohexane:Et3N= 6:4:0.01], Rf 0.35, yielding 2c (400 mg, 60%) as a colourless oil.
(Found: C, 62.93; H, 6.61; N, 9.19; S, 10.69. C16H20N2O2S requires C, 63.13; H, 6.62; N, 9.20; S, 10.53); δH(300 MHz; CDCl3) 1.40 (9H, s), 2.26 (3H, s), 4.95 (2H, s), 6.99–7.02 (2H, m), 7.22–7.29 (3H, m); δC(75 MHz; CDCl3) 13.7, 28.6, 50.5, 85.6, 125.1, 127.1, 127.2, 128.6, 129.5, 135.8, 146.9, 168.3
1-Benzyl-4-(benzylsulfanyl)-2-methyl-1H-imidazole 2a
Compounds 2a and 2b were obtained by the same procedure than for 2c, except that 1.3 molar equiv of the alkylating agent (PhCH2Cl or 4-methoxybenzyl chloride) was used instead of Boc2O.(Found: C, 73.28, H, 6.14, N, 9.25, S, 10.7. C18H18N2S requires C, 73.43; H, 6.16; N, 9.51; S, 10.89); δH(300 MHz; CDCl3) 2.22 (3H, s), 3.90 (2H, s), 4.83 (2H, s), 6.53 (1H, s), 6.86–6.88 (2H, m), 7.08–7.25 (8H, m); δC(75 MHz; CDCl3) 13.6, 40.3, 50.1, 124.3, 127.0, 127.1, 128.4, 128.6, 129.36, 129.39, 130.3, 136.2, 138.9, 146.3; [EtOAc:cyclohexane= 3:2, 1% Et3N]: Rf 0.38; m/z (DCI) 295 (100%), 189 (90%).
1-Benzyl-4-(4-methoxybenzylsulfanyl)-2-methyl-1H-imidazole 2b
δ H(300 MHz; CDCl3) 2.32 (3H, s), 3.76 (3H, s), 3.95 (2H, s), 4.94 (2H, s), 6.64 (1H, s), 6.76 (2H, d, J 8.7), 6.96–6.99 (2H, m), 7.11 (2H, d, J 8.7), 7.29–7.34 (3H, m); δC(75 MHz; CDCl3) 13.6, 39.8, 50.1, 55.6, 114.0, 124.2, 127.0, 128.4, 129.4, 130.5, 130.6, 131.0, 136.3, 146.3, 158.8; m/z (DCI) 325 (100%), 121 (30%).N-Boc-Glutamic anhydride 3
Commercially available N-Boc glutamic acid (8 g, 32 mmol) was stirred for 15 min with 80 cm3acetic anhydride at 55 °C. Toluene (100 cm3) was added and the solution was concentrated to dryness. Traces of acetic acid were removed by drying under vacuum over KOH to afford 7.41 g (100%) of anhydride 3.δ H(300 MHz; CDCl3) 1.45 (9H, s), 1.85–2.0 (1H, m), 2.42–2.46 (1H, dd, J 12.8, 6.2), 3.01 (1H, dd, J 5.5, 2.4), 4.39–4.43 (1H, m), 5.34 (1H, br s); δC(75 MHz; CDCl3) 23.6, 28.3, 29.8, 50.9, 81.1, 155.4, 165.3, 167.2; [α]D20−21 (c 1 in CH2Cl2)
(S)-Methyl-4-(tert-butoxycarbonylamino)-5-(N-cyanomethyl, N-methyl)amino)-5-oxopentanoate 4
A solution of sarcosine nitrile (1.36 g, 19 mmol, obtained from its hydrochloride, by ether extraction from an ice-cold basic solution) in dioxane (5 cm3) was added dropwise at 10 °C to the anhydride 3 (2.98 g, 13 mmol) in 25 cm3dioxane. After 14 h stirring at RT, water (50 cm3) and ethyl acetate (30 cm3) were added. The aqueous layer was treated with ice-cold 1M HCl to reach pH 3. The acid was extracted with ethyl acetate and the organic layer dried over MgSO4. After solvent evaporation and drying under vacuum, the residue (2.74 g, 9.1 mmol) was dissolved in absolute methanol (70 cm3). To this solution were added EDCI (1.92 g, 10.0 mmol) and DMAP (110 mg, 0.91 mmol). Stirring was kept during 5 h at RT. After solvent evaporation, extractive workup with ethyl acetate/satd NH4Cl, and drying over MgSO4, the ester 4 (2.3 g, 56%) was obtained as a pale brown oil.δ H(300 MHz; CDCl3), major rotamer: 1.39 (9H, s), 2.31–2.50 (4H, m), 3.30 (3H, s), 3.71 (3H, s), 4.20–4.41 (3H, m), 5.26–5.34 (1H, m); δC(75 MHz; CDCl3) 27.7, 27.9, 28.3, 35.2, 35.3, 35.7, 52.5, 80.1, 115.4, 155.6, 172.3, 172.8
(S)-Methyl 5-(N-(2-amino-2-thioxoethyl), N-(methyl)amino)-4-(tert-butoxycarbonylamino)-5-oxopentanoate 5
Starting from compound 4 (2.0 g, 6.38 mmol), the thioamide 5 (1.44 g, 65%) was obtained following the procedure described for 1.(Found: C, 48.32, H, 7.39, N, 12.35, S, 9.16. C14H25N3O5S requires C, 48.40; H, 7.25; N, 12.09; S, 9.23); δH(300 MHz; CDCl3): 1.38 (9H, s), 1.57–1.65 (1H, m), 2.26–2.61 (3H, m), 2.98 (3H, s), 3.78 (3H, s), 3.96 (1H, d, J 17.1), 4.38–4.45 (1H, m), 5.03 (1H, d, J 17.0), 5.43 (1H, d, J 8.1), 7.98 (1H, br s), 8.63 (1H, br s); δC(75 MHz; CDCl3) 28.2, 28.3, 29.0, 36.0, 52.1, 52.7, 59.0, 80.4, 156.1, 172.4, 173.1, 203.5; [α]D20 +20.61 (c 0.97 in CH2Cl2); m/z (DCI) 348 (100%).
(S)-Methyl 4-(tert-butoxycarbonylamino)-4-(4-(tert-butoxy carbonylthio)-1-methyl-1H-imidazol-2-yl)butanoate 6
Following the procedure given for 2c and starting from 5 (1.21 g, 3.48 mmol), 6 (790 mg, 53%) was obtained as a pale yellow oil. (Found: C, 53.08; H, 7.39; N, 9.86; S, 7.89. C19H31N3O6S requires C, 53.13; H, 7.27; N, 9.78; S, 7.47); δH(300 MHz; CDCl3): 1.27 (9H, s), 1.42 (9H, s), 2.11–2.21 (1H, m), 2.30–2.42 (1H, m), 2.72 (2H, d, J 8.0), 3.55 (3H, s), 3.69 (3H, s), 4.30–4.37 (1H, m), 5.34 (1H, d, J 7.6), 7.06 (1H, s); δC(75 MHz; CDCl3) 28.3, 28.4, 29.8, 30.2, 32.9, 52.6, 53.2, 80.0, 85.4, 124.6, 127.6, 148.6, 155.6, 168.2, 172.7; [α]D20 +9.41 (c 1.2 in CH2Cl2)(S)-4-Amino-4-(4-mercapto-1-methyl-1H-imidazol-2-yl)butanoic acid hydrochloride 7
Protected compound 6 (95 mg, 0.22 mmol) was dissolved in dioxane under argon. A degased solution of 3M hydrochloric acid was added and the reaction mixture stirred 13 h at 40 °C. Evaporation to dryness followed by freeze-drying from water afforded 56 mg (100%) of the free thiol 7.δ H(300 MHz; D2O) 2.32–2.39 (2H, m), 3.06–3.24 (2H, m), 3.75 (3H, s), 4.11 (1H, t, J 6.0), 7.51 (1H, br s); δC(75 MHz; D2O) 21.4, 26.8, 34.8, 52.8, 123.3, 149.1, 172.1; [α]D20 +24.3 (c 1 in abs EtOH).
(S)-Methyl 2-(cyanomethylamino)-4-methylpentanoate 8
A 1 M aqueous solution of NaOH (27.6 cm3) was added dropwise to a suspension of valine methyl ester (5 g, 27.6 mmol) and benzotriazol (3.61 g, 30.3 mmol) in methanol (50 cm3). Formaline (2.1 cm3, 5.5 mmol) was added and the reaction medium was stirred 4 hr at RT. Extraction with petroleum ether followed by drying over MgSO4, filtration and evaporation gave an oil (6.87 g) which was redissolved in DMSO (50 cm3). Sodium cyanide (1.64 g, 33.6 mmol) was added. After stirring 40 hr at RT, ethyl acetate was added, the organic layer was decanted off and washed with Na2CO3, brine and dried over MgSO4. The aqueous residues were treated with bleach before discarding.(Found: C, 58.36; H 8.81; N 15.94. C9H16N2O2 requires C, 58.67; H, 8.75; N, 15.31); δH(300 MHz; CDCl3) 0.92 (6H, d, J 6.6), 1.41–1.58 (2H, m), 1.67–1.82 (2H, m), 3.39 (1H, dd, J 8.1, 6.2), 3.54 (1H, d, J 17.4), 3.64 (1H, d, J 17.4), 3.75 (3H, s); δC(75 MHz; CDCl3) 21.8, 22.7, 24.6, 35.9, 42.1, 52.0, 58.7, 117.5, 174.8; [α]D= −32.0 (c 1, CH2Cl2).
(S)-Methyl 2-(N-(cyanomethyl)acetamido)-4-methyl pentanoate 9a
Aminonitrile 8 (700 mg, 3.78 mmol) in CH2Cl2 (5 cm3) was treated with 640 mg (6.22 mmol) of acetic anhydride. After 2 h stirring at RT, evaporation and flash chromatography through silica gel, eluent [EtOAc:cyclohexane = 6:4], Rf 0.55 gave compound 9a (425 mg, 50%).(Found: C, 58.43; H 8.29; N 12.55. C11H18N2O3 requires C, 58.39; H, 8.02; N, 12.38); δH(300 MHz; CDCl3): two rotamers, ratio 3:2 in this solvent: major: 0.95–1.02 (6H, m), 1.64–1.89 (3H, m), 2.30 (3H, s), 3.72 (3H, s), 4.09 (1H, d, J 15.0), 4.36 (1H, d, J 15.0), 4.45 (1H, dd, J 9.1, 6.0); minor: 0.95–1.02 (6H, m), 1.64–1.89 (3H, m), 2.20 (3H, s), 3.77 (3H, s), 4.28 (2H, s), 5.41 (1H, dd, J 9.9, 5.4); δC(75 MHz; CDCl3) 24.7, 30.4, 38.2, 52.6, 54.0, 115.7, 171.0, 171.1; [α]D20−28.4 (c 1.09 in CH2Cl2).
(S)-Methyl 2-(N-(cyanomethyl)formamido)-4-methyl pentanoate 9b
An equimolar mixture of formic acid (9.7 g, 212 mmol) and acetic anhydride was heated at 65 °C during 10 minutes. After cooling to 0 °C, this solution was added to aminonitrile 8 (1.5 g, 8.14 mmol) in CH2Cl2 (5 cm3). After 10 min stirring at RT, treatment with ice-cold water (240 cm3) and extractive work up with CH2Cl2 followed by washing with satd NaHCO3 gave compound 9b (1.6 g, 95%).(Found: C, 56.51; H, 7.55; N, 13.69. C10H16N2O3 requires C, 56.59; H, 7.60; N, 13.20); δH(300 MHz; CDCl3) two rotamers, ratio 4:1 in this solvent: major: 0.95–1.02 (6H, m), 1.59–1.69 (1H, m), 1.76–1.87 (2H, s), 3.78 (3H, s), 4.19–4.39 (3H, m), 8.15 (1H, s); δC(75 MHz; CDCl3) 21.0, 22.7, 29.2, 37.9, 52.8, 58.2, 114.9, 162.8, 170.9; [α]D20−24.4 (c 0.53 in CH2Cl2).
(S)-Methyl 2-(N-(2-amino-2-thioxoethyl)acetamido)-4-methylpentanoate 10a
Following the procedure described for the formation of 1, compound 9a (250 mg, 1.10 mmol) was treated with thioacetic acid, leading to 10a (223 mg, 78%).(Found: C, 45.66; H, 7.21; N, 8.71; S, 8.20. C11H20N2O3S requires C, 50.75; H, 7.74; N, 10.76; S, 12.32); δH(300 MHz; CDCl3): two rotamers, ratio 6.7:1 in this solvent: major: 0.93–0.99 (6H, m), 1.41–1.58 (1H, m), 1.80 (2H, t, J 7.4), 2.08 (3H, s), 3.8 (3H, s), 4.27 (1H, t, J 7.0), 4.36 (1H, d, J 19.4), 4.52 (1H, d, J 19.4), 7.83 (1H, br s), 10.1 (1H, br s); δC(75 MHz; CDCl3) 22.0, 22.1, 23.2, 25.2, 38.0, 53.3, 59.4, 172.2, 175.0, 203.5; [α]D20−63.8 (c 0.8 in CH2Cl2).
(S)-Methyl 2-(N-(2-amino-2-thioxoethyl)formamido)-4-methylpentanoate 10b
Following the procedure described for the formation of 1, compound 9b (1.6 g, 7.54 mmol) was treated with thioacetic acid, leading to 10b (1.31 g, 71%).(Found: C, 48.68; H, 7.39; N, 11.33; S, 12.67. C11H14N2OS requires C, 48.76; H, 7.37; N, 11.37; S, 13.02; δH(300 MHz; CDCl3): two rotamers, ratio 1.25:1 in this solvent: major: 0.82–0.92 (6H, m), 1.55–1.65 (3H, m), 3.60 (3H, s), 3.98–4.34 (2H, m), 4.78 (1H, m), 8.14 (1H, s), 9.37 (1H, br s), 9.80 (1H, br s); minor: 0.82–0.92 (6H, m), 1.55–1.65 (3H, m), 3.65 (3H, s), 3.98–4.34 (2H, m), 4.43 (1H, m), 8.24 (1H, s), 8.91 (1H, br s), 9.70 (1H, br s).
(S)-Methyl 2-(4-(tert-butoxycarbonylthio)-2-methyl-1H-imidazol-1-yl)-4-methylpentanoate 11a
Following the procedure described for the formation of 2, compound 10a (160 mg, 0.62 mmol) led to protected mercaptoimidazole 11a (127 mg, 60%).(Found: C, 55.49; H, 7.78; N, 7.76; S, 9.25. C16H26N2O4S requires C, 56.12; H, 7.65; N, 8.18; S, 9.36); δH(300 MHz; CDCl3): 0.92 (6H, m), 1.47 (9H, s), 1.47 (1H, m), 1.84–2.03 (2H, m), 2.39 (3H, s), 3.73 (3H, m), 4.67 (1H, dd, J 9.0, 6.6), 7.23 (1H, s); δC(75 MHz; CDCl3) 13.6, 21.8, 22.7, 24.6, 28.3, 41.2, 53.1, 56.9, 85.3, 123.7, 125.6, 146.3, 167.6, 170.2; [α]D20−3.27 (c 0.61 in CH2Cl2); m/z (DCI) 343 (100%), 299 (10%), 287 (25%).
(S)-Methyl 2-(4-(tert-butoxycarbonylthio)-1H-imidazol-1-yl)-4-methylpentanoate 11b
Following the procedure described for the formation of 2, compound 10b (299 mg, 1.21 mmol) led to protected mercaptoimidazole 11b (262 mg, 66%).(Found: C, 55.04; H, 7.51; N, 8.57; S, 9.56. C15H24N2O4S requires C, 54.86; H, 7.37; N, 8.53; S, 9.76); δH(300 MHz; CDCl3): 0.90–0.94 (6H, m), 1.38–1.47 (1H, m), 1.47 (9H, s), 1.91–1.97 (2H, m), 3.74 (3H, m), 4.74 (1H, dd, J 8.7, 7.2), 7.29 (1H, d, J 1.3), 7.62 (1H, d, J 1.3); δC(75 MHz; CDCl3) 21.6, 22.6, 24.6, 28.2, 41.7, 53.1, 60.5, 85.4, 124.6, 127.6, 137.8, 167.3, 170.1; m/z (DCI) 329 (100%), 273 (90%), 229 (20%).
(S)-2-(4-Mercapto-2-methyl-1H-imidazol-1-yl)-4-methyl pentanoic acid 12a
Hydrolysis was carried out by stirring a solution of compound 11b (86 mg, 0.26 mmol) in 1.5 M HCl in H2O:dioxane 1:1 at 40 °C overnight. After concentration in vacuo, the residue was taken up in water and freeze-dried. 56 mg (100%) of compound 12b were obtained as a pale yellow foam.δ H(300 MHz; DMSO) 0.85 (3H, d, J 6.6), 0.88 (3H, d, J 6.6), 1.17–1.20 (1H, m), 1.81–2.02 (2H, m), 2.40 (3H, s), 4.62 (1H, dd, J 9.8, 6.0), 7.20 (1H, s); δC(75 MHz; DMSO) 11.6, 21.3, 22.6, 24.3, 58.0, 126.2, 148.3, 170.0.
(S)-2-(4-Mercapto-1H-imidazol-1-yl)-4-methyl pentanoic acid 12b
Hydrolysis was carried out by stirring a solution of compound 11b (86 mg, 0.26 mmol) in 1.5 M HCl in H2O:dioxane 1:1 at 40 °C overnight. After concentration in vacuo, the residue was taken up in water and freeze-dried. 56 mg (100%) of compound 12b were obtained as a pale yellow foam.δ H(300 MHz; D2O) 0.76 (3H, d, J 6.6), 0.78 (3H, d, J 6.6), 1.38–1.47 (1H, m), 1.88–2.05 (2H, m), 5.15 (1H, dd, J 10.4, 5.5), 7.51 (1H, d, J 1.3), 8.79 (1H, s); δC(75 MHz; D2O) 20.5, 22.2, 24.7, 39.0, 62.1, 125.5, 127.7, 138.8, 141.4, 147.5, 172.9. [α]D20 +72.6 (c 1.22 in EtOH).
(S)-Methyl 2-(2-(cyanomethylamino)-3-phenyl propanamido) acetate 13
The dipeptide Phe-Gly-OMe was treated as described for 8.(Found: C, 60.57; H, 6.17; N, 15.86. C14H17N3O3 requires C, 61.08; H, 6.22; N, 15.26); δH(300 MHz; DMSO + TFA) 3.11 (2H, d, J 6.6), 3.63 (3H, s), 3.92 (2H, d, J 5.6), 4.17 (2H, s), 7.24–7.31 (5H, m), 7.40 (1H, dd, J 6.4, 3.0), 7.87 (1H, dd, J 6.2, 3.0), 9.11 (1H, t, J 5.6); δC(75 MHz; DMSO + TFA) 36.2, 41.0, 52.1, 60.7, 125.6, 128.8, 129.8, 134.3, 167.4, 169.9; [α]D20−28.9 (c 0.94 in EtOH).
(S)-Methyl 2-(2-(N-(cyanomethyl)formamido)-3-phenyl propanamido)acetate 14
Aminonitrile 13 was treated with HCOOH/Ac2O, using the same protocol than for 9b.(Found: C, 59.62; H 5.14; N 13.68. C15H17N3O4 requires C, 59.40; H, 5.65; N, 13.85); δH(300 MHz; DMSO + TFA) 3.01 (1H, dd, J 14.3, 10.1), 3.21 (1H, dd, J 14.3, 5.5), 3.61 (3H, s), 3.89 (1H, d, J 5.9), 4.19 (1H, d, J 17.5), 4.34 (1H, d, J 17.5), 4.64–4.69 (1H, m), 7.18–7.27 (5H, m), 7.92 (1H, s), 8.79 (1H, t, J 5.7); 13C NMR (CDCl3, 75 MHz) 29.8, 36.2, 41.4, 52.7, 62.6, 115.3, 127.7, 129.1, 129.3, 135.5, 168.6, 168.9, 170.0; [α]D20−62.6 (c 1 in CH2Cl2).
(S)-Methyl 2-(2-(N-(2-amino-2-thioxoethyl)formamido)-3-phenylpropanamido)acetate
Obtained by treatment of the aminonitrile 14 with thioacetic acid (see procedure for 1).(Found: C, 53.42; H 5.82; N 12.32, S 9.43. C15H19N3O4S requires C, 53.40; H, 5.68; N, 12.45; S, 9.50); δH(300 MHz; DMSO + TFA) two rotamers, ratio 3:1, major: 3.05–3.26 (2H, m), 3.60 (3H, s), 3.84 (2H, d, J 5.7), 4.00 (1H, d, J 17.5), 4.09 (1H, d, J 17.5), 4.54 (1H, t, J 7.7), 7.19–7.30 (5H, m), 8.07 (1H, s), 8.77 (1H, t, J 5.6), 8.87 (1H, br s), 9.70 (1H, br s); δC(75 MHz; CDCl3) 37.3, 41.9, 52.7, 54.7; 63.6, 127.9, 129.6, 130.1, 137.7, 164.8, 170.8, 171.3, 204.2; [α]D20−95.1 (c 1 in CH2Cl2); m/z (DCI) 338 (100%), 304, 265, 90, 76
(S)-Methyl 2-(2-(4-(tert-butoxycarbonylthio)-1H-imidazol-1-yl)-3-phenylpropanamido)acetate 15a
Thioamide resulting from treatment of 14 (250 mg, 0.74 mmol) was dehydrated to give the S-Boc mercaptoimidazole 15a (131mg, 42%), using the procedure described for 2c.(Found: C, 57.69; H, 5.76; N, 9.89; S, 7.75. C20H25N3O5S requires C, 57.26; H, 6.01; N, 10.02; S, 7.64); δH(300 MHz; CDCl3), two rotamers, ratio 9:1, major: 1.48 (9H,s), 3.17 (1H, dd, J 14.0, 9.4), 3.50 (1H, dd, J 14.0, 5.7), 3.71 (3H, s), 3.99 (2H, t, J 5.5), 4.81 (1H, dd, J 9.2, 5.8), 6.99–7.02 (2H, m), 7.16–7.22 (3H, m), 7.29 (1H, s), 7.32 (1H, d, J 1.1); δC(75 MHz; CDCl3) 28.3, 39.3, 41.5, 52.5, 62.9, 85.8, 124.7, 127.4, 127.7, 128.9, 129.0, 135.8, 138.5, 154.8, 168.4, 169.7; [α]D20−55.3 (c 0.85 in CH2Cl2); m/z (DCI) 420, 376, 320, 222, 79
(S)-Methyl 2-(2-(4-(methylthio)-1H-imidazol-1-yl)-3-phenyl propanamido)acetate 15b
The procedure for the formation of 2c was used, except that methyl iodide (2 molar equiv) was used instead of Boc2O.δ H(300 MHz; CDCl3): 2.35 (3H, s), 3.17 (1H, dd, J 14.1, 9.6), 3.51 (1H, dd, J 14.1, 5.7), 3.70 (3H, s), 4.00 (2H, d, J 5.4), 4.90 (1H, dd, J 9.4, 5.5), 6.97–7.00 (2H, m), 7.06 (1H, s), 7.19–7.21 (3H, m), 7.26 (1H, s), 7.88 (1H, t, J 5.4); δC(75 MHz; CDCl3) 18.1, 39.1, 41.3, 52.4, 62.5, 118.3, 127.3, 128.7, 128.8, 135.9, 136.1, 137.6, 168.9, 169.9; m/z (ESI) 334; [α]D20−54.5 (c 0.22 in CH2Cl2)
(S)-2-(2-(4-Mercapto-1H-imidazol-1-yl)-3-phenyl propanamido)acetic acid 16a
The procedure used for compound 7 was applied.δ H(300 MHz; D2O) 3.32 (1H, dd, J 13.9, 10.0), 3.54 (1H, dd, J 13.9, 5.9), 3.93 (1H, d, J 17.9), 4.02 (1H, d, J 17.9), 5.38 (1H, dd, J 9.0, 6.0), 7.14–7.17 (2H, m), 7.29–7.31 (3H, m), 7.44 (1H, s), 8.65 (1H, s); δC(75 MHz; CDCl3) 38.4, 41.5, 63.7, 127.9, 128.0, 129.0, 129.2, 129.3, 134.9, 138.5, 169.6, 173.0; [α]D20−96.9 (c 0.8 in CH2Cl2); m/z (ESI) 304
(S)-Methyl 4-(tert-butoxycarbonylamino)-4-(1-methyl-4-(methylthio)-1H-imidazol-2-yl)butanoate 17
Following the procedure described for the formation of 15b, thioamide 5 (444 mg, 1.28 mmol) led to protected mercaptoimidazole 17 (198 mg, 45%).δ H(300 MHz; CDCl3): 1.43 (9H, s), 2.16–2.38 (2H, m), 2.39 (3H, s), 2.71 (2H, t, J 7.4), 3.51 (3H, s), 3.72 (3H, s), 4.35 (1H, m), 5.77 (1H, br d, J 7.0), 6.77 (1H, s); δC(75 MHz; CDCl3) 19.0, 23.3, 28.7, 29.9, 32.9, 52.7, 53.9, 80.1, 121.9, 133.2, 148.3, 156.0, 173.1
(S)-Methyl 4-(tert-butoxycarbonylamino)-4-(1-methyl-1H-imidazol-2-yl)butanoate 18
Refluxing sulfide 17 (99 mg, 0.29 mmol) in ethanol (15 cm3), in the presence of 50 mg of Raney nickel (50% in water) during 14 h, followed by filtration through celite, gave 82 mg (95%) of compound 18.δ H(300 MHz; CDCl3): 1.43 (9H, s), 2.11–2.37 (2H, m), 2.72 (2H, t, J 7.7), 3.54 (3H, s), 3.70 (3H, s), 4.35 (1H, m), 5.60 (1H, br d, J 7.7), 6.77 (1H, s), 6.89 (1H, s); δC(75 MHz; CDCl3) 22.8, 28.4, 30.1, 32.6, 52.5, 53.3, 80.2, 120.8, 127.1, 147.0, 155.7, 172.9
Acknowledgements
We are grateful to the FONGECIF, CNAM and the Région Haute Normandie for generous financial support to Alain Crépin and Sylvain Petit, respectively. The Centre Universitaire Normand de Chimie Organique (CRUNCh) is also gratefully acknowledged. Dr Christian A. G. N. Montalbetti (Evotec, U.K.), is gratefully acknowledged for helpful discussions.References
- S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Tetrahedron, 1997, 53, 12789 CrossRef CAS; J. Gante, Angew. Chem. Int. Ed. Engl., 1994, 33, 1699 CrossRef.
- R. E. Melendez and W. D. Lubell, J. Am. Chem. Soc., 2004, 126, 6759 CrossRef CAS.
- S. Couve-Bonnaire, D. Cahard and X. Pannecoucke, Org. Biomol. Chem., 2007, 5, 1151 RSC.
- M. H. V. Ramana Rao, E. Pinyol and W. D. Lubell, J. Org. Chem., 2007, 72, 736 CrossRef and references cited therein.
- B. P. Roques, M.-C. Fournie-Zaluski, E. Soroca, J.-M. Lecomte, B. Malfroy, C. Llorens and J.-C. Schwartz, Nature, 1980, 288, 286 CrossRef CAS; B. P. Roques, F. Noble, V. Dauge, M.-C. Fournie-Zaluski and A. Beaumont, Pharmacol. Rev., 1993, 45, 87 CAS; C. David, L. Bischoff, H. Meudal, A. Mothe, N. De Mota, S. DaNascimento, C. Llorens-Cortes, M.-C. Fournie-Zaluski and B. P. Roques, J. Med. Chem., 1999, 42, 5197 CrossRef CAS.
- B. W. Matthews, Acc. Chem. Res., 1988, 21, 333 CrossRef CAS.
- T. P. Holler, F. Ruan, A. Spaltenstein and P. B. Hopkins, J. Org. Chem., 1989, 54, 4570 CrossRef CAS; C. E. Hand and J. F. Honek, J. Nat. Prod., 2005, 68, 293 CrossRef CAS.
- P. Deprez, E. Mandine, A. Vermond and D. Lesuisse, Bioorg. Med. Chem. Lett., 2002, 12, 1287 CrossRef CAS.
- A. Spaltenstein, T. P. Holler and P. B. Hopkins, J. Org. Chem., 1987, 52, 2977 CrossRef CAS.
- V. Zoete, F. Bailly, J.-P. Catteau and J.-L. Bernier, J. Chem. Soc. Perkin Trans., 1997, 1, 2983 Search PubMed.
- F. Bailly, N. Azaroual and J.-L. Bernier, Bioorg. Med. Chem., 2003, 11, 4623 CrossRef CAS.
- K. Ikeda, S. Kitani, K. Sato, T. Suzuki, C. Hosokawa, Y. Suzuki, K. Tanaka and M. Sato, The protocol described in the following article, Carbohydrate Res., 2004, 339, 1367 CrossRef CAS claims 1 molar equiv of AcSH, whereas the amount used therein corresponds to 8 molar equiv.
- T. P. Holler and P. B. Hopkins, J. Am. Chem. Soc., 1988, 110, 4837 CrossRef CAS.
- X. Huang, X. Luo, Y. Roupioz and J. Keillor, J. Org. Chem., 1997, 62, 8821 CrossRef CAS.
- H. Tokuyama, T. Kuboyama, A. Amano, T. Yamashita and T. Fukuyama, Synthesis, 2000, 9, 1299 CrossRef.
- A. R. Katritzky, N. Kirichenko, B. V. Rogovoy and H.-Y. He, J. Org. Chem., 2003, 68, 9088 CrossRef CAS.
- S. Zaman, K. Mitsuru and A. D. Abell, Org. Lett., 2005, 7, 609 CrossRef CAS.
- O. A. Attanasi, P. Davoli, G. Favi, P. Filippone, A. Forni, G. Moscatelli and F. Prati, Org. Lett., 2007, 9, 3461 CrossRef CAS.
- Y. Tominaga, Y. Shiroshita, M. Kawabe, H. Goto, Y. Oniyama and Y. Matsuda, Heterocycles, 1985, 23, 2531 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/b810678a |
| This journal is © The Royal Society of Chemistry 2009 |