Photoswitchable rotaxanes using the photolysis of alkoxyacridanes†
Werner Abraham*, Andre Wlosnewski, Karin Buck and Sabine Jacob
Institute of Chemistry, Humboldt-Universitaet zu Berlin, Brook-Taylor-Str. 2, D-12489, Berlin, Germany. E-mail: abraham@chemie.hu-berlin.de
First published on 10th November 2008
Abstract
9-Aryl-9-alkoxy-acridanes and their counterparts, 9-aryl-acridinium ions, have been incorporated into the axles both of one- and two-station [2]rotaxanes. The ring component of the rotaxanes consists of the tetracationic ring cyclobis(paraquat-4,4′-bisphenylene). The electron-rich acridanes represent suitable recognition sites for the electron-poor ring because charge transfer interaction plays an important role. The 9-aryl groups at the acridane unit bearing substituents such as the alkoxy and the amino groups influence the strength of the recognition site. Photoexcited acridanes bearing a suitable leaving group such as the methoxy substituent in the 9-position undergo heterolysis, resulting in the formation of the acridinium methoxides. The acridanes are regenerated by the nucleophilic attack of the methoxide ion at the acridinium ion formed. The lifetime of the ionic state is strongly dependent on the solvent composition. Because the positively charged acridinium ions repel the positively charged ring component of the interlocked molecules, a movement of the ring is initiated provided the molecular axle contains an evasive recognition site. Two-station rotaxanes presented here possess as the second station an anisol unit. Both the photoreaction and the thermal back-reaction render the thermodynamic driving force of the interaction of the ring with one of the two recognition stations. Accordingly, movement of the ring forward and back, driven by Brownian motion, occurs. The switching cycle can also be triggered by acid–base titration. The photoexcitation of the acridane unit present in one-station rotaxanes leads to a very unfavourable acridinium recognition station. However, because of the absence of a second station, the ring remains at the unfavourable acridinium station having interaction with 9-aryl group.
Introduction
Rotaxanes are mechanically interlocked molecules composed of a threadlike molecule (axle) incorporating recognition stations and a ring. Bistable rotaxanes, having at least two recognition stations, are molecular shuttles in which the ring position may be altered. Energy for the shuttling process stems from the Brownian motion.1 Provided that the two stations have sufficiently different interaction strength with the ring, the ring will reside preferentially on one of the two recognition stations. If an outer stimulus weakens the strong interaction of the station on which the ring resides, the ring will move to the station with the originally weaker interaction. Thus, a complete rearrangement of the mechanically bonded components will occur, e.g. the co-conformation,2 and the shuttle is referred to as a switch.1Solvents,3 chemicals,4 electrons5 or light can all serve as stimuli for the switching process, the advantage of the light-driven systems being that no chemical fuels are necessary and no waste products are formed.
The way in which a photoreaction may control the interaction between the ring and the photoactive recognition station is an important consideration. The usefulness of a type of photoreaction is related to the type of the noncovalent interaction. Photoinduced E/Z-isomerisation (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N or CC double bonds) and photoinduced electron transfer are often used.6 The hydrophobic interactions in rotaxanes based on cyclodextrins (CDs) can be disturbed by simple isomerisation of stilbene- or azobenzene-units. Due to steric interference of the Z-isomers with CDs, an alternative recognition station can interact with the CD.7 In comparison with the steric interference, the isomerisation of fumaramide plays a more active role. The hydrogen bonding between a tetramide-based macrocycle and the formed maleamide station is drastically reduced because the intramolecular hydrogen bonds within the maleamide predominate. The reset can be accomplished either photochemically or thermally.8 Recently, the photoisomerisation of a spiropyrane was shown to switch the movement of a macrocycle due to the strong hydrogen-bond acceptor property of the opened merocyanine isomer.9
N or CC double bonds) and photoinduced electron transfer are often used.6 The hydrophobic interactions in rotaxanes based on cyclodextrins (CDs) can be disturbed by simple isomerisation of stilbene- or azobenzene-units. Due to steric interference of the Z-isomers with CDs, an alternative recognition station can interact with the CD.7 In comparison with the steric interference, the isomerisation of fumaramide plays a more active role. The hydrogen bonding between a tetramide-based macrocycle and the formed maleamide station is drastically reduced because the intramolecular hydrogen bonds within the maleamide predominate. The reset can be accomplished either photochemically or thermally.8 Recently, the photoisomerisation of a spiropyrane was shown to switch the movement of a macrocycle due to the strong hydrogen-bond acceptor property of the opened merocyanine isomer.9
Photoinduced electron transfer is most suitable for controlling the strength of charge-transfer interaction. The bipyridinium unit either in the threadlike component or in the ring of rotaxanes introduced by the Stoddart group10 is the most often-used strong electron acceptor. However, direct excitation at the charge-transfer absorption band results in formation of an intimate radical ion pair which undergoes rapid charge recombination that is faster than the ring movement.11 A detailed study of time resolved absorption spectra of the rotaxane in the presence of an external electron donor as a dual-action redox reagent has shown the function of an autonomous mechanism which allows about 10% ring movement in competition with back electron transfer. Even in the absence of the external donor, about 4% ring movement could be estimated.12 It is also worth noting that a hydrogen-bonded rotaxane can be switched by photoinduced electron transfer.13 In each case, the lifetime of the charge separated state of the rotaxanes is very short.
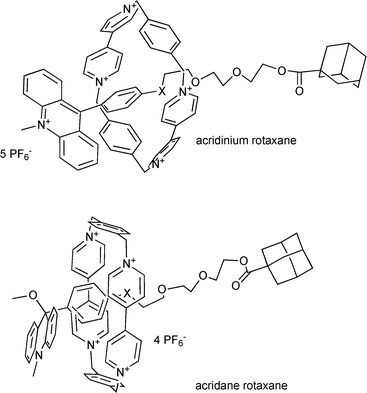
We have introduced an alternative switching principle for rotaxanes based on charge-transfer interaction of the tetracationic ring cyclobis(paraquat-4,4′-bisphenylene) with a photoactive electron donor. The switch is characterized by the generation of cations by photoheterolysis of alkoxyarylcycloheptatrienes which transform an electron donor (arylcycloheptatriene) into an electron acceptor (aryltropylium ion).14 The lifetime of the rotaxanes in their tropylium state is in the range of seconds. However, side reactions such as the hydrogen shift disturbed the photoheterolysis.15 In order to improve the functionality of the photoswitchable rotaxanes, we have recently presented a preliminary communication which described the introduction of another photoswitch based upon photoheterolysis of 9-alkoxy-9-aryl-N-methylacridanes which form acridinium ions with methoxide counterions. The photoactive alkoxyphenyl acridanes bearing the methoxide leaving group in the 9-position are used as recognition station in rotaxanes such as 1.16 The [2]rotaxane 1 (Scheme 1) based on the acridane photoswitch has two different recognition sites, namely the 9-alkoxy-9-phenyl-acridane (A), which is also a stopper unit, and the photoinactive alkoxyphenyl station (B).
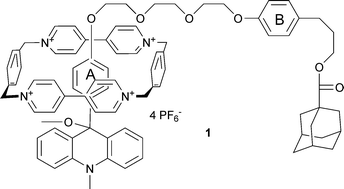 | ||
| Scheme 1 Rotaxane 1. | ||
Surprisingly, the interaction of the tetracationic ring with the alkoxyphenyl unit A within the acridane moiety is favored over the alkoxyphenyl station B. This finding was attributed to the additional edge-to-face interaction between the ring and the acridane unit.16 The only observed photoreaction of the acridane is photoheterolysis. Consequently, these photoswitchable rotaxanes are an alternative for the direct electron transfer used by the groups of Balzani and Stoddart.12 9-Arylacridinium are Janus molecules because the acridinium part is an electron acceptor while the aryl group in the 9-position is an electron donor. Due to the torsional angle between both subunits they are decoupled from each other. Therefore the question arises as to whether the 9-aryl acridinium station is able to act as recognition station for the tetracationic cyclophane competing with the photoinactive station present in the molecular axle of bistable rotaxanes. In particular, this problem may become relevant if the electron donor strength of the 9-aryl group is increased, as in the 9-aminophenyl group. One-station rotaxanes containing only the 9-arylacridinium station should be suitable for studying the interaction between the subunits of the acridinium station and the tetracationic cyclophane with the help of NMR-spectroscopy. In this paper we report on newly designed one- and two-station rotaxanes incorporating 9-alkoxy- and 9-aminophenyl-9-methoxy-9,10-dihydroacridines (acridanes) and their acridinium counterparts. By using the aminophenyl group as station A, a further improvement of the selectivity may result due to the increased donor capability of the aryl group at 9-position of the acridane moiety. We compare the efficiency of the photoreaction of the acridane rotaxanes and their free molecular axles, and we discuss the influence of the tetracationic cyclophane present in rotaxanes on the reactivity and photophysical properties of the acridinium and acridane stations.
Results and discussion
The photoswitches 9-aryl-9-methoxy-N-methyl-9,10-dihydroacridines (9-aryl-9-methoxyacridanes)
Until now the photochemistry of compounds 2 and 4 (Scheme 2) has not been studied.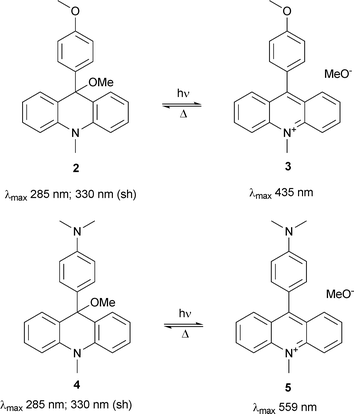 | ||
| Scheme 2 Two types of photoswitches used in rotaxanes. | ||
The UV-vis spectra of 2 and 4 recorded after consecutive irradiations in acetonitrile solution (Fig. 1 of the ESI†) exhibit three isosbestic points indicating a single step photoreaction resulting in the formation of the corresponding acridinium salts 3 and 5, respectively. The complete recovery of the acridanes by nucleophilic attack of methanol is only observed in acetonitrile solution containing at least 5% methanol. The formed methoxide supports the reaction by abstracting the proton (see Scheme 3). Therefore, the thermal reaction is pseudo first order.
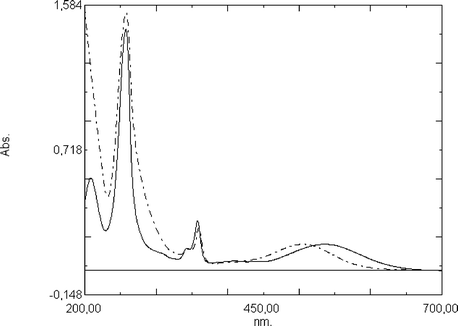 | ||
| Fig. 1 UV-Vis spectra of the molecular thread 15 (—) and the rotaxane 29 (– · –) in acetonitrile solution. | ||
 | ||
| Scheme 3 Thermal back-reaction of acridinium methoxides. | ||
The analysis of the exponential decay of the absorption of the intermediate acridinium ions at 360 nm yields the lifetime (1/k) of the ionic state. It depends strongly on the composition of the solvent. For example, the lifetime in acetonitrile solution containing 20% methanol is 990 s for 3 and 900 s for 5. The lifetime of 5 is shortened to 25 s in methanol solution (see ESI). Compounds 2 and 4 represent photochromic systems with large differences between the UV-vis absorption maxima of the involved compounds (see Scheme 2).
Syntheses of molecular axles
There are two possibilities for connecting the acridine unit with the various threadlike ethylene glycol chains. (i) In the case of the 9-aryl-N-methyl-acridinium compounds 7 and 9, N-methylacridinone was reacted with lithium-organic compounds. The primarily formed acridanes are converted to the acridinium compounds during the purification procedure with solvents containing NH4PF6 (Scheme 4). (ii) The aniline compounds are able to react directly with N-methylacridinium iodide17 to yield the acridinium salts 15–18 (Scheme 5). The corresponding acridane compounds are easily available by reacting the acridinium salts with methanol in the presence of a base (Scheme 6).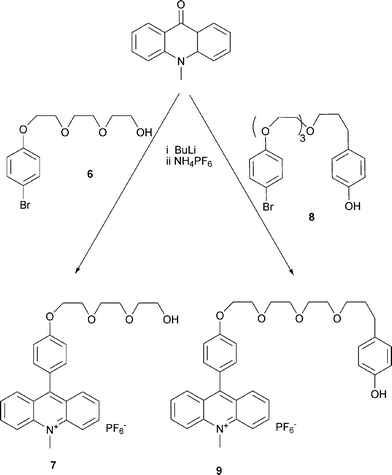 | ||
| Scheme 4 Synthesis of molecular axles 7 and 9. | ||
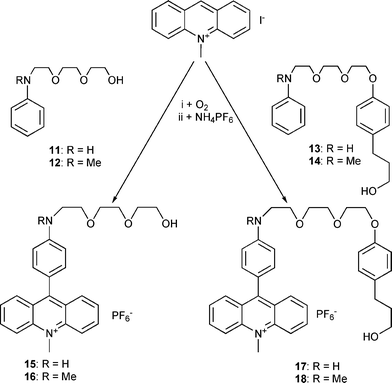 | ||
| Scheme 5 Synthesis of molecular axles incorporating 4-aminophenyl acridinium moieties. | ||
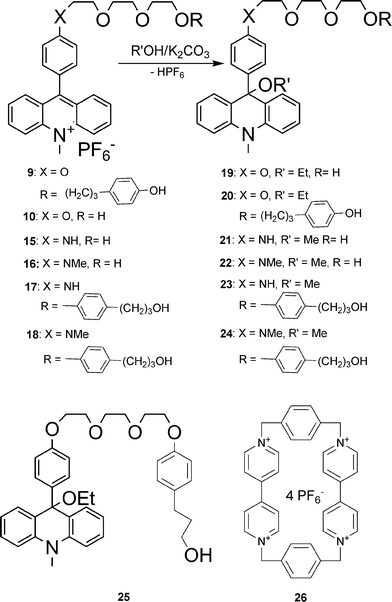 | ||
| Scheme 6 Axle molecules incorporating the acridane moiety and cyclophane 26. | ||
Rotaxanes
The compound 20 differs from the previously reported molecular axle of rotaxane 1 by flipping the photoinactive station within the two-station molecular thread. Therefore, in the course of the rotaxane preparation, the electron donor strength is diminished by esterification of the phenolic OH-group (see Scheme 8). We questioned whether the weaker electron donor capacity of the photoinactive station B has any influence on the distribution between the two stations.
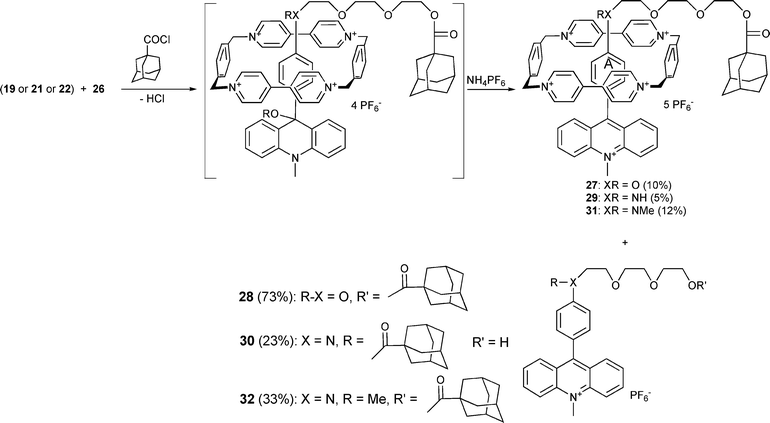 | ||
| Scheme 7 Synthesis of one-station rotaxanes. | ||
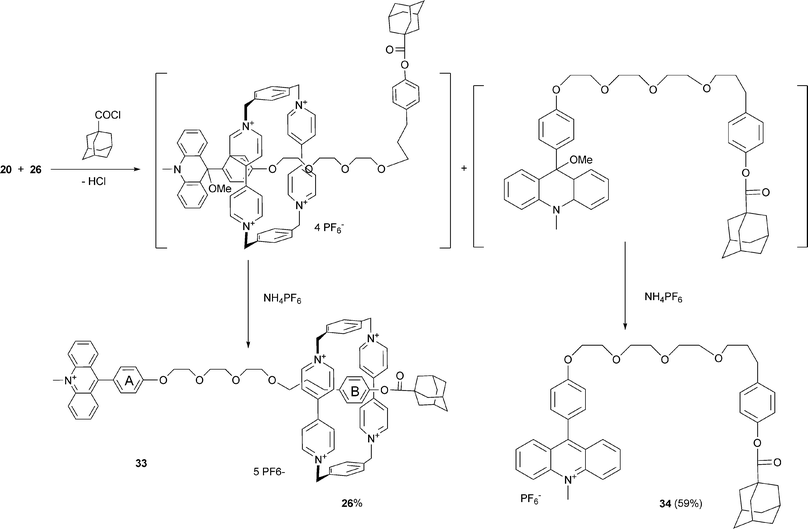 | ||
| Scheme 8 Synthesis of the two-station rotaxane 33. | ||
Despite the comparable association constants obtained for one- and two-station pseudorotaxanes the yield of the one-station rotaxanes 27, 29 and 31 (10, 5 and 12%, respectively) is considerably lower than that of the two-station rotaxanes 33 (26%), 1 (28%) and 37 (22%). Obviously the second recognition station supports the rotaxane formation. In most cases the acylated free axles (28, 30, 32, 34 and 38) were formed as the main products. The acylation of 21 takes place only at the more nucleophilic amino group yielding compound 30. In contrast, only the rotaxane 29 with the free amino group was formed, indicating that the ring resides on this group during the synthesis, thus protecting the amino group. In contrast to the synthesis of the one-station rotaxane 29, the amino group of the axle 23 was acylated during the synthesis of the two station rotaxane 36. Obviously both the station A and the station B are populated by the cyclophane 26 in pseudorotaxanes formed as precursors during the rotaxane synthesis. The rotaxanes 35 and 36 were formed in a 1:1 ratio, but due to their very similar properties, the separation of both compounds was not successful. In order to avoid the acylation of the amino group the axle 24 containing the methylated amino group has been used to prepare the rotaxane 37 (Scheme 9).
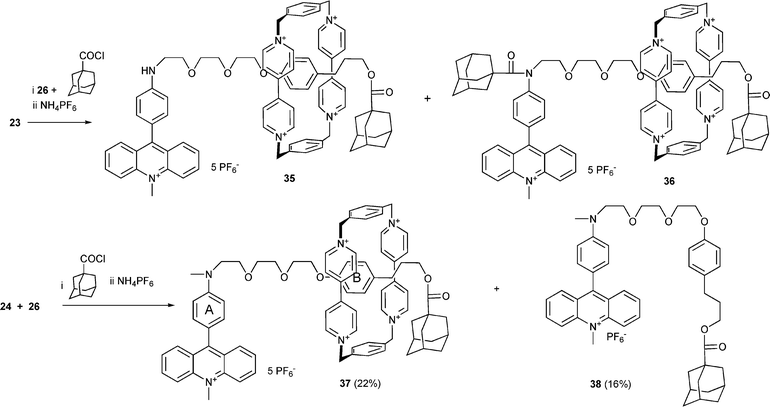 | ||
| Scheme 9 Synthesis of two-station rotaxanes. | ||
In order to prepare the photoactive acridane rotaxanes, the corresponding acridinium rotaxanes were treated with a small volume of methanol in acetonitrile solution in the presence of weak bases such as NaHCO3 or di-i-propylethylamine (Scheme 10). During the reaction, the solution turns colourless because both the acridane component and the ring absorb only UV-light.
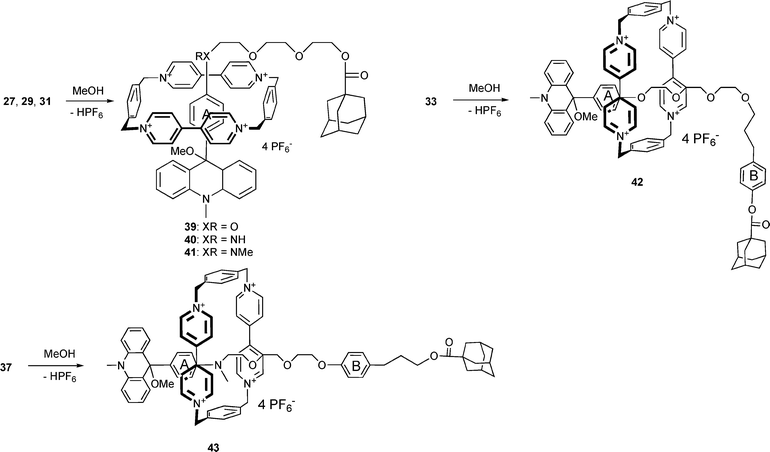 | ||
| Scheme 10 Synthesis of acridane rotaxanes. | ||
It is worthy to note that the acridane rotaxanes are also formed without a base simply upon dissolution of the acridinium rotaxanes in methanol (2–5 × 10−5 M).
Whereas 29 and 31 are quantitatively transformed into the acridanes 40 and 41, 18% of 27 remains in equilibrium with 39. In the case of the two-station rotaxane 37, the interaction energy between the cyclophane and the acridane station causes the formation of the acridane upon dissolution in methanol containing 20% acetonitrile (eqn 1).
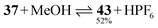 | (1) |
The pseudo acid – pseudo base equilibrium is ∼1:1 in diluted solution (2–5 × 10−5 M). The presence of the second station gives rise to only 50% turnover while the one-station rotaxane 31 reacts quantitatively to produce the acridane rotaxane (see above). In contrast, the molecular axle 24 reacts as a pseudo base to give 60% acridinium methoxide in acetonitrile/methanol 1:1 solution (eqn 2).
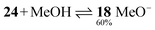 | (2) |
The free molecular axle 21 incorporating the acridane station is completely transformed to the acridinium compound in dilute solution (methanol/acetonitrile 1:1). This emphasises the higher stability of the acridinium compound 15 with respect to 21. The acridane rotaxanes do not react with methanol. The transformation of the acridinium rotaxanes to the acridane rotaxanes is obviously driven by the energy won by the stronger interaction of the acridane moiety with 26 compared with the interaction with the acridinium unit. Accordingly, acridinium compounds 9, 15 and 16 do not react with methanol to give the acridane. Thus, the acridane unit within the rotaxane and the acridane unit present in the corresponding molecular axle exhibit opposite behaviour in their reactivity with the nucleophile methanol.
Rotaxane properties
Acridinium rotaxanes. Rotaxanes with only one recognition station should exist in only a single co-conformation with the ring residing on this station. Therefore, the strongest possible interaction should exist between the cyclophane and the station since there would be no disturbances due to shuttling between two or more stations. The one-station rotaxanes 27, 29 and 31 consist of two parts: the acridinium station A and the flexible polyether chain. We expected that this chain would be a relatively high barrier against the ring location. The question arises as to whether the strongly twisted 9-aryl substituent18 of the acridinium unit aryl group is able to accommodate the tetracationic ring despite the presence of the positively charged acridinium subunit. The chemical induced shift (CIS) of specific proton signals both of the cyclophane and of the axle manifested in the 1H-NMR spectra indicated the strength of the interaction between specific regions of the axle with the ring compound. The resonances of both the ortho-protons (with respect to the substituent in the 4-position) of the aryl group attached to the acridinium moiety and of the adjacent methylene group of the chain are up-field shifted by more than 3 ppm with respect to those arising from the free axle compound (see ESI). Other resonances of the chain protons are not shifted. In contrast, the aryl protons adjacent to the acridinium subunit are upfield-shifted by less than 1 ppm. These spectral shifts clearly demonstrate that the positively charged ring 26 resides on the aryl group at the 9-position of the acridinium moiety. Further evidence comes from correlation-peaks arising from protons in the ring component and from both the ortho-protons of the aryl group and the adjacent protons of the chain observed in ROESY-spectra. The proton resonance of the N-methyl group present in rotaxane 31 is also upfield shifted by 2 ppm. Taken together, the ring resides on the aryl group of the acridinium station and the adjacent chain as far away as possible from the positively charged acridinium moiety. The N-methyl-group does not disturb the interaction of the aryl group with the ring. In conclusion, while pseudorotaxanes of the acridinium axles with 26 dissociate, the stopper units in rotaxanes formed from acridane rotaxanes prevent dissociation into the two components. Therefore the cyclophane has to arrange with in the unfavorable situation.
The situation is completely changed for the two-station rotaxanes. The distribution between the acridinium station and the second station having weak electron donor capacity depends on the interaction of the tetracationic cyclophane 26 with the two stations. According to the CIS-values observed for the two stations in all two-station acridinium rotaxanes the cyclophane resides exclusively on the station B. The proton resonances of the acridinium unit are not shifted with respect to those of the free axles. In contrast, the interactions between the ring and the station B are accompanied by significant upfield shifts of the aromatic proton resonances and the adjacent protons of the polyether chain caused by strong diamagnetic shielding due to the π-stacking of the bipyridinium ring and station B aromatic systems. The assignments of proton signals were made by using standard COSY and NOE (ROESY) data (see ESI). Cross-peaks of the ROESY spectrum reveal the interaction between the cyclophane and the station B (see ESI). The exclusive occupation of the station B holds true also for those acridinium rotaxanes containing the aminophenyl substituent at the 9-position (rotaxanes 35–37).
Rotaxanes 35 and 36 could not be separated. Nevertheless, the complete assignment of proton resonances to the two rotaxanes was possible with the help of two dimensional NMR spectra such as COSY and ROESY. Because only the proton resonances of station B are exclusively upfield shifted by the same amount (see ESI), this station is populated by the ring. Due to the voluminous adamantoyl group at the acridane moiety the population of the aniline subunit is not possible.
Also the proton resonances of station B within the rotaxane 37 appear at the same values as seen for 35 and 36. NOEs taken from ROESY spectra prove the population of station B by the cyclophane within the rotaxane 37. In conclusion, the aryl groups of the acridinium moieties are not capable of competing with the rather weak electron-donating second station even in the case of the ester group at this station in the rotaxane 33.
The proton resonances of the ring 26 are well resolved in the 1H NMR spectra both of the one- and two-station rotaxanes. While the β-protons (with respect to N+) of the ring are upfield shifted, the proton resonances of the aromatic groups of the xylylene spacers are low-field shifted. Due to the asymmetry of the axle, the proton resonances of the methylene groups of 26 are split into two doublets of an AB-system.
Acridane rotaxanes. Because the acridane station is more suitable to accommodate the tetracationic ring than the acridinium station,16 the ring does not change its position by transforming the acridinium rotaxanes to the one-station acridane rotaxanes 39–41. For the acridane rotaxanes, the upfield shifts of the aryl protons are comparable with those of the same group of the acridinium rotaxanes. Because there is no competition between the different stations, the observed upfield shift should approach the maximum value that can ever be achieved due to the interaction between the acridane units and the ring component. Therefore, the observed proton resonances of the protons of the aryl group at the 9-position of the acridane unit can be used as a probe to monitor the localisation of the ring in two-station rotaxanes. The population of the aryl group of station A by 26 is also indicated by NOEs (according to correlation peaks between the aromatic protons of the benzylic spacer of 26 and the aromatic protons of the aryl group of station A in the ROESY spectra).
While the cyclophane 26 of acridinium rotaxanes 27, 29 and 31 does not interact with the acridinium part (see above), all proton resonances of the acridane core of the rotaxanes 39–41 are low-field shifted (see ESI), indicating an edge-to-face interaction between the aromatic rings of 26 and the plane of the acridane subunit enabled by the shape of this part of the molecular axle (see Scheme 11). Obviously, with respect to the acridinium counterpart, this additional interaction is the driving force of the occupation of the acridane station as an entity while also accounting for the formation of the acridane rotaxanes from the corresponding acridinium rotaxanes in methanol solution (see above). The proton resonance of the N-methyl group present in rotaxane 41 is upfield shifted by 1.1 ppm. This finding indicates that the N-methyl group does not disturb the interaction of the aryl group with the cyclophane.
 | ||
| Scheme 11 Calculated structures (MM2-level) of one-station rotaxanes represent the changes in the shape of the molecular axle. | ||
In two-station acridane rotaxanes, the acridane station A competes with the station B. Any distribution between the two stations by shuttling of the cyclophane 26 should result in a lower CIS-value of the acridane station compared with the value observed in one-station rotaxanes. The 1H NMR spectra clearly show that the acridane station A is exclusively populated by the cyclophane 26. The upfield shifts of the aromatic protons of the aryl group at the 9-position of the acridane moiety of rotaxane 42 are exactly as that observed with the one-station rotaxane 39 (see above and the ESI). The aromatic proton resonances of the second station B are not shielded. Surprisingly, the proton resonances of the adjacent propyl chain are moderately upfield shifted (see ESI). Possibly a back-folding of the whole chain occurs, bringing the CH2 groups in contact with the ring from outside.
The acridane rotaxane 43 exhibits very broad signals corresponding to the proton resonances both of the ring and of the molecular axle at room temperature. Only the proton signals of the chain adjacent to station B are sharp and assignable with the help of two dimensional methods such as HH- and CH-COSY. The population of station A can be concluded from the proton resonances of station B. The signals of the propoxy chain appear at the same values as observed for compound 24, and this finding is an appropriate probe for the failed threading of this part of the axle.16 At moderately increased temperature (343 K), the proton resonances of the phenyl group of station A appear at δ = 4.4 and 2.5 ppm indicating the occupation of station A by the ring. The assignment of these signals was done with the help of two-dimensional C-H-COSY spectra at 343 K. Again the observed CIS-values match those observed with the one-station rotaxane 41. The proton resonances of the phenyl group of station B are shifted slightly up-field by 0.5 and 0.3 ppm. This shift might be caused by an interaction of station B with the outside ring by back-folding of the molecular axle. CIS-values of other proton resonances are given in the ESI. The proton resonances of the aryl group attached to the acridane moiety that accommodates the ring also appear at low temperature as two broad sets of signals. This indicates that the asymmetry of the two sites of the 9-aryl group due to both the slow rotary motion of the ring and the 9-aryl substituent. The ROESY spectrum of the rotaxane 43 recorded at 233 K reveals the occupation of station A because correlation peaks between the cyclophane proton signals (β-protons) and the protons of the acridane ring (protons in 1- and 8-positions) are present.
Dynamic processes in acridane rotaxanes. Apart from the aromatic protons of the benzylic groups, cyclophane signals are very broad for all acridane rotaxanes in both CD3CN and CD3OD/CD3CN (5:1) solutions at room temperature. The β-protons (with respect to N+) of the bipyridinium moiety are even merged into the base line.
Broadening of the ring signals indicates dynamic processes. By lowering the temperature to 233 K and 213 K, respectively, the broad signals of the α- and β-protons of the ring (with respect to N+) observed for the rotaxanes 40 and 43 are split into eight signals of two protons each. The signals of the benzylic protons exhibit the opposite behavior. At room temperature, a AB system is observed, indicating that the two sites of the ring are non-equivalent due to the asymmetry of the molecular axle. At 213 K, this signal is broadened (see ESI), indicating that the dynamic process is slow on the NMR time scale. Two different rotational processes may be responsible for these observed phenomena: 1) the rotation of the ring around the axle, which is frozen at low temperature, making the ring protons above and below the axle non-equivalent; and 2) the internal rotation of the bipyridinium units is rapid at room temperature and slow at 213 K with respect to the NMR time scale (see Scheme 12).
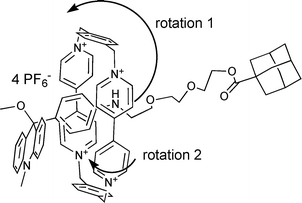 | ||
| Scheme 12 Depiction of two rotational phenomena in 40 that influence spectral data. | ||
With respect to rotaxane 42 upon lowering the temperature, the α- and β-proton signals (with respect to N+) of the ring appear as four broad singlets, indicating that a thermally activated motion of the bipyridinium units is frozen (see ESI). The same behaviour was observed with the rotaxane 1. The α-proton resonances are split by 0.6 ppm and the β-proton resonances by 1 ppm. If the ring resides on the 9-aryl-9-methoxy-acridane station, one side of the bipyridinium units is involved in edge-to-face interaction with the dihydroacridine plane. The other side of the bipyridinium plane is directed toward the part of the axle with the second station B. The protons of the methylene groups of the ring are also topologically non-equivalent and appear as two doublets of an AB-system in rotaxanes 1 and 42 even at room temperature. Because only the α- and β-protons act as a probe for the dynamic process, we may exclude the possibility that the entire ring rotates around station A. Therefore it seems likely that the rotation of the bipyridinium unit around itself exchanges the protons of the two sides, thereby enabling the alternating interaction with the acridane plane. Accordingly, ROESY spectra at 233 K exhibit exchange cross-peaks between the α- and β-proton signals in addition to the NOE cross peaks (see ESI).
Photochemistry
The encapsulation of the aryl group of the acridinium unit by the ring gives rise to a number of interesting spectroscopic properties. While the longest wavelength band of the rotaxane 27 is not different from that of the molecular axle 9, the longest wavelength absorption bands of rotaxanes 29 and 31 are blue shifted with respect to the corresponding compounds 15 and 16 (Fig. 1). The different characters of the electronic transitions of 27 on one side and 29 and 31 on the other side give rise to this different behavior. The longest wavelength absorption bands of rotaxanes 29 and 31 monitored in acetonitrile solution are blue-shifted by 1.2 × 103 and 1.7 × 103 cm−1, respectively, with respect to the absorption bands of the corresponding molecular axles. The most likely explanation of these findings is the reduced electron-donating strength of the aryl groups due to the interaction with the electron acceptor 26 and the polar microenvironment provided by the tetracationic cyclophane. The internal charge-transfer transition is more sensitive to the solvent polarity than the localised transition of the rotaxane 27.
In summary, the longest wavelength absorption band of the rotaxanes 29 and 31 may serve as a probe for the localisation of the ring. Accordingly, the absorption spectrum of the two-station rotaxane 37 corresponds to that of axle 18 and reveals the population of station B.
Provided that the acridinium ions carry electron-donating substituents at the 9-position, the locally excited state occupied by light absorption is accompanied by a very fast electron transfer to a charge shift state which quenches the fluorescence.19 Due to the interaction of 26 with the electron-rich phenyl group of the acridinium unit, the electron-donating strength of this subunit is reduced. Accordingly, in contrast with the molecular axles 9, the rotaxane 27 exhibits fluorescence characteristic of the acridinium subunit (see ESI).
In 9-arylacridanes, the phenyl group is electronically decoupled from the acridane core. For this reason, the absorption spectra of all acridane derivatives are nearly identical, exhibiting absorption bands at around 320 nm (as shoulder) and 280 nm (Fig. 2).
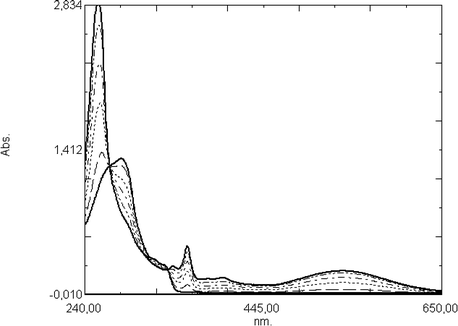 | ||
| Fig. 2 UV-vis spectra recorded after consecutive irradiation periods (0.5, 2, 4, 8, 16 min) at 313 nm of compound 21 (8.4 × 10−5 M) in acetonitrile solution (the acridinium ion 15 is formed). | ||
The quantum yield of the photoreaction of compound 21 in acetonitrile solution is 0.4. Using a mixture of acetonitrile and methanol, the nucleophilic attack of the methoxide ion on the formed acridinium ion is relatively fast. The lifetime of the ionic state is 50 s observed in acetonitrile/methanol solution (5:1). We monitored the transient absorption spectrum by subtracting the absorptions spectrum recorded before irradiation from that recorded immediately after irradiation with light of a wavelength >300 nm. The spectrum consists of two parts: The negative absorption corresponds to the decomposition of the acridane while the positive absorption clearly matches that of the acridinium ion 15 (compare Figs. 2 and 3).
The one-station rotaxanes 39–41 also undergo photoheterolysis upon excitation with light between 254 and 330 nm in acetonitrile solution containing methanol. Due to the charge-transfer interaction16 between the acridane station and the ring component, the efficiency of the photoreaction is decreased. Furthermore, the absorption spectrum of the rotaxanes is superimposable upon the absorbance spectrum of the cyclophane and the acridane station.
Because both subunits absorb light in the same wavelength range, the ring acting as an inner light filter. From the comparison of the absorbance of the transient absorption at 360 nm after photoexcitation of the molecular axle 21 and the rotaxane 40, respectively, a quantum yield of about 0.03 can be estimated for 40.
Transient absorption spectra observed upon photoexcitation of rotaxanes 40 and 41 are similar and exhibit two characteristic features (see Fig. 4 for an example).
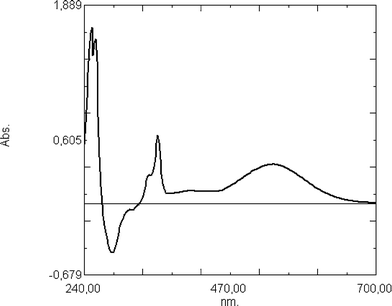 | ||
| Fig. 3 Transient absorption spectrum recorded immediately after the irradiation of compound 21 (1 × 10−4 M) for 5 s in acetonitrile/methanol (5:1) solution. | ||
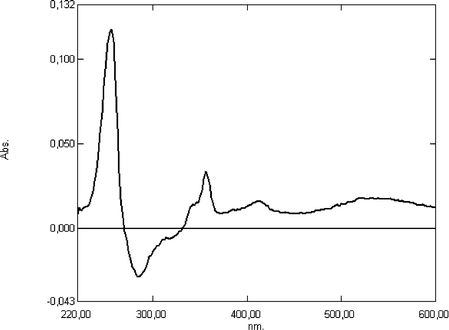 | ||
| Fig. 4 Transient absorption spectra observed upon photoexcitation of the rotaxane 40 (3 × 10−5 M) in acetonitrile/methanol (5:1) solution. | ||
The typical spectrum of an acridinium salt with the two characteristic transitions around 260, 360, 410 and 550 nm besides the depletion in the region of the acridane unit absorption (270 nm) was observed. The long wavelength absorption bands are red-shifted by 30 nm with respect to the absorption band of the stable rotaxanes 29 and 31, which have hexafluorophospate anions. After the photochemical conversion of the acridane moiety into the acridinium methoxide, the ring is in a unfavorable high-energy position. According to the co-conformation found for one-station acridinium rotaxanes, the cyclophane has only to move from a position close to the acridinium core to a position at the end of the 9-aryl substituent. Since this is also an energy-rich co-conformation the driving force for changing the co-conformation should be small. If we assume a slow movement of the ring, the absorption spectrum of the transient acridinium methoxide rotaxane would be recorded with a non-relaxed intermediate state. Here, the electronic transition may be of lower energy compared with the same transition within the relaxed acridinium rotaxane with hexafluorophosphate counter ions. Acidifying the solution of the rotaxanes 29 and 31 with trifluoroacetic acid (TFA) restored the blue-shifted acridinium absorption band.
The lifetime of the intermediate acridinium methoxides obtained from the rotaxanes 39–41 depends on the content of methanol in the solvent mixture MeCN/MeOH. Some values are given in Table 2 of the ESI.
The two-station rotaxanes have low-energy co-conformation both in the acridinium and in the acridane state. Accordingly, the transient absorption spectra recorded after the photoexcitation both of the acridane rotaxanes 42 and 43 match that of the axle 24 (see Fig. 5 for 43).
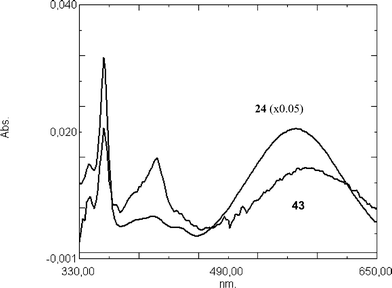 | ||
| Fig. 5 Transient absorption spectra recorded upon photolysis of molecular axle 24 and two-station acridane rotaxane 43 (in each case 3 × 10−5 M) in methanol/acetonitrile solution (1:1). | ||
The lifetime of the acridinium rotaxane 33 (as methoxide) is 110 s in methanol solution; it is increased to 1000 s in MeCN/MeOH 5:1. The efficiency of the photoreaction is only 10% as good as that of axle 34.
The quantum yield of the photoheterolysis of the rotaxane 43 is reduced to about 0.02. The lifetimes of the intermediate acridinium rotaxanes formed from 43 are 60 s in methanol solution and 220 s in a mixture of methanol and acetonitrile (1:1). The same lifetime of the transient acridinium ion was observed for compound 24.
Because the NMR spectra of both acridinium rotaxanes with hexafluorophosphate anions have shown that the ring resides on station B, it is beyond doubt that the ring will move from station A to B upon photolysis of the acridane rotaxanes. In the case of the rotaxane 43, support for this idea comes from the transient UV-vis spectra. The longest wavelength absorption band matches that of the molecular axle 24 (see Fig. 5). A blue-shift of the longest-wavelength absorption band would be expected if the ring resided on station A (in the acridinium state) as it was found for the one-station rotaxane 31 (see above).
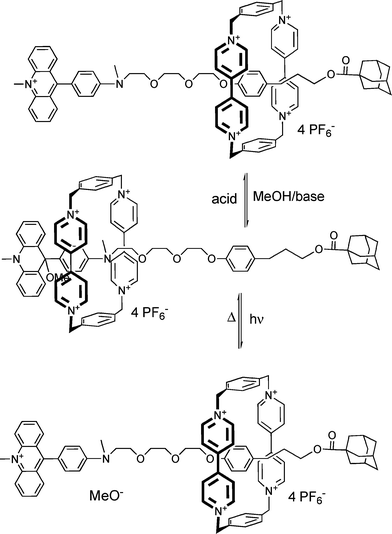
The preparation and isolation of the acridane rotaxanes was deemed unnecessary. They can be prepared in situ by the reaction of the corresponding acridinium rotaxanes in diluted solution with a base such as N-ethyldiisopropylamine or NaHCO3. By acidifying with trifluoroacetic acid the acridinium rotaxanes are regenerated. This procedure can be repeated at least ten times. The transformation can be easily followed by UV-vis spectroscopy. This means that the switch cycle can also be carried out by acid–base titration (see Scheme 13).
Conclusions
One- and two-station rotaxanes incorporating the 9-aryl-acridinium station and the corresponding 9-methoxy-acridane station, respectively, have been synthesised. Two station rotaxanes contain an alternate anisol or phenol ester recognition station. Due to charge-transfer and edge-to-face interactions, the acridane interacts more strongly with the tetracationic cyclophane 26 than the alternate station. The situation is reversed in the acridinium state of the two-station rotaxanes. Despite the positive charges at both the ring and the acridinium moiety, the ring resides on the 9-aryl-acridinium station of the one-station rotaxanes. The occupation of the 9-aryl group by the ring results in changed chemical and photophysical properties of the acridinium subunit with respect to the reference compounds without the ring 26.Shuttling processes between a 9-aryl-9-methoxy acridane recognition station and an alternate station can be initiated both photochemically by photoheterolysis followed by the thermal back-reaction and by acid/base titration.
Experimental
General methods
Commercially available chemicals and solvents (UVASOL, Merck) were used as received unless otherwise noted; solvents were dried according to standard procedures. Column chromatography (CC) was carried out on 200 mesh silica gel (Merck). Melting points (m.p.) were determined with a Boetius heating microscope.ESI mass spectroscopy was carried out on LTQ FT, Finnigan MAT, Bremen, Germany) equipped with an electrospray ion source (Thermo Electron).
NMR spectra were recorded on a Bruker DPX 300 (300 MHz) and a Bruker Advance 400 (400 MHz). The proton signals were attributed to the different subunits with the aid of two-dimensional NMR spectroscopy, such as C-H-COSY, H-H-COSY, and ROESY.
UV/Vis measurements were performed with a Shimadzu UV 2101 PC spectrometer.
Absorption spectra of the solutions were recorded before and after repeated irradiations in order to detect irreversible degradation. Transient absorption spectra between 220 and 650 nm were recorded with the UV 2101 PC spectrometer using the fastest scan mode.
Steady state photolysis experiments were carried out with acetonitrile solutions using a 500 W or 200 W high pressure mercury lamp operated in conjunction with a glass filter (cut off 300 nm) or a metal interference filter (Carl Zeiss Jena) (313 nm). The solutions were irradiated and measured in 1 cm quartz cuvettes. The rate constant of the formation of acridinium ion 15 from 21 was calculated by the tangent method from plots of conversion vs. the time of irradiation. The concentration of 15 was obtained by measuring the optical absorption of 15 at 538 nm (ε = 8140 dm3 mol−1 cm−1). 7-(4-dimethylaminophenyl)-cyclohepta-1,3,5-triene was used as a secondary actinometer (Φr313nm = 0.45, EtOH, 296 K)21 in order to determine the intensity of incoming light.
The thermal reactions were followed by UV-Vis-spectroscopy using the kinetics-program of the spectrometer. The decay curves were analysed by nonlinear regression fits (Origin 6.0, Microcal Software, Inc.).
Binding studies were carried out by means of 1H NMR titrations of solutions of 26 usually at a concentration of 1 mmol. Increasing amounts of 19, 22 or 25 were added up to ring: axle of 0.2:1–10:1 ratio (R) (20–80% saturation). Upfield shifts of the resonances of different protons of the ring were monitored in order to calculate binding constants K and the upfield shift of the protons of the ring fully saturated by the axle δGC.
Titration data points δ = f(R) were fitted to the equation 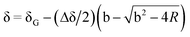 22 with
22 with 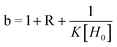 and
and 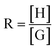 where H = host, G = guest, and Δδ = δG−δGC.
where H = host, G = guest, and Δδ = δG−δGC.
Best-fit parameters K and δGC were obtained in a nonlinear least-square fitting procedure using the curve fitting program “Sigma Plot” (Jandel Scientific Software).
Syntheses
Full experimental data are given in the ESI†. Typical procedures are listed below.The model compounds 2 and 3, the rotaxane 1 and the axle 25 were available from previous studies.16,20
The dichloromethane solution was evaporated to give the mixture of compounds 21 and 30. The separation and purification was performed by column chromatography (1 g NH4PF6 in 440 mL acetone/cyclohexane 10:1). Besides 21 (0.070 g) 30 (0.10 g, 23%) was obtained as oily compound; δH (400 MHz, CDCl3, TMS) 1.6 (6 H, m, adamantane), 1.8 (6 H, br s, adamantane), 1.9 (3 H; br s, adamantane), 3.5 (2 H, m, OCH2), 3.6 (6 H, m, ethoxy), 3.7 (2 H, m, ethoxy), 3.8 (2 H, m, N-CH2), 4.83 (3 H, s, NMe), 7.54 (2 H, d, J 8.3, Ar), 7.70 (2 H, d, J 8.3, Ar), 7.85 (2 H, m, acridinium, H-2,7), 8.02 (2 H, dd, J 1.1 and 7.6 Hz acridinium, H-1,8), 8.39 (2 H, m, acridinium, H-3,6), 8.61 (2 H, d, J 9.2, acridinium, H-4,5); m/z (ESI) 579.3219 [M − PF6−]+. C37H43N2O4 requires 579.3217.
General procedure. NaHCO3 (0.2 g) was added to the acridinium rotaxane (0.02 mmol) dissolved in acetonitrile (4mL) under addition of methanol (0.1 mL). The suspension was stirred for several hours until decolouration of the solution. NaHCO3 was filtered off. The solution was evaporated. The remaining oily solid was extracted with chloroform (3 × 5 mL). The remaining solid was used without further purification.
The dichloromethane solution was evaporated to give the compound 38. The separation and purification was performed with the help of column chromatography [MeOH/H2O/NH4Cl (saturated aqueous solution) 200:60:1]. The blue fractions were collected, the solvents were evaporated, and the remaining blue solid was dissolved with a mixture of H2O (30 mL)/dichloromethane (30 mL)/NH4PF6 (0.2 g). The organic phase was dried. The solvent was evaporated to give compound 38 (0.044 g, 16%) as blue viscous oil; δH (400 MHz, CD3CN, TMS) 1.7 (6 H, m, adamantane), 1.8 (8 H, m, adamantane, CH2), 1.9 (3 H, br s, adamantane), 2.50 (2 H, t, J 7.4, CH2), 3.1 (3 H, s, NCH3), 3.6 (4 H, m, ethyleneoxy), 3.7 (2 H, m, ethyleneoxy), 3.72 (4 H, m, ethyleneoxy), 3.88 (2 H, t, J 6.3, O(CO)CH2), 3.99 (2 H, m, ethyleneoxy), 4.71 (3 H, s, N+Me), 6.73 (2 H; d, J 8.3, Ph), 6.98 (2 H, d, J 8.5 Hz, Ph), 7.02 (2 H, br d, J 8.8, Ar), 7.36 (2 H, d, J 8.7, Ar), 7.8 (2 H, m, acridinium, H-2,7), 8.23 (2 H, d, J 8.7, acridinium, H-1,8), 8.29 (2 H, m, acridinium, H-3,6), 8.52 (2 H, d, J 9.2 Hz, acridinium, H-4,5), 8.50 (2 H, d, J 9.2, acridinium, H-4,5); m/z (ESI) 727.4105 [M − PF6−]+. C47H55N2O5 requires 727.4099.
Acknowledgements
We gratefully acknowledge the Deutsche Forschungsgemeinschaft for financial support.References
- J. F. Stoddart, Acc. Chem. Res., 2001, 34, 410 CrossRef CAS; C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS; V. Balzani, M. Venturi, and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim 2003 Search PubMed; E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem. Int. Ed. Engl., 2007, 46, 72 Search PubMed.
- M. C. T. Fyfe, P. T. Glink, S. Menzer, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem. Int. Ed. Engl., 1997, 36, 2068 CrossRef CAS.
- W. Clegg, C. Gimenez-Saiz, D. A. Leigh, A. Murphy, A. M. Z. Slawin and S. J. Teat, J. Am. Chem. Soc., 1999, 121, 4124 CrossRef CAS.
- R. A. Bissel, E. Cordova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133 CrossRef.
- H.-R. Tseng, S. A. Vignon and J. F. Stoddart, Angew. Chem. Int. Ed. Engl., 2003, 42, 1491 CrossRef CAS; U. Létinois-Halbes, D. Hanss, J. M. Beierle, J.-P. Collin and J.-P. Sauvage, Org. Lett., 2005, 7, 5733 CrossRef CAS.
- S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77 RSC.
- H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605 CrossRef CAS; C. A. Stanier, S. J. Alderman, T. D. W. Claridge and H. L. Anderson, Angew. Chem. Int. Ed. Engl., 2002, 41, 1769 CrossRef; Q.-C. Wang, D.-H. Qu, J. Ren, K. Chen and H. Tian, Angew. Chem. Int. Ed. Engl., 2004, 43, 2661 CrossRef CAS; H. Murakami, A. Kawabuchi, R. Matsumoto, T. Ido and N. Nakashima, J. Am. Chem. Soc., 2005, 127, 15891 CrossRef CAS.
- A. Altieri, G. Bottari, F. Dehez, D. A. Leigh, J. K. Y. Wong and F. Zerbetto, Angew. Chem. Int. Ed. Engl., 2003, 42, 2296 CrossRef CAS; E. M. Pérez, D. T. F. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210 CrossRef CAS; M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058 CrossRef CAS.
- W. Zhou, D. Chen, J. Li, J. Xu, J. Lv, H. Liu and Y. Li, Org. Lett., 2007, 9, 3929 CrossRef CAS.
- P. L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. F. Kaifer, D. Philp, M. Pietraszkiecz, L. Prodi, M. V. Reddington, M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1999, 114, 193.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, K. R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart, M. Venturi and S. Wenger, Chem. J. Eur., 2000, 6, 3558 CrossRef; V. Balzani, M. Clemente-León, A. Credi, M. Semeraro, M. Venturi, H.-R. Tseng, S. Wenger, S. Saha and J. F. Stoddart, Aust. J. Chem., 2006, 59, 193 CrossRef CAS.
- V. Balzani, M. Clemente-León, A. Credi, B. Ferrer, M. Venturi, A. H. Flood and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 1178 CrossRef CAS.
- A. M. Brower, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124 CrossRef CAS.
- W. Abraham, L. Grubert, U.-W. Grummt and K. Buck, Chem. Eur. J., 2004, 10, 3562–3568 CrossRef CAS.
- U. Pischel, W. Abraham, W. Schnabel and U. Müller, Chem. Commun., 1997, 1383 RSC; V. A. Kharlanov, W. Abraham and U. Pischel, J. Photochem. Photobiol. A: Chem., 2004, 162, 213 CrossRef CAS.
- W. Abraham, K. Buck, M. Orda-Zgadzaj, S. Schmidt-Schäffer and U.-W. Grummt, Chem. Commun., 2007, 3094 RSC.
- V. A. Trofimov, O. N. Chupakhin and Z. Pushkareva, Khim. Geterotsikl. Soedin., 1971, 612.
- S. A. Jonker, S. I. van Dijk, K. Goubitz, C. A. Reiss and J. W. Verhoeven, Mol. Cryst. Liq. Cryst., 1990, 183, 273 CAS.
- G. Jones II, M. S. Farahat, S. R. Greenfield, D. J. Gosztola and M. R. Wasielewski, Chem. Phys. Lett., 1994, 229, 40 CrossRef CAS.
- L. Grubert and W. Abraham, Tetrahedron, 2007, 44, 10778 CrossRef.
- W. Abraham, H. Otto and D. Kreysig, J. Photochem., 1981, 16, 261 CAS.
- R. S. Macomber, J. Chem. Educ., 1992, 69, 375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental information, UV-vis and NMR spectra of selected compounds and transients, absorption maxima of rotaxanes and molecular axles observed in different solvents, assignment of protons of rotaxanes, fluorescence spectrum of rotaxane 29. See DOI: 10.1039/b815848g |
| This journal is © The Royal Society of Chemistry 2009 |