The kinetics and mechanism of the acid-catalysed detritylation of nucleotides in non-aqueous solution
Mark A. Russell, Andrew P. Laws, John H. Atherton and Michael I. Page*
Department of Chemical and Biological Sciences, University of Huddersfield, Queensgate, Huddersfield, UK HD1 3DH
First published on 6th November 2008
Abstract
The kinetics and mechanism of the deprotection (detritylation) of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine nucleoside catalysed by dichloroacetic acid to give a 4,4′-dimethoxytrityl carbocation have been studied in toluene, dichloromethane and acetonitrile. There is little or no effect of solvent polarity on the equilibrium and rate constants. Entropies of activation are highly negative ∼−105 J K−1 mol−1 and similarly show little variation with solvent. Addition of small amounts of water to the reaction medium reduces the detritylation rate, presumably through its effect on the solution acidity. All observations are compatible with detritylation occurring through a concerted general acid-catalysed mechanism rather than a stepwise A1 process.
Introduction
The acid-catalysed removal of the 4,4′-dimethoxytrityl protecting group from the 5′-oxygen of the solid-support-bound nucleotide during oligonucleotide synthesis is commonly referred to as ‘detritylation’. The reaction yields a hydroxyl function at the 5′-position of the nucleotide and a relatively stable 4,4′-dimethoxytrityl carbocation (Scheme 1). The large scale synthesis of oligonucleotides requires quantitative deprotection of the nucleotide as incomplete detritylation causes uncoupled nucleosides in the subsequent coupling steps, reducing the purity and overall yield of the final product.1,2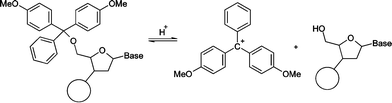 | ||
| Scheme 1 | ||
High concentrations of strong acids are required for rapid and quantitative removal of the trityl protecting group, but under these conditions depurination of the guanosine, and to a lesser extent the adenosine nucleosides, is frequent and highly undesirable when product purity is extremely important. The acid catalysed depurination of 2′-deoxyguanosine cleaves the N-glycosyl bond between the purine base and the ribose sugar and occurs through the intermediate formation of an oxenium cation (Scheme 2).3–5
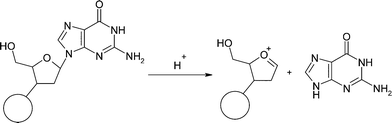 | ||
| Scheme 2 | ||
The rate of detritylation is generally faster than the rate of depurination and to minimise the amount of depurination, detritylation is often performed as quickly as possible.3,6 To achieve this objective strong protic acids such as benzenesulfonic acid and trichloroacetic acid have been used, but due to continuing complications of depurination and an increasing desire for product purity over yield, less acidic alternatives have recently been sought with dichloroacetic acid being amongst the most popular. The use of dilute solutions of weaker acids minimises the extent of depurination but often leads to incomplete detritylation.1–3,6–8
Although there is a large amount of literature describing the types of acid and conditions required for optimum detritylation there is little or no mechanistic data available for the deprotection reaction under the conditions usually adopted for large scale synthesis i.e. in non-aqueous organic solvents. There have been studies in aqueous and aqueous–acetonitrile solutions by Maskill and co-workers on the deamination of 4,4′-dimethoxytritylamine9,10 and on the ionisation of 4,4′-dimethoxytrityl alcohol11,12 to the trityl carbocation, but these results are not easily extrapolated to non-aqueous organic solvents.
Herein we report mechanistic studies of the detritylation reaction in organic solvents and investigate the equilibrium between various intermediate species.
Results and discussion
The reaction equilibrium
One obvious potential difference between the reaction in non-aqueous solvents compared with water is the stability of ions, and the extent of ion-pair formation. The first step in the investigation was to explore the equilibrium of the detritylation reaction of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine (Scheme 3). The product 4,4′-dimethoxytrityl carbocation absorbs visible radiation at a λmax which shows a bathochromic shift on decreasing solvent polarity if the dielectric constant is taken as a measure of solvent polarity (Table 1). This implies that the stability of the cation is related to the solvent polarity as λmax is related to the energy gap between the HOMO and LUMO, with a lower λmax indicative of a higher energy gap and therefore a more stable carbocation. The absorbance is a good measure of concentration of the carbocation and equilibrium and kinetic studies were undertaken by measuring changes in the absorbance at the λmax of the carbocation in the corresponding solvent. As will be described later, the presence of trace amounts of water in the reaction systems has a profound effect on the reaction. As a consequence steps were taken to ensure all solvents were dry and water was excluded. The concentration of water in the systems was measured by Karl Fischer titration in each experiment before and after reaction.| Solvent | λmax | Dielectric constant |
|---|---|---|
| Toluene | 510 | 2.4 |
| Dichloromethane | 505 | 4.8 |
| Acetonitrile | 497 | 37.5 |
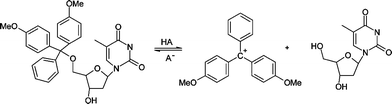 | ||
| Scheme 3 | ||
The reaction of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine to generate the carbocation (Scheme 3) only occurs in non-aqueous solvents with sufficiently high concentration of acid.
The extent of reaction is easily measured by titration of the nucleoside against dichloroacetic acid in toluene, dichloromethane and acetonitrile, which give sigmoidal absorbance–dichloroacetic acid profiles (Fig. 1).The relationship between acidity and extent of reaction was not as may have been expected. Carbocation/ion formation may be anticipated to be favoured in the more polar solvent due to charge stabilisation. However, the best solvent for trityl carbocation formation is dichloromethane and the most polar solvent, acetonitrile, is intermediate between toluene and dichloromethane. The concentration of dichloroacetic acid required to give 50% conversion to the trityl cation is 0.007 M in dichloromethane, 0.125 M in acetonitrile and 0.230 M in toluene. As well as the 4,4′-dimethoxytrityl cation being formed more readily in dichloromethane with a lower concentration of dichloroacetic acid required for complete conversion, the molar extinction coefficient also appears to be slightly greater in this solvent than in acetonitrile or toluene, in which it is identical. The 4,4′-dimethoxytrityl carbocation is a very stable species due to its highly delocalised charge, and so stabilisation by either solvent or counter-ion is less important.
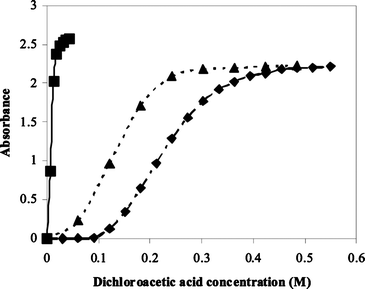 | ||
| Fig. 1 Plot of absorbance against acid concentration for the detritylation of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine (3.6 × 10−5 M) with dichloroacetic acid in ✦ toluene (0.035 M water), ■ dichloromethane (0.034 M water) and ▲ acetonitrile (0.044 M water) at 30 °C. | ||
Differences also arise because on the varying stabilities of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine, 2′-deoxythymidine and the acid and its conjugate base in different solvents (Scheme 3).
The presence of small amounts of water in the reaction systems can cause difficulties when comparing the different solvents. The concentration of water present during detritylation has a significant effect upon the shape of the absorbance versus acid concentration profiles (Fig. 2).
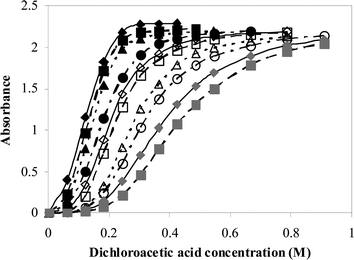 | ||
| Fig. 2 Plot of absorbance against acid concentration for the detritylation of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine (3.6 × 10−5 M) with dichloroacetic acid in acetonitrile with increasing concentrations of water at 30 °C. Water concentration increases from left to right (0.011–0.34 M). | ||
Increasing the concentration of water in the reaction solution causes the profiles to become more sigmoidal in shape with a greater initial range of acid concentration where there is apparently no carbocation formation. As a consequence the concentration of acid required to cause complete cation formation is increased. There is also a small decrease in the molar extinction coefficient at the measured wavelength as the concentration of water is increased, which is due to a slight shift in the λmax of the 4,4′-dimethoxytrityl cation, presumably due to an increase in solvent polarity on water addition. The effects are the same in all three solvent systems.
It is apparent that the presence of water in the reaction has a significant effect on the detritylation equilibrium. One possible explanation is reaction of water with the 4,4′-dimethoxytrityl carbocation to form 4,4′-dimethoxytrityl alcohol. Evidence for the presence of this reaction was obtained from treating 4,4′-dimethoxytrityl alcohol with dichloroacetic acid in acetonitrile, which gave absorbance–acid concentration profiles sigmoidal in shape similar to those observed previously, as expected from the equilibrium formation of the 4,4′-dimethoxytrityl carbocation (Scheme 4).
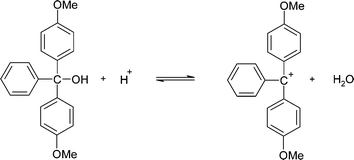 | ||
| Scheme 4 | ||
Addition of water to this solution reduced the absorbance due to the carbocation, indicating the reversible formation of the alcohol, which was confirmed by 13C NMR spectra that show distinct chemical shifts of the central carbon of the 4,4′-dimethoxytrityl group (Table 2).
| 4,4′-Dimethoxytrityl species | 13C NMR of central carbon in deuterated acetonitrile δ/ppm |
|---|---|
| Thymidine | 87.3 |
| Cation | 197.3 |
| Alcohol | 81.6 |
It appears that the observed ‘lag’ in the absorbance–acid concentration profiles (Fig. 1 and Fig. 2) at low concentrations of acid is due to trapping of the cation formed from the detritylation of the trityl ether with any available water to form 4,4′-dimethoxytrityl alcohol, thus keeping the measured absorbance close to zero. This occurs until a sufficient concentration of acid is added to force the equilibrium towards formation of the carbocation (DMTr+) by removal of both the thymidine group from the ether (DMTrOTh) and the hydroxyl group from the alcohol (DMTrOH). If the concentration of water is increased, a greater concentration of dichloroacetic acid is required to cause quantitative cation formation (Scheme 5).
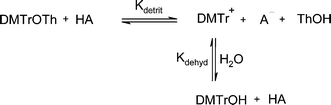 | ||
| Scheme 5 | ||
The two equilibrium constants, Kdetrit and Kdehyd (Scheme 5), are given by eqn (1) and eqn (2) and were determined for detritylation in acetonitrile.
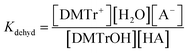 | (1) |
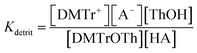 | (2) |
For the dehydration reaction, Kdehyd (Scheme 5) was calculated assuming that at high acid concentration complete conversion to DMTr+ occurs and its concentration is equal to the concentration of the ether DMTrOTh used (3.6 × 10−5 M). If water is added to reduce the concentration of DMTr+ by half then there will be equal concentrations of DMTr+ and alcohol, DMTrOH (1.8 × 10−5 M). Eqn (1) is then simplified to eqn (3).
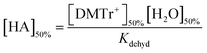 | (3) |
Using the data in Fig. 2, a plot of [HA]50% as a function of [H2O]50% is linear with a slope of 1.70, which is equal to [DMTr+]50%/Kdehyd and from which Kdehyd = 1.06 × 10−5 M.
Reversing the reaction of Scheme 3 by adding thymidine to the solution of the 4,4′-dimethoxytrityl carbocation could not be achieved due to the poor solubility of thymidine in acetonitrile. Consequently, an estimate of Kdetrit was determined using the data obtained from the addition of propan-1-ol to the reaction prior to acid titration to simulate the reverse reaction of detritylation; the formation of ether from the cation (Fig. 3).
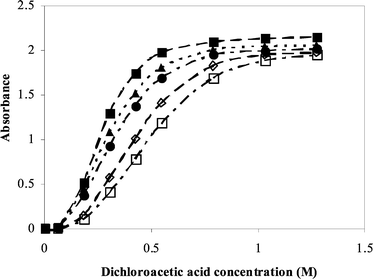 | ||
| Fig. 3 Plot of absorbance against acid concentration for the reaction of 4,4′-dimethoxytritylthymidine (3.6 × 10−5 M) with dichloroacetic acid in acetonitrile with varying concentrations of propan-1-ol at 30 °C. Propan-1-ol concentration increases from left to right (0.502–0.903 M). | ||
Using the same assumptions used for the determination of Kdehyd, eqn (2) simplifies to eqn (4).
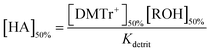 | (4) |
A plot of [HA]50% as a function of [ROH]50% is linear with a slope of 0.65, which is equal to [DMTr+]50%/Kdetrit and from which Kdetrit = 2.75 × 10−5 M.
The ratio of the two equilibrium constants can then be used to calculate the equilibrium between the ether and the alcohol, Ktrans = 2.59 (Scheme 6). This could indicate that the 4,4′-dimethoxytrityl alcohol is, marginally, thermodynamically more stable than the ether, but there will also be thermodynamic contributions from the relative stabilities of the thymidine alcohol and water in acetonitrile.
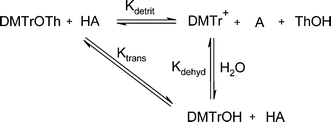 | ||
| Scheme 6 | ||
The effect of the magnitude of Ktrans is evident by comparing Fig. 2 and Fig. 3 where, for example, to reduce the amount of 4,4′-dimethoxytrityl carbocation corresponding to an absorbance of 0.5 at an acid concentration of 0.2 M, requires approximately 0.27 M water or 0.72 M propan-1-ol.
Kinetics
The kinetics of the detritylation of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine (Scheme 3) were studied spectrophotometrically by stopped-flow and conventional techniques in toluene, dichloromethane and acetonitrile. Pseudo-first-order rate constants kobs were determined from the change in absorbance due to the 4,4′-dimethoxytrityl carbocation with time. The concentration of acid used was greater than that needed to achieve complete conversion to the carbocation. The dependence of kobs on the dichloroacetic acid concentration for each solvent gave a good linear correlation from which the second-order rate constants were calculated (Table 3).There is a remarkably small dependence of the rate of detritylation on the solvent and, perhaps contrary to expectation, the second-order rate constant in acetonitrile is the smallest despite the presumed development of some charge in the transition state and the better charge stabilisation in the more polar solvent. Similarly one would expect the acid strength of dichloroacetic acid to vary with solvent and so the small dependence of rate is indicative of a small dependence on acidity and possibly an ‘early’ transition state.
Increasing the concentration of water in acetonitrile decreases the rate of formation of the carbocation. As before, good linear correlations are obtained for plots of kobs against dichloroacetic acid concentration from which the corresponding second-order rate constants were obtained (Table 4). As the initial concentration of water is increased in the reaction medium the rate of reaction decreases, which is presumably due to a decrease in the acidity of the solution.
| Initial water concentration/M | k/M1 s−1 |
|---|---|
| 0.022 | 4.7 |
| 0.078 | 3.7 |
| 0.110 | 2.2 |
| 0.130 | 1.1 |
The activation energy parameters were determined for detritylation in toluene, dichloromethane and acetonitrile, which all gave good linear correlations using the Eyring equation. Given the large differences in polarity of the solvents, the activation parameters are remarkably similar (Table 5). Reactions that involve the generation of charge on going to the transition state are normally susceptible to large solvent effects, and entropies of activation normally vary enormously.
Mechanism
It is usually assumed that the mechanism for the acid-catalysed detritylation reaction is A1 in nature with initial protonation of the 5′-oxygen followed by unimolecular C–O bond cleavage to give the carbocation and the displaced alcohol (Scheme 7).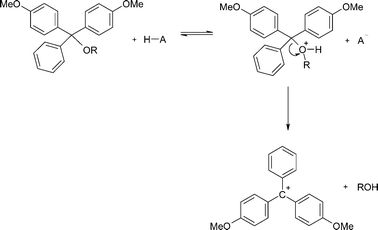 | ||
| Scheme 7 | ||
This assumption is supported by studies on the deamination of 4,4′-dimethoxytritylamine9,10 and on the ionisation of 4,4′-dimethoxytrityl alcohol11,12 to the carbocation in aqueous solutions. The A1 mechanism is a simple stepwise process and the second order rate constant kHA is equal to the product of the equilibrium constant for the first step K1 and the rate constant for the second step k2 (Scheme 8).
 | ||
| Scheme 8 | ||
In acetonitrile the second-order rate constant is 4.7 M−1 s−1. Estimation of K1 can be obtained from the ratio of the dissociation constants of dichloroacetic acid, KaHA, and that for the protonated ether, KaROR, as described below.
The pKa of dichloroacetic acid in acetonitrile is 15.8.13 There are no pKa values available for protonated ethers in acetonitrile, but in water they range between −2 and −6.14 Based on the relationship between pKa values in water and acetonitrile, those for ether conjugate acids can be estimated to be between 3 and 6, suggesting that K1 = 10−10–10−13.
The value of the unimolecular rate constant for the breakdown of the protonated trityl ether, k2, for the hypothetical A1 mechanism (Scheme 8) can be calculated from the ratio of the actual second-order rate constant for detritylation and K1 indicating that k2 = kHA/K1 = 1011–1014 s−1. This extremely high value indicates that the lifetime of the conjugate acid intermediate (Scheme 8) is close to or less than that expected for an intermediate to exist,15 and therefore the formation of this intermediate and consequently the A1 mechanism seem unlikely. A more likely mechanism for detritylation in acetonitrile and in other non-aqueous solvents is a concerted general acid-catalysed process (Scheme 9). The microscopic reverse of this reaction is the concerted general base-catlysed addition of water to the carbocation, which is the mechanism that has been proposed for triarylmethyl carbocations in water.16
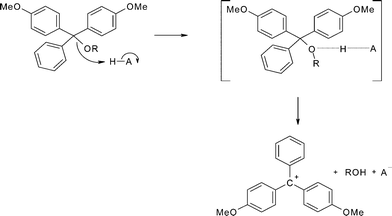 | ||
| Scheme 9 | ||
Jencks17 and Fife18,19 have both proposed a number of requirements to determine whether a mechanism will be stepwise or concerted. Jencks suggests that for a reaction to occur by a concerted general acid-catalysed mechanism there must be a large pKa change during the course of the reaction and the catalyst must have a pKa intermediate between the initial and final pKa values of the substrate site. During the course of the detritylation reaction in acetonitrile there is a large change in the pKa of the protonated ether from 3–6 to that of the alcohol liberated (thymidine) of > 22 while the pKa of 16 for the proton donor, dichloroacetic acid, is intermediate between those of the ether conjugate acid and the alcohol. Fife has also proposed a number of requirements for a concerted general acid-catalysed mechanism based mainly on the study of the mechanisms of hydrolysis of acetals and ketals. These state that concerted mechanisms occur when either the leaving group is very good and/or the product cation is very stable. A concerted general acid-catalysed mechanism for the detritylation of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine is compatible with all of these criteria.
Experimentally, the small dependence of the rate constants for the acid-catalysed detritylation of 5′-O-(4,4′-dimethoxytrityl) thymidine on solvent (Table 3), the negative entropies of activation and their small variation with solvent (Table 4) are all compatible with a bimolecular process leading to an early transition state with little charge development (Scheme 9). A concerted mechanism for proton transfer coupled to C–O bond fission is anticipated for the formation of the stable 4,4′-dimethoxytrityl carbocation.
Typically, the formation of relatively unstable carbocations would be more favoured in polar solvents due to increased stabilisation of the charged species. The equilibrium and kinetic studies reported here where solvent polarity is not having these expected effects on either the rate or equilibrium, are the result of the 4,4′-dimethoxytrityl carbocation being a stable species due to extensive delocalisation.
Experimental
Equilibrium studies were performed on a Cary 1E UV-Visible spectrophotometer with 1 cm quartz cuvettes. All solvents were dried over 4 Å molecular sieves under dry argon for at least 24 hours prior to use. In a typical experiment, solutions of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine (3.6 × 10−5 M, 2 ml) were pipetted into the cuvette and dichloroacetic acid (12 M, 1–20 μl) was then titrated. After each aliquot of acid was added the sample was shaken and left to equilibrate for five minutes prior to recording of the UV spectrum over a wavelength range of 250–600 nm.1H and 13C NMR spectra were recorded on a Bruker 400 MHz or Bruker 500 MHz spectrometer with deuterated acetonitrile as solvent.
Kinetic measurements were performed on an Applied Photophysics SX.18 MV-R1 Stop-flow spectrophotometer with a 25 : 1 syringe ratio. In a typical experiment, a solution of 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine (9 × 10−4 M) was loaded into the smaller of the two syringes and dichloroacetic acid (0.2-3.5 M) was loaded into the larger syringe. Injection of the contents of the two syringes into the reaction cell initiated the reaction, which was followed by measurement of the increase in the absorbance of the produced carbocation, an average of 20–30 results being taken for each run.
Synthesis of 4,4′-dimethoxytrityl alcohol
Sodium hydroxide (10 ml, 2.95 mmol) was added to a solution of 4,4′-dimethoxytritylchloride (1 g, 2.95 mmol) in acetonitrile (25 ml). As the reaction proceeded the solution was kept basic with further additions of sodium hydroxide (approx. 3 ml). On addition of water (20 ml) to the solution, a yellow–brown oil, which was found to be the crude form of the 4,4′-dimethoxytrityl alcohol, and a white precipitate was formed. The oil was dissolved in ether (25 ml) and water (25 ml) and the organic layer extracted with ether (3 × 15 ml). After drying over sodium sulfate and filtration, the solvent was removed and the resulting very viscous orange oil was the 4,4′-dimethoxytrityl alcohol (0.76 g, 81%). 1H NMR: δ (CD3CN) 7.23 (5H, m, aromatic H), 7.12 (4H, d, aromatic H, J = 2.2 Hz), 6.82 (4H, d, aromatic H, J = 2.2 Hz), 4.24 (1H, s, OH) (lost on D2O shake), 3.57 (6H, s, OCH3). 13C NMR: δ (CD3CN) 159.9, 149.0, 140.9, 130.0, 128.6, 127.7, 113.8, 81.6, 55.8. IR: ν/cm−1 3485.0 (OH), 3058.1 (Ph-H), 2835.3 (OCH3), 1607.5, 1583.1, 1508.7 (Ph) cm−1.References
- C. H. Paul and A. T. Royappa, Nucleic Acids Res., 1996, 24, 3048 CrossRef CAS.
- S. P. Adams, K. S. Kavka, E. J. Wykes, S. B. Holder and G. R. Galluppi, J. Am. Chem. Soc., 1983, 105, 661 CrossRef CAS.
- A. H. Krotz, B. McElroy, A. N. Scozzari, D. L. Cole and V. T. Ravikumar, Org. Proc. Res. Dev., 2003, 7, 47 Search PubMed.
- M. H. Baik, R. A. Friesner and S. J. Lippard, J. Am. Chem. Soc., 2002, 124, 4495 CrossRef CAS.
- M. Roger and R. D. Hotchkiss, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 653 CrossRef CAS.
- M. Septak, Nucleic Acids Res., 1996, 24, 3053 CrossRef CAS.
- A. H. Krotz, D. L. Cole and V. T. Ravikumar, Nucleosides Nucleotides, 1999, 18, 1207 CAS.
- B. S. Sproat and W. Bannwarth, Tetrahedron Lett., 1983, 24, 5771 CrossRef CAS.
- J. Crugeiras and J. Maskill, J. Chem. Soc., Perkin Trans., 1998, 2, 1901 Search PubMed.
- M. L. Canle, I. Demirtas and H. Maskill, J. Chem. Soc., Perkin Trans., 2001, 2, 1748 Search PubMed.
- J. Crugeiras and H. Maskill, J. Chem. Soc., Perkin Trans., 2000, 2, 441 Search PubMed.
- J. Crugeiras and H. Maskill, Can. J. Chem., 1999, 77, 530 CrossRef CAS.
- M. K. Chantooni and I. M. Kolfhoff, Anal. Chem., 1979, 51, 133 CrossRef CAS.
- G. Perdoncin and G. Scorrano, J. Am. Chem. Soc., 1977, 6983 CrossRef CAS.
- W. P. Jencks, Acc. Chem. Res., 1980, 13, 161 CrossRef CAS.
- J. R. Gandler, J. Am. Chem. Soc., 1985, 107, 8218 CrossRef CAS.
- W. P. Jencks, J. Am. Chem. Soc., 1972, 94, 4731 CrossRef CAS.
- T. H. Fife, Acc. Chem. Res., 1971, 5, 264.
- T. H. Fife and E. Anderson, J. Org. Chem., 1971, 36, 2357 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |