Switchable electrostatic interactions between gold nanoparticles and coiled coil peptides direct colloid assembly†
Sara C. Wagnera, Meike Roskampb, Helmut Cölfenc, Christoph Böttcherd, Sabine Schlecht*b and Beate Koksch*a
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: koksch@chemie.fu-berlin.de; Fax: +49 30 838 55644; Tel: +49 30 838 55344
bInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Fabeckstraße 34–36, 14195 Berlin, Germany. E-mail: schlecht@chemie.fu-berlin.de; Fax: +49 30 838 53310; Tel: +49 30 838 52423
cDepartment of Colloid Chemistry, Max-Planck-Institute of Colloids and Interfaces, 14424 Potsdam, Germany
dResearch Centre for Electron Microscopy, Freie Universität Berlin, Fabeckstraße 36a, 14195 Berlin, Germany
First published on 17th November 2008
Abstract
The nanoparticle–peptide interaction described here is based on electrostatic forces and the pH value can act as a trigger to direct the organization of functionalized nanoparticles in a reversible and repeatable manner. The ability of the peptide to interact with the charged gold nanoparticles is directly related to its helical structure and was not found for a random coil peptide with the same net charge. Interestingly, the interaction with nanoparticles seems to induce a fibrillation of the coiled coil peptide.
Introduction
Metal and semiconductor nanoparticles possess unique and size-dependent magnetic, optical, and electronic properties.1 Their size and shape can be controlled by different synthetic routes. The directed assembly of nanoparticles by means of biomolecules is of great current interest to the materials science and medical technology fields due to their potential use in sensing2a and cell targeting.2b Several recent studies have described the use of DNA and proteins for the generation of hybrid materials. Both of these classes of biomolecules have specific and selective binding properties3a and unique functionalities that are valuable for catalytic,3b optic,3c and switching3d applications. Several techniques including physical adsorption, electrostatic binding, and covalent coupling have been used to immobilize biomolecules on inorganic supports.4 The application of electrostatic forces for the organization of nanoparticles by peptides has been little explored. In contrast, the organisation of charged particles by oppositely charged particles of about the same size has been explored in different size regimes.5 Therefore, this also represents a promising strategy for the adjustment of the peptide–nanoparticle interaction e.g. by variation of the pH in a switchable manner. Further decisive aspects for the strength and the effectiveness of the electrostatic interactions will be the shape of the peptide and the charge distribution on its surface.Results and discussion
The α-helical coiled coil folding motif combines the broad functionality of proteins with molecular recognition properties and structural stability similar to those of DNA, thus, providing an attractive system for the organization of functionalized nanoparticles.6 This motif consists of two to seven α-helices wound around one another to form a left-handed coil.7 In general, the sequence follows a regular pattern of the so-called heptad repeat denoted (a-b-c-d-e-f-g)n. The thermodynamic stability of this quaternary structure is mainly driven by the formation of a hydrophobic core between the amphiphilic α-helices. For that reason, positions a and d are mainly occupied by hydrophobic amino acids. Interhelical electrostatic interactions between positions e and g determine the specificity of folding as well as the helix orientation.8 The solvent-exposed positions b, c, and f exert only minor influences on the stability of the coiled coil structure and are occupied primarily by hydrophilic residues.In previous studies, we reported the de novo design of a coiled coil peptide model system for electrostatic binding to charged membranes.9 Here, we apply the same design principles for the interaction with negatively charged Au nanoparticles containing a shell of mercaptoundecanoic acid (MUA) (Fig. 1a). We chose the coiled coil peptide here referred to as VW05, in which positions a and d are exclusively occupied by leucine and positions e and g contain glutamic acid and lysine, respectively (Fig. 1b). Furthermore, arginine, a highly basic amino acid, is present in position f yielding a solvent-exposed positively charged edge on the helical cylinder. The peptide was N-terminally labelled with anthranilic acid (Abz) to facilitate concentration determination and to enable visualization of the peptide after gel electrophoresis. An attractive electrostatic interaction with Au/MUA nanoparticles is only expected under conditions that provide for a net positive charge of peptide. In contrast, a net negative charge of the peptide should preclude this attractive interaction. Since the coiled coil structure provides a defined multivalent template, the positively charged peptide mediates an assembly of nanoparticles as shown for one dimension in Fig. 1a. Due to the size ratio between peptide helix and nanoparticle, this assembly has more than one dimension. In order to determine the importance of the coiled coil structure as a structural template, we also constructed a peptide VW05-ref that has the same amino acid composition as VW05 but a primary structure that does not follow a heptad repeat (Fig. 1b). In particular, leucine residues were placed such that the formation of a “knobs into holes” packing is not possible and, thus, the thermodynamic driving force for coiled coil formation is reduced. Therefore, this control peptide was designed to exist primarily as a random coil monomer in solution.
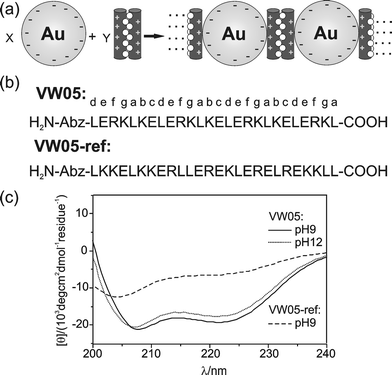 | ||
| Fig. 1 (a) General scheme of coiled coil peptide-induced assembly of Au/MUA nanoparticles, shown for one dimension. The length of the peptide helix and the size of nanoparticle can be estimated to be 3 nm and 6.2 nm, respectively. (b) Sequence of coiled coil peptide VW05, and non-coiled coil peptide VW05-ref. (c) CD spectra of 100 μM VW05 (pH 9 and 12) and VW05-ref (pH 9). | ||
Circular dichroism (CD) spectroscopy reveals that VW05 adopts an α-helical coiled coil structure at pH 9.0 and 12.0, as indicated by two minima at 208 and 222 nm (Fig. 1c).10 Between pH 9.0 and 12.0, the peptide's calculated net charge changes from +3.6 to −4.9, respectively (Fig. S2†). The CD spectrum of VW05-ref at pH 9.0 does not reveal a minimum at 200 nm which would be characteristic of a completely unfolded structure, but reveals an α-helical content of 21%, calculated from the molar ellipticity at 222 nm.10,11 The fact that there is a finite α-helical content may be a consequence of glutamic acid's and leucine's propensity for forming α-helices.12 Importantly, the oligomerization of peptide VW05-ref was ruled out by analytical ultracentrifugation measurements, whereas a trimeric complex was observed in the case of VW05 (Fig. S4†). A detailed discussion of ultracentrifugation measurements is given in the supporting information.
UV/Vis absorption spectroscopy was used to monitor the surface plasmon resonance band of Au/MUA nanoparticles. A distinct red-shift and broadening of the surface plasmon resonance maximum (λmax) are associated with a decrease in the interparticle distance for a given dielectric environment.13 In the following results, it turns out that several factors such as pH, peptide concentration (cp) and incubation time affect the value of λmax. At pH 9.0, an absorption maximum of Au/MUA nanoparticles at 522.0 nm is observed in the absence of peptide and for extended incubation times (Fig. 2a). Upon the addition of VW05, the λmax becomes red shifted (Fig. 2a). This spectroscopic change increases with cp and yields an initial λmax of 536.0 nm at 30 μM. With regard to the effect of incubation time, these values vary directly with one another; for example, at a cp of 10 μM, the λmax changes from 530.1 nm to 536.8 nm over a period of six hours. Fig. 2b shows the pH dependence of the absorption maximum at a fixed VW05 concentration of 10 μM and an incubation time of six hours. Interestingly, although the calculated pI value of VW05 is 10.4, a greater value of λmax of 524.7 nm compared to the λmax in the absence of peptide was determined at pH 10.5. This result implies that the net charge of the peptide is not the only factor that determines attraction between peptide and nanoparticle. The presentation of the solvent-exposed arginine surface upon coiled coil formation, considering a pKA value of arginine of 12.514, may still support electrostatic interactions at a pH of 10.5. Nevertheless, the addition of 10 μM VW05 at pH 12.0 results in the same value of λmax observed at pH 9.0 in the absence of peptide.
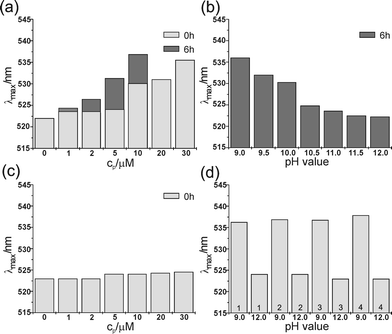 | ||
| Fig. 2 The position of the absorption maxima of 0.05 μM Au/MUA nanoparticles (a) in the presence of different concentrations of VW05 at pH 9.0 after 0 h and 6 h of incubation; at cp >10 μM, strong scattering effects hinder absorption measurements after 6 hours of incubation; (b) at 10 μM VW05 and various pH values; (c) at different concentrations of VW05-ref and pH 9.0; (d) four cycles of pH variation between 9.0 and 12.0 in the presence of 30 μM VW05. The sample was allowed to incubate for 5 min after each pH change. | ||
In contrast to the coiled coil peptide VW05, incubation of the nanoparticles with the control peptide VW05-ref does not lead to a noticeable change in the absorption maximum at pH 9.0. In this case, the slight increase in λmax may be due to a change in the ionic strength of the solution with increasing cp.
Gel electrophoresis was used to investigate whether an interaction between both peptides and the Au/MUA nanoparticles takes place at pH 9.0. Au/MUA nanoparticles showed a band of relatively high mobility that decreases with increasing cp of VW05 and is dramatically reduced at cp≥ 10 μM (Fig. 3a). To monitor the position of peptide next to the Au/MUA nanoparticles, the agarose gel was also visualized by fluorescence emission following excitation at 314 nm, corresponding to the absorption wavelength of the peptide's UV-label (Fig. 3b). Although, at cp of 50 μM, VW05 clearly shows a well-defined low-mobility band that corresponds to a net positive charge, no peptide can be visualized at the same cp in the presence of Au/MUA nanoparticles. We assume that the peptide is contained in the nanoparticle aggregates and that quenching effects caused by the absorption of nanoparticles interfere with the detection of peptide. An increase in cp up to 500 μM unveils an electrophoretic separation of the excess peptide from the nanoparticle–peptide aggregate. These results confirm that an interaction takes place between VW05 and the nanoparticle, one that cannot be observed under the same conditions for VW05-ref; in this case, electrophoretic separation of colloids and peptides occurs independently of cp (Fig. 3c–d). The greater electrophoretic mobility of VW05-ref compared to VW05 is a consequence of their different oligomerization states, monomeric in the case of VW05-ref and trimeric in the case of VW05.
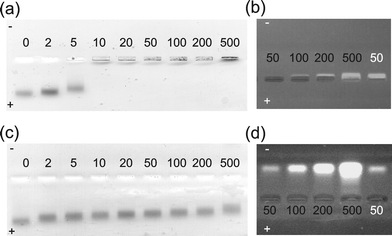 | ||
| Fig. 3 Agarose gels of VW05 (a, b) and VW05-ref (c, d) at pH 9.0, as visualized by (a, c) visible light and (b, d) fluorescence emission following excitation at 314 nm. The numbers refer to the peptide concentration in μM in the presence (black) and in the absence (white) of 0.05 μM Au/MUA nanoparticles. | ||
The peptide-induced assembly of Au/MUA nanoparticles was examined more precisely by transmission electron microscopy (TEM). The average diameter of the Au core of these nanoparticles is 5.3 ± 0.5 nm. At pH 9.0, mainly isolated Au nanoparticles are detected that are evenly spread if prepared on the surface of hydrophilized carbon covered grids (Fig. S5a†). Infrequently, regions of more densely packed particle monolayers are observed, and we attribute this to the sample blotting procedure and subsequent drying of the grids (see ESI†). At pH 12.0, the spatial distribution of Au/MUA nanoparticles is comparable to that described for pH 9.0. However, in the presence of VW05 at pH 9.0, the morphology is dominated by particle assemblies that vary in diameter from 100 nm to few μm (Fig. S5c–d).
Neither the addition of VW05 at pH 12.0 nor the addition of VW05-ref at pH 9.0 appears to induce this type of nanoparticle organization (Fig. S6 and S7). Here, the observed structures resemble the features of the pure nanoparticles, which is consistent with the results of the UV/Vis and gel electrophoretic studies.
In order to obtain direct information on the assemblies’ native solution state, we additionally employed cryo-TEM measurements. We expected to learn about the structural aspects of the peptide nanoparticle interaction as the peptide could not be visualized in the dried samples. Cryo-TEM measurements of pure Au/MUA at pH 9.0 confirmed the above observation that the particles are isolated in solution in the absence of the peptide. No objects other than pure gold nanoparticles were detectable. The stabilizing layer of MUA is not visible in the images. The number of nanoparticles per area (per volume actually) was significantly lower in the cryo-specimen (see overview in Fig. S5†) if compared to the dried samples. This can be understood if one keeps in mind that freezing aliquots of the sample for a direct microscopical observation retains the particle concentration in solution which otherwise is dramatically increased if the sample is dried on a surface leading to a more dense arrangement of particles. Therefore the aggregation effect of nanoparticles in the presence of VW05 becomes even more impressive. The overview in Fig. 4c shows a large assembly of particles spanning the complete micrometre hole of the perforated carbon film. At higher magnification the participation of the peptide is clearly visible (Fig. 4d). Ultrathin fibers connect the nanoparticles in a network-like manner and multiple linear connections between the particles are visible. The diameter of the fibers ranges from ultrathin single helices of about 1 nm diameter up to slightly larger bundles of 3 nm diameter, which corresponds to a monomeric and trimeric helical organization, respectively. The fiber length in the assembly network is difficult to determine, however, peptide fibers not involved in the assemblies show dimensions in the micrometre range. In contrast, such fibers cannot be observed in the absence of nanoparticles. Due to the low concentration of the peptide the unambiguous identification of its prefiber organization in cryo-TEM images is nearly impossible. However, fibril formation of 10 μM VW05, in the absence of nanoparticles, can be ruled out by ultracentrifugation measurements. We therefore assume that the high local peptide concentration on the surface area of the nanoparticles decreases the lag time for nucleation and assists the α-helical fibrillation of peptide. Such a potential of nanoparticles to induce protein fibrillation has recently been reported for amyloid forming peptides.15
 | ||
| Fig. 4 (a and b) Cryo-TEM micrographs of 0.05 μM Au/MUA nanoparticles at pH 9.0 and at different magnifications. The particles are mostly isolated and, as is obvious from (b), absent from any further visibly adherent material. Scale bars are 500 nm (a) and 50 nm (b), respectively. (c) Cryo-TEM image showing a large assembly (overview) of 0.05 μM Au/MUA nanoparticles in the presence of 10 μM of the peptide VW05 (scale bar is 200 nm). (d) The magnified detail from (c) indicates the multilayered organization of the VW05 mediated assembly as well as multiple fiber-like features (thought to correspond to α-helices) interconnecting the gold nanoparticles in a network-like manner (arrows). Scale bar is 25 nm. | ||
Conclusions
In summary, we have investigated the suitability of the α-helical coiled coil structure for the tunable structural organization of charged Au/MUA nanoparticles mediated by electrostatic interactions. Two peptides, VW05 and VW05-ref, were studied, that both contain the same composition of amino acids and, thus, possess the same net charge at a given pH. However, significant differences in their primary structure led to specific conformational properties: an α-helical coiled coil for VW05 due to its heptad repeat pattern, but mainly random coil for VW05-ref.We were able to demonstrate the superiority of the α-helical coiled coil folding motif not only as a defined structural template but also as a self-organizing system for the assembly of Au/MUA nanoparticles. Since the nanoparticle–peptide interaction is based on electrostatic forces, we further demonstrated that pH control can be applied as a trigger to direct the organization of functionalized nanoparticles. Remarkably, a cyclic change in the pH of solution can be used to reversibly direct the assembly of colloids and peptides, as shown by UV/Vis spectroscopy and TEM (Fig. 2d and Fig. S8). A key feature of the system described here is that organization of the nanoparticles into defined networks can be directed by the external factor of pH, and that the organization is reversible and repeatable.
We believe that this study contributes valuable insight into the de novo design of small peptides for the development of bionanostructures. Our results offer a novel route to nanoparticle–peptide hybrid systems whose assembly can be controlled in a switchable manner by pH conditions. The system described here is currently being further developed to yield nanoparticle networks that have defined geometries and can be applied to the production of useful hybrid materials with tunable optical properties.
Experimental
Au/MUA nanoparticle synthesis
Au nanoparticles were synthesized according to a procedure published by Stucky et al.16 0.435 g of tert-butylamine-borane complex was added to a solution of 0.247 g AuPPh3Cl (0.5 mmol, 1 eq) and 0.25 mL dodecanethiole (1.0 mmol, 2 eq) in 40 mL of benzene. The mixture was stirred at 55 °C for 1 hour before cooling to room temperature. Then 40 mL of ethanol were added and the precipitant separated by centrifugation. The black solid powder was washed with an excess of ethanol, dried under vacuum, and redissolved in 25 mL of chloroform. 44 mg of mercaptoundecanoic acid (0.2 mmol) were solved in a solution of 10 mL triple-distilled water and 200 μL of tetramethylammonium hydroxide (TMAH). 1 mL of a 3 μM solution of dodecanethiole-protected gold nanoparticles was added and the mixture was stirred at room temperature for 2 hours, which led to a complete phase transfer of the colloids from the organic into the water phase. The organic phase was removed and the aqueous phase concentrated to approximately 7 mL under reduced pressure. In order to remove the ligand's excess, the colloid solution was dialysed against 300 mL of triple-distilled water three times. The further transfer of the colloids into 10 mM Tris/HCl buffer (pH 9.0) or 10 mM phosphate/NaOH buffer (pH 12.0) was done by dialysis for three times. For all analytic studies, freshly prepared Au/MUA nanoparticles were used.Peptide synthesis and sample preparation
The peptides were synthesized on an automated Syro XP synthesizer (MultiSynTech GmbH, Witten, Germany) using a standard fluorenyl-methoxycarbonyl (Fmoc) chemistry protocol. The peptides were cleaved from the resin with 95% trifluoroacetic acid, purified by reversed HPLC, and identified from the MALDI-TOF spectra. For concentration determination, the peptides were N-terminally labelled with anthranilic acid (Abz). The peptide solutions were prepared in freshly filtered Tris/HCl buffer (10 mM, pH 9.0) or phosphate/NaOH buffer (10 mM, pH 12.0). The peptide concentration was determined by measuring the absorbance at λ = 320 nm. As a reference, a calibration curve using Abz-Glycine (Bachem GmbH, Weil am Rhein, Germany) in the appropriate buffer at different concentrations was recorded.CD spectroscopy
CD spectra were recorded on a J-810 spectrophotometer (Jasco GmbH, Gross-Umstadt, Germany) using a quartz cuvette of 1 mm path length at 20 °C. The spectra were taken as the average of 3 scans by collecting data from 240 to 200 nm at 0.5 nm intervals, a 2 nm bandwidth, and 1 s response time. All CD spectra were corrected by the buffer spectra and converted into mean residue ellipticity [θ].UV/Vis spectroscopy
UV/Vis spectra were collected on a CARY 50 Bio spectrophotometer (VARIAN, Darmstadt, Germany). The concentration of Au nanoparticles was chosen to provide an absorption value of ∼1.1 at 522.0 nm and was not changed upon the addition of different amounts of peptide. The experiments were carried out at room temperature; the spectra were taken from 400 to 800 nm in steps of 0.25 nm intervals. All spectra were corrected for the absorbance of buffer.Agarose gel electrophoresis
Gel electrophoresis was performed at room temperature with a Gel XL Ultra V-2 Electrophoresis System (Labnet International, Windsor, Great Britain) in a 1% agarose gel in Tris/HCl buffer (0.1 M, pH 9.0). The output voltage was held constant at 100 V. Electrophoresis was stopped when the bands began to broaden (after approximately 10–15 minutes).Transmission electron microscopy (TEM)
6 μL aliquots of the corresponding solution (0.05 μM Au/MUA nanoparticles in the absence or presence of 10 μM peptide) were placed for 60 s on glow-discharged (60 s plasma treatment at 8 W in a BALTEC MED 020) carbon-coated collodium support films covering 400-mesh copper grids (BAL-TEC, Lichtenstein). After blotting the supernatant fluid with a filter paper, the grids were left to air-dry. TEM images were recorded with a Philips CM12 transmission electron microscope (FEI company, Oregon, USA) at 100 kV accelerating voltage and at different calibrated primary magnifications (between 13 k and 58 k) on Kodak SO-163 negative film. Image J (version 1.38x, Wayne Rasband, USA) was used for the determination of the Au core of the nanoparticle by counting at least 300 particles. The standard deviation of the average diameter was calculated using the Origin Software (version 7.0, Microcal, USA).Cryo-TEM preparation and microscopy
Droplets of the sample (5 μL) were applied to perforated (1 μm hole diameter) carbon film covered 200 mesh grids (R1/4 batch of Quantifoil Micro Tools GmbH, Jena, Germany), which had been hydrophilized before use by 60 s plasma treatment at 8 W in a BALTEC MED 020 device. The supernatant fluid was removed with a filter paper until an ultrathin layer of the sample solution was obtained spanning the holes of the carbon film. The samples were immediately vitrified by propelling the grids into liquid ethane at its freezing point (90 K) by operating a guillotine-like plunging device.The vitrified samples were subsequently transferred under liquid nitrogen into a Tecnai F20 FEG transmission electron microscope (FEI Company, Oregon, USA) using the Gatan (Gatan Inc., California, USA) cryoholder and -stage (Model 626). Microscopy was carried out at 94 K sample temperature using the microscopes low dose protocol at a calibrated primary magnification of 62 k× and an accelerating voltage of 160 kV (FEG - illumination). Images were recorded using an EAGLE 2k-CCD device (FEI Company, Oregon, USA) at full 2048 by 2048 pixel size. The defocus was chosen in all cases to be 980 nm.
Ultracentrifugation studies
Average molecular weights were determined by sedimentation equilibrium with an Optima XL-I analytical ultracentrifuge (Beckman, Coulter, Palo Alto, CA) equipped with absorbance and Rayleigh interference optics at 25 °C. Peptide solutions were dialyzed against 10 mM Tris/HCl buffer (pH 9.0) for two days. Each peptide was spun at three different speeds (35,000; 40,000 and 45,000 rpm for VW05 and 40,000; 45,000 and 50,000 rpm for VW05-ref) at six concentrations in the range of 300 to 50 μM (VW05) and five concentrations in the range 400 to 75 μM (VW05-ref). Absorbance was monitored at 320 nm. Partial specific volumes were calculated from the known amino acid sequence to be 0.7756 ml g−1 for both peptides using the program SEDNTERP v. 1.06 (http://www.jphilo.mailway.com/download.htm).The equilibrium experiments have been evaluated with the model independent M* Program17 (Kristian Schilling, Nanolytics 2007) to determine the weight average molar mass for the different loading concentrations.
In addition, the sedimentation equilibrium concentration profiles have been fitted globally to a non-interacting ideal single species model over the entire investigated concentration range and three speeds (WINNONLIN v. 1.06.).18
Additionally the molar mass of the peptides was independently determined from their molar mass distribution, which was calculated from sedimentation velocity experiments using the program SEDFIT v. 10.09 beta.19 The molar mass average was calculated by integration of the distribution. The sedimentation velocity experiments were performed at 60,000 rpm and 25 °C using absorption optics at 320 nm and three different concentrations (300, 100 and 50 μM for VW05 and 400, 200 and 50 μM for VW05-ref).
Acknowledgements
The authors thank Dr Allison Berger and Dr Pamela Winchester for proof reading of the manuscript. We also thank Antje Völkel for the skilfully performed ultracentrifugation experiments. This work was supported by the Fonds der Chemischen Industrie (research fellowship of S. C. Wagner) and the Deutsche Forschungsgemeinschaft (SFB 765).References
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS.
- (a) S. I. Stoeva, J.-S. Lee, J. E. Smith, S. T. Rosen and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 8378–8379 CrossRef CAS; (b) R. Hong, G. Han, J. M. Fernandez, B.-J. Kim, N. S. Forbes and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 1078–1079 CrossRef CAS.
- (a) X. Mo, Y. An, C.-S. Yun and S. M. Yu, Angew. Chem., Int. Ed., 2006, 45, 2267–2270 CrossRef CAS; (b) A. Gole, C. Dash, C. Soman, S. R. Sainkair, M. Rao and M. Sastry, Bioconjugate Chem., 2001, 12, 684–690 CrossRef CAS; (c) P. Hazarika, F. Kukolka and C. M. Niemeyer, Angew. Chem., Int. Ed., 2006, 45, 6827–6830 CrossRef CAS; (d) D. Liu, A. Bruckbauer, C. Abell, S. Balasubramanian, D.-J. Kang, D. Klenerman and D. J. Zhou, J. Am. Chem. Soc., 2006, 128, 2067–2071 CrossRef CAS.
- A. F. Collings and F. Caruso, Rep. Prog. Phys., 1997, 60, 1397–1445 CrossRef CAS.
- A. M. Kalsin, M. Fialkowski, M. Paszewski, S. K. Smoukov, K. J. M. Bishop and B. A. Grzybowski, Science, 2006, 312, 420–424 CrossRef CAS; M. E. Leunissen, C. G. Christova, A.-P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. Van Roij and A. Van Blaaderen, Nature, 2005, 437, 235–240 CrossRef CAS.
- (a) M. M. Stevens, N. T. Flynn, C. Wang, D. A. Tirell and R. Langer, Adv. Mater., 2004, 16, 915–918 CrossRef CAS; (b) Y. Fillon, A. Verma, P. Gosh, D. Ernenwein, V. M. Rotello and J. Chmielewski, J. Am. Chem. Soc., 2007, 129, 6676–6677 CrossRef CAS; (c) M. Lundqvist, P. Nygren, B.-H. Jonsson and K. Broo, Angew. Chem., Int. Ed., 2006, 45, 8169–8173 CrossRef CAS; (d) D. Aili, K. Enander, J. Rydberg, I. Lundström, L. Baltzer and B. Liedberg, J. Am. Chem. Soc., 2006, 128, 2194–2195 CrossRef CAS.
- (a) P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS; (b) J. Liu, Y. Deng, C.-S. Cheng, N. R. Kallenbach and M. Liu, Proc. Natl. Acad. Sci. USA, 2006, 103, 15457–15462 CrossRef CAS.
- K. Pagel, K. Seeger, B. Seiwert, A. Villa, A. E. Mark, S. Berger and B. Koksch, Org. Biomol. Chem., 2005, 3, 1189–1194 RSC.
- T. Vagt, O. Zschöring, D. Huster and B. Koksch, ChemPhysChem, 2006, 7, 1361–1371 CrossRef CAS.
- The folding of both peptides VW05 and VW05-ref is not affected by the addition of Au/MUA nanoparticles as can be seen in Fig. S1 and S3.
- J. T. Yang and K. H. Chan, Biochemistry, 1974, 13, 3350–3359 CrossRef CAS.
- P. Y. Chou and G. D. Fasman, Biochemistry, 1974, 13, 222–245 CrossRef CAS.
- (a) K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS; (b) I. E. Sendroiu, S. F. L. Mertens and D. J. Schiffrin, PhysChemPhys, 2006, 8, 1430–1436 Search PubMed.
- R. M. C. Dawson, D. C. Elliot, W. H. Elliot and K. M. Jones, Data for Biochemical Research, 3rd edition, Oxford Science Publications, 1986, pp. 1–31 Search PubMed.
- S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. USA, 2007, 104, 8691–8696 CrossRef CAS.
- N. Zheng, J. Fan and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 6550–6551 CrossRef CAS.
- H. Cölfen and S. E. Harding, Eur. Biophys. J., 1997, 25, 333–346 CrossRef.
- M. L. Johnson, J. J. Correia, D. A. Yphantis and H. R. Halvorson, Biophys. J., 1981, 36, 575–588 CrossRef CAS.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures. See DOI: 10.1039/b813429d |
| This journal is © The Royal Society of Chemistry 2009 |