Hydrogen atom abstraction from C–H bonds of benzylamides by the aminoxyl radical BTNO: A kinetic study
Alessandra
Coniglio
,
Carlo
Galli
*,
Patrizia
Gentili
* and
Raffaella
Vadalà
Dipartimento di Chimica, Università‘La Sapienza’, and IMC-CNR Sezione Meccanismi di Reazione, P.le A. Moro 5, 00185, Roma, Italy. E-mail: carlo.galli@uniroma1.it; Fax: +39 06 490421
First published on 7th November 2008
Abstract
The aminoxyl radical BTNO (benzotriazole-N-oxyl; >N–O˙) is generated from HBT (1-hydroxybenzotriazole; >N–OH) by oxidation with a CeIV salt. BTNO presents a broad absorption band with λmax 474 nm that lends itself to investigate the kinetics of H-abstraction from H-donor substrates by spectrophotometry. Thus, rate constants (kH) of H-abstraction by BTNO from CH2-groups α to the nitrogen atom in X-substituted-(N-acetyl)benzylamines (X-C6H4CH2NHCOCH3) have been determined in MeCN solution at 25 °C. Correlation of the kHX data with the Hammett σ+ parameters gives a small value for ρ (−0.65) that is compatible with a radical H-abstraction step. The sizeable value (kH/kD = 8.8) of the kinetic isotope effect from a suitably deuteriated amide substrate further confirms H-abstraction as rate-determining. Evidence is acquired for the relevance of stereoelectronic effects that speed up the H-abstraction whenever the scissile C–H bond is co-linear with either the nitrogen lone-pair of the amide moiety or an adjacent aromatic group. An assessment of the dissociation energy value of the benzylic C–H bond in ArCH2NHCOMe is accordingly reported.
Introduction
The aminoxyl radicals (R2NO˙) are valuable and versatile reactive intermediates in numerous applications.1 In particular, the synthetic importance of aminoxyl radicals is highlighted by their use as catalysts in oxidation reactions endowed with low environmental impact.1–4 Among these radicals PINO (phthalimideN-oxyl), derived from HPI (N-hydroxyphthalimide), offers a prominent case.5PINO removes a hydrogen atom from a substrate (by hydrogen atom transfer, HAT), enabling subsequent interaction with O2 and therefore an oxidation outcome (Scheme 1).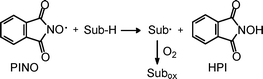 | ||
| Scheme 1 Oxidation of an H-donor substrate (Sub-H) by radical PINO in the presence of dioxygen. | ||
In principle, aminoxyl radicals can be obtained from the parent hydroxylamines by either H-abstraction or electron-abstraction followed by deprotonation (Scheme 2).1
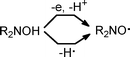 | ||
| Scheme 2 Possible pathways of generation of aminoxyl radicals. | ||
For example, PINO is generated from HPI by the action of O2 and Co(OAc)2,4,5 or else by oxidation with Pb(OAc)4;6PINO has alternatively been generated electrochemically.7 Kinetic studies of the H-abstraction reaction from substrates having C–H or O–H bonds of appropriate energy have provided information upon the reactivity of PINO towards alkylarenes or phenols.6a,8 The synthetic value of the oxidation reactions induced by PINO has stimulated us to assess the reactivity features of this, as well of other aminoxyl radicals more closely.
We have recently reported on the H-abstraction reactivity of another aminoxyl radical, that is, BTNO (benzotriazoleN-oxyl).9 This radical has been generated from oxidation of HBT (1-hydroxybenzotriazole) with a CeIV salt (Scheme 3) and characterized by UV-Vis and EPR spectra, laser flash photolysis and cyclic voltammetry.9a Rate constants of H-abstraction (kH) from C–H bonds of H-donor substrates, including benzyl alcohols and alkylarenes, have been determined spectroscopically and found to be in the 10−3 to 102M−1 s−1 range at 25 °C in MeCN solution.9b With selected substrates, activation parameters for the H-abstraction step have been measured, and the activation energy successfully correlated to the dissociation energy of the C–H bond undergoing cleavage.9b Whenever comparing BTNOvsPINO, the radical reactivity of PINO has always been found higher than that of BTNO towards similar substrates. The explanation provided is that PINO gives rise to a stronger NO–H bond (in HPI) upon H-abstraction from a substrate, as compared to the weaker NO–H bond in HBT (88 vs. ca. 85 kcal/mol, respectively), this representing a crucial factor for a radical process where bond cleavage and formation dominate the reactivity.1,9b
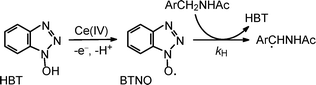
The present paper deepens our understanding of the reactivity of aminoxyl radicals in the H-abstraction reaction, a topic recently reviewed.1 It also extends this knowledge to the oxidation of amides, a class of substrates never explored kinetically so far. In fact, we report on a kinetic study of the H-abstraction by BTNO from the C–H bond α to the nitrogen atom of amides (Scheme 3), a reaction investigated for synthetic purposes by the use of PINO.10
During our kinetic study, evidence for the important contribution from stereoelectronic effects upon the H-abstraction reactivity has appeared. More specifically, the co-linearity between the α-C–H bond undergoing cleavage and the nitrogen lone-pair of the amide comes out as a key structural requirement.11 Likewise, for substrates similarly bearing a lone-pair, such as alcohols or ethers or amines, the H-abstraction by t-BuO. (or other radicals as well) had been described to be more rapid than H-abstraction from simple hydrocarbons as a result of the specific contribution from stereoelectronic effects.11,12 We report here on our attempt to give quantitative support to these issues. Activation parameters for H-abstraction from the α-C–H bond in two amide substrates have been determined, and enable to assess the contribution from stereoelectronic effects in decreasing the energy value of the scissile C–H bond.
Results and discussion
H-abstraction reactivity
Addition of a stoichiometric amount of the monoelectronic oxidant cerium(IV) ammonium nitrate (NH4)2Ce(NO3)6 (i.e., CAN; E° 1.3 V vs the normal hydrogen electrode, NHE)13 to HBT (E° 1.08 V vsNHE)14 in MeCN solution at 25 °C, both compounds being at 0.5 mM initial concentration, gives rise to a broad absorption band in the 350–600 nm range (Fig. 1)9 having λmax at 474 nm and ε 1840 M−1 cm−1.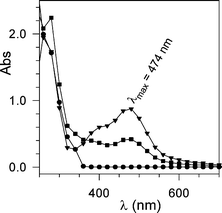 | ||
| Fig. 1 UV-Vis spectrum of a 0.5 mM solution of HBT in MeCN: (●) before the addition of CAN; after the addition of CAN (0.5 mM): (▼) 15 ms after the addition, (■) 110 s after the addition. | ||
The band pertains to the formation of the BTNO species: electron transfer from HBT to CAN is in fact exoergonic and occurs quantitatively (cf. Scheme 3).9 The spectrum of BTNO is not stable but decays according to an almost first-order exponential curve (kdecay = 5.1 × 10−3 s−1 in MeCN at 25 °C), with a half life of 140 s at 0.5 mM.9b The spontaneous decay of BTNO is strongly accelerated in the presence of purposely added H-donor substrates. By following the progress of the reaction at 474 nm through stopped-flow or conventional spectrophotometry, the reactivity of BTNO in the H-abstraction from the CH2-group in α to nitrogen is investigated here for a series of amides (ArCH2NHCOMe, cf. Schemes 3 and 4). Previous synthetic investigations had shown that the radical oxidation performed by either PINO or BTNO in the presence of O2 gives rise to carbonyl end-products (imide + aldehyde) from a common α-amido carbon radical intermediate originating from cleavage of the α-C–H bond of the amide substrate (Scheme 4).10,15
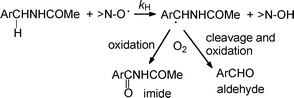 | ||
| Scheme 4 Pathways of products formation in the HAT route of oxidation of amides by an aminoxyl radical. | ||
For kinetic purposes, the solution of the C-H bearing substrate is employed here at initial concentrations higher than the concentration of BTNO, in order to fulfil the pseudo first-order kinetic conditions, and the drop of absorbance at 474 nm is followed. The pseudo-first-order rate constants k′, determined at 25 °C at three-to-four initial concentrations of amide from at least duplicated experiments, are converted into second-order H-abstraction rate constants (kH) by determining the slope of a k′vs. [SubH]° plot, as shown in Fig. 2 for the case of 4-chloro-N-benzylacetamide. Typical uncertainty of the kinetic data ranges from 3 to 6%.
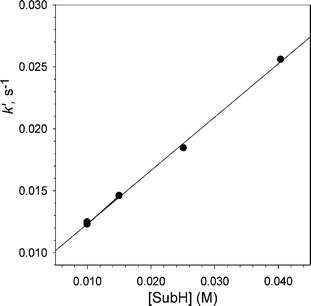 | ||
| Fig. 2 Determination of the second order rate constant kH for reaction of BTNO with 4-chloro-N-benzylacetamide in MeCN at 25 °C, from the plot of the pseudo-first-order rate constants k′ at various initial concentration of the substrate. | ||
The k′ constants are faster than the spontaneous decay of BTNO in all cases investigated, as the linearity of the plot and the minor y-intercept in Fig. 2 demonstrates, and exhibit first-order dependence on the excess substrate concentration. The second-order rate constants (kH) are given in Table 1 for the substrates of this study, which includes the α,α-bisdeuterio-N-benzylacetamide purposely synthesized (vide infra).
| Substrate | k H (M−1 s−1) |
|---|---|
| a Initial conditions: [BTNO] 0.5 mM, [SubH] 5–40 mM. Determinations in triplicate; typical errors 3–6%. | |
| PhCH2NHCOMe | 0.49 |
| PhCD2NHCOMe | 0.056 |
| 4-MeO-C6H4CH2NHCOMe | 2.54 |
| 4-Me-C6H4CH2NHCOMe | 1.17 |
| 4-Cl-C6H4CH2NHCOMe | 0.43 |
| 4-CF3-C6H4CH2NHCOMe | 0.26 |
| 4-NO2-C6H4CH2NHCOMe | 0.33 |
| PhCH(Me)NHCOMe | 0.11 |
|
|
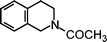
| N-Acetyl-tetrahydroisoquinoline | 26 |
Hammett correlation
The effect of the substituents upon reactivity is reckoned by correlating the kHX data for reaction of the 4-X-N-benzylacetamides with BTNO (in Table 1) vs the σ constants of the X-substituents according to the Hammett equation (log kX/kH = ρσ). A slightly better linearity is obtained in the plot (Fig. 3) when using the σ+ parameters, which take into account the resonance contribution from electron-donor substituents more effectively.16 The correlation parameter ρ, obtained from the slope of the plot, is −0.65. This is a small value, as expected for a radical process, and its negative sign besides the better fit to the σ+ parameters confirms the polar character of the aminoxyl radical.8b,9,10 The present ρ value compares very well with the one previously determined (i.e., −0.55 vsσ+) for reaction of BTNO with another series of benzylic substrates, that is, the 4-X-substituted benzyl alcohols (4-X-C6H4CH2OH).9a A common selectivity trend for H-abstraction by BTNO from benzylic C–H bonds in the two series of substrates emerges.1 This is likely to be due to the analogous stabilization offered by the X-substituents of the aromatic ring to an incipient benzylic radical in the transition state: electron-donor groups make the H-abstraction faster whereas electron-withdrawing ones slow it down.17 This is tantamount to saying that the C–H bond undergoing cleavage in the two series of substrates is made weaker or stronger by the X-substituents, causing a faster or slower H-abstraction, respectively. Earlier and strictly related studies of the reactivity of the radical PINO with benzylic substrates such as 4-X-C6H4CH2OH had provided ρ values of −0.68 in MeCN and of −0.41 in AcOH, once again giving better correlation with σ+.1,6a,8b The consistency of these ρ values stresses the uniform pattern of selectivity of the electrophilic >N–O˙ species (either PINO or BTNO) in the HAT reaction. Finally, the relative reactivity from competitive oxidations for a series of X-N-benzylacetamides in reaction with alkylperoxy-λ3-iodane, i.e., a radical HAT agent, had been reported in the literature,18 and a ρ value (i.e., −0.56) very close to our finding given. This supports the same extent of stabilization/destabilization offered by X-substituents to an incipient benzyl radical α to nitrogen in the rate-determining H-abstraction step.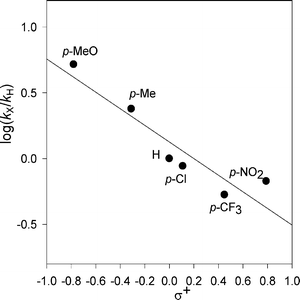 | ||
| Fig. 3 Hammett plot for the reaction of 4-X-substituted N-benzylacetamides with BTNO in MeCN at 25 °C. | ||
Kinetic isotope effect
The ratio of the rate constants for reaction of BTNO with PhCH2NHCOMe and PhCD2NHCOMe (in Table 1) gives kH/kD = 8.8. This sizeable value is consistent with a rate-determining H-abstraction step, and supports the contribution from tunnelling already commented on for reaction of either PINO or BTNO with various H-donors (kH/kD values in the 11–27 range).6a,8b,9Both the ρ and kH/kD data (−0.65 and 8.8) obtained here with the CAN/HBT oxidizing system compare favourably with the corresponding ones (i.e., −0.64 and 6.4, respectively) obtained in the oxidation of X-substituted benzyl alcohols by the copper enzyme laccase as the oxidant in the presence of HBT.19 The close agreement provides clear-cut support to the involvement of the very >N–O˙ radical BTNO as the reactive intermediate in that kind of chemo-enzymatic oxidation, in keeping with the necessary monoelectronic oxidation of mediator HBT by laccase as outlined in Scheme 5.14
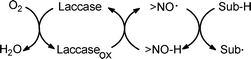 | ||
| Scheme 5 The aerobic chemo-enzymatic oxidation of an H-donor substrate (Sub-H) by laccase with a >NO-H mediator. | ||
Therefore, the present kinetic study corroborates our trust in the mechanism of the chemo-enzymatic oxidation by laccase and mediator compounds having the >NO-H functionality.14
Evaluation of the BDE(C-H) for the benzylic CH2-group in ArCH2NHCOMe
In the radical HAT route investigated here, the dissociation energy of the C–H bond undergoing cleavage, i.e.BDE(C-H), embodies a crucial reactivity feature.1 Accordingly, the weaker the C–H bond the faster is expected the HAT route, either with BTNO or other radical species. We had corroborated this expectation by correlating the energies of activation of the BTNO-induced HAT route with an homologous series of benzylic substrates (alkylarenes and benzyl alcohols) vs the corresponding BDE(C-H) data according to the Evans-Polanyi equation (Ea = α BDE(C-H) + const).9b Unfortunately, not always are thermochemical data available in order to attempt a similar correlation and the ensuing rationalization of the reactivity trend. This in part is the case of the present study because BDE(C-H) data for amide substrates are scarce.20What energy value could the benzylic C–H bond of PhCH2NHCOMe have? The kH rate constant determined here (Table 1) for reaction of BTNO with PhCH2NHCOMe is 0.49 M−1 s−1; the kH determined under the same conditions for reaction of BTNO with PhCH2OH is very close in value (i.e., 0.94 M−1 s−1).9b Because we extrapolated a BDE(C-H) of 79 kcal/mol for PhCH2OH,9b the corresponding BDE(C-H) of PhCH2NHCOMe ought to be pretty close in value. Strangely enough, the reactivity of BTNO with the benzylic C–H bond of the amine PhCH2NEt2, despite a reported BDE(C-H) of 89 kcal/mol,20 was too fast to measure (> 100 M−1 s−1) with our kinetic device.9b The BDE(C-H) of the latter compound is 10 kcal/mol larger than the extrapolated BDE(C–H) of PhCH2OH, the kH of which was slow enough to measure.9b Either our extrapolated BDE(C-H) of PhCH2OH is too low, or the literature value of BDE(C-H) for PhCH2NEt2 is too high!
In the attempt to settle this point, we have determined the activation parameters for reaction of BTNO with PhCH2NHCOMe. The rate constants, measured in the temperature range of 12.5 to 49.8 °C according to the experimental procedure described above, are given in Table 2. From use of the Arrhenius equation (lnkH = lnA − Ea/RT),16 an Ea of 4.1 ± 0.2 kcal/mol is obtained. An analogous determination yields an Ea value of 3.1 ± 0.3 kcal/mol for reaction of BTNO with 4-MeO-C6H4CH2NHCOMe (Table 2).
| PhCH2NHCOMe | 4-MeO-C6H4-CH2NHCOMe | ||
|---|---|---|---|
| k H (M−1 s−1) | T (°C) | k H (M−1 s−1) | T (°C) |
| 0.364 | 12.5 | 0.72 | 12.7 |
| 0.393 | 18.1 | — | — |
| 0.490 | 25.0 | 0.80 | 25.5 |
| 0.680 | 41.3 | 1.13 | 38.5 |
| 0.832 | 49.8 | — | — |
By entering the newly acquired Ea value (i.e., 4.1 kcal/mol) of PhCH2NHCOMe in the Evans-Polanyi plot already obtained for reaction of BTNO with a few benzylic substrates (Fig. 4),9b a BDE(C-H) of 71 kcal/mol can be extrapolated; analogously, for 4-MeO-C6H4CH2NHCOMe (Ea of 3.1 kcal/mol) a BDE(C–H) of 68 kcal/mol is extrapolated. On averaging these two numbers, one gets 70 (±2) kcal/mol for the dissociation energy of these particular C–H bonds: this certainly represents a small BDE value, particularly when compared to the archetypal benzylic BDE(C-H) of 88.5 kcal/mol of toluene.20 Such a conspicuous drop of C–H bond energy on going from PhCH3 to ArCH2NHCOMe, whatever the precision of our determinations is, is likely to be ascribed to the operation of stereoelectronic effects upon the amide substrate, as we comment on in the next section.
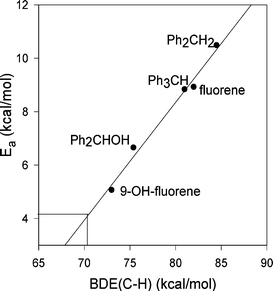 | ||
| Fig. 4 Evans-Polanyi plot for reaction of BTNO with benzylic substrates (from ref. 9b), and extrapolation of the BDE(C-H) of PhCH2NHCOMe from the Ea value obtained (see text). | ||
Actually, the issue could be even more complex than this, as a referee has correctly pointed out. First, we neglet the effect of the solvent when comparing kinetic data vs.BDE data. Hydrogen bonding between our solvent (MeCN) and substrates such as PhCH2NHCOMe or PhCH2OH can certainly affect the rate constants. However, the Evans-Polanyi plot of Fig. 4 shows good linearity for five substrates encompassing alkylarenes and benzylic alcohols, regardless their apolar or polar nature. Therefore, the hydrogen bonding issue provides only a partial explanation for the discrepancy between rate constants and BDE values stressed above. Secondly, we can not exclude that the high rate constant obtained9b for reaction of BTNO with PhCH2NEt2 is rather due to the incursion of a mechanism different from the HAT one. For example, the abstraction of electron (ET) by BTNO from the electron-rich PhCH2NEt2 could be alternatively envisioned. A similar ET route is documented for reaction of the aminoxyl radical PINO (0.92 V/NHE for the PINO/PINO- redox couple) with 4-X-substituted-N,N-dimethylanilines (redox potentials in the 0.5–1.1 V/NHE range), as reported recently.1,21 Benzyl amines are however more difficult to remove an electron from (redox potentials > 1 V/NHE)22 than anilines, and an ET route between BTNO and PhCH2NEt2 could be thought as less favoured. For sure, an ET route between BTNO and the present series of amides can be disregarded in view of redox potential considerations, as the one-electron oxidation of PhNHCOCH3 has a redox potential >1.8 V/NHE.23 Anyhow, we take the ET point as a sound suggestion, and work has already been planned in order to better investigate the boundaries of the ET/HAT dichotomy for reaction of aminoxyl radicals with benzylamines.
Stereoelectronic effects
Griller et al.11 have convincingly demonstrated that hyperconjugation from the lone-pair of a heteroatom weakens an adjacent C–H bond, and stabilizes an intervening C-radical generated by H-abstraction. The contribution from this weakening effect on the energy of an adjacent C–H bond is the greater the more co-linear are the C–H bond and the lone-pair,11 as shown in Fig. 5 for the case of an amine.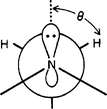 | ||
| Fig. 5 Stereoelectronic interaction between the lone-pair of an amine and a C–H bond. Reprinted with permission from Griller et al. (ref. 11). Copyright (1981) American Chemical Society. | ||
This interaction reportedly accounts for a 7–9 kcal/mol weakening of the C–H bond.12 For torsion angles (θ) wider than 30°, the effect drastically fades away.11 Our above finding of a substantial reduction of BDE(C-H) when passing from PhCH3 (88.5 kcal/mol) to PhCH2NHCOMe (71 kcal/mol) is likely due to the stereoelectronic interaction from the nitrogen lone-pair of the amide with the scissile C–H bond.
Additional data obtained from specific substrates enable us to further address the stereoelectronic issue. In a previous synthetic investigation,15 the oxidation of N-acetyl-tetrahydroisoquinoline by BTNO gave evidence for exclusive functionalization at C1, i.e., at the C–H bonds α to nitrogen, to produce an imide compound (cf. Scheme 4 and Fig. 6. Our present kinetic data (Table 1) point out that H-abstraction from N-acetyl-tetrahydroisoquinoline is indeed much faster (ca. 20–40 fold) than from the structurally comparable but acyclic C6H5CH2NHCOMe or 4-Me-C6H4CH2NHCOMe. Simple modelling with Hyperchem shows that in N-acetyl-tetrahydroisoquinoline the benzylic C–H bond of C1 is almost co-linear with both the p-orbitals of the aromatic ring and with the nitrogen lone pair (Fig. 6), being kept in this profitable conformation by steric restrictions caused by the heterocyclic system. This double beneficial effect is likely to weaken that C–H bond, as well as to stabilize the intermediate benzyl radical at C1 (Fig. 6). In the acyclic counterparts, instead, free rotation of the chain partially disrupts a similarly profitable conformation of the corresponding C–H bond, and lessens the extent of stabilisation of the intermediate radical ArCH(˙)NHCOMe.
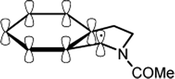 | ||
| Fig. 6 Stabilising interaction from both the π-system and the nitrogen lone-pair to the C1 radical resulting from H-abstraction in N-acetyl-tetrahydroisoquinoline by BTNO. | ||
As another example, the kH rate constants of PhCH(Me)NHCOMe and PhCH2NHCOMe with BTNO (Table 1), when normalized for the number of equivalent H-atoms, are 0.11 and 0.24 M−1 s−1 respectively (or 0.46:1 in relative terms), thereby indicating an unfavourable contribution from the α-Me-substitution upon reactivity. In principle, H-abstraction from the secondary benzylic C–H bond of PhCH(Me)NHCOMe ought to be easier and faster than for the primary C–H bond of PhCH2NHCOMe. This is consistent with the relative reactivity of H-abstraction from ArCH2CH3 and ArCH3 (normalised for the number of equivalent H-atoms)9b by BTNO, that is 4.2:1 (see also below), as well as with the BDE trend of a secondary vs primary benzylic C–H bond.20 The specific unfavourable effect from α-Me substitution in PhCH(Me)NHCOMe offsets the expected easier cleavage of its secondary C–H bond, and accounts for an overall depression of the H-abstraction rate by ca. 9 fold (i.e., 4.2/0.46). A plausible explanation for this unfavourable effect would be that PhCH(Me)NHCOMe is conformationally more stable (Fig. 7, left structure) whenever the larger groups in α, either Me- or–NHCOMe, are co-linear with the aromatic p-orbitals in order to lessen clash with the aromatic ortho C–H bonds.
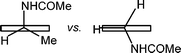 | ||
| Fig. 7 Limiting conformations of PhCH(Me)NHCOMe and PhCH2NHCOMe. The section of the aromatic ring is represented as a rectangle. | ||
Consequently, the benzylic C–H bond is preferentially directed in the ring plane, where any incipient α-amido carbon radical resulting from H-abstraction would not experience π-stabilisation. This is going to slow down the oxidation with respect to PhCH2NHCOMe (Fig. 7, right structure), where co-linearity of one α-C–H bond with respect to the plane of the aromatic ring is always feasible.
In order to support this conformational issue, the hydrogen abstraction rate constants by BTNO from a series of benzyl alcohols have been determined in MeCN at 25 °C, and are reported in Scheme 6.
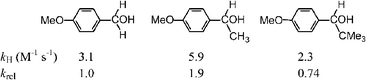 | ||
| Scheme 6 Absolute (normalised) and relative rate constants of H-abstraction by BTNO. | ||
Once again, whereas abstraction of a secondary H-atom is 1.9 fold faster than that of a primary one, in keeping with the corresponding BDE values,20 co-linearity of the t-Bu-group with the aromatic p-orbitals in the tertiary alcohol is preferred because lessens steric hindrance (cf. Fig. 7, left structure), thereby pushing the scissile C–H bond in the ring-plane where no π-stabilization is experienced, and resulting in a depression of reactivity (krel = 0.74).
Conclusions
The reciprocal orientation between a C–H bond undergoing cleavage and the lone pair of an adjacent heteroatom, or else an adjacent aromatic ring, emerges as an important structural feature upon the reactivity of an homolytic C–H cleavage, and is responsible for substantial weakening of the C–H bond energy. We have attempted to highlight these structural features upon the HAT reactivity of the aminoxyl radical BTNO with a few examples of amide substrates.Experimental
Reagents
HBT (Aldrich) was used as received; most of the substrates were obtained commercially and used without further purification. A few starting amides were synthesized from the parent amines by conventional acetylation with Ac2O in pyridine,24 and characterised by NMR and GC-MS.15Cerium(IV) ammonium nitrate (CAN; Erba RPE) was oven-dried before use. Reagent grade acetonitrile (Erba RPE) was used as the solvent. N-Acetyl-α,α-bisdeuteriated benzylamine was obtained from LiAlD4reduction of PhCONH2 to PhCD2NH2 in a 1:2 THF:diethyl ether mixed solvent, followed by acetylation.24 The purity was checked by GC-MS.Instrumentation
The kinetic study was carried out with a Hi-Tech SFA-12 stopped flow instruments, interfaced to a HP 8453 diode array spectrophotometer, and having a thermostated cuvette holder. A conventional UV-Vis spectrophotometer (Perkin Elmer Lambda 18) was alternatively used. A thermocouple was employed for reading the temperature in the cuvette.Kinetic Procedure
The BTNO species was generated in MeCN in a thermostated quartz cuvette, by adding a 0.5 mM solution of CAN to a 0.5 mM solution of HBT. A broad absorption band developed almost immediately (15 ms) in the 350–600 nm region (λmax at 474 nm, ε 1840 M−1 cm−1).9 The A474 reading was not stable, but decayed with a half-life of ca. 140 s. The rate of decay was unaffected by the use of deareated MeCN. Rate constants of H-abstraction from H-donor substrates were determined at 25 °C in MeCN (Table 1), by following the decrease of the A474 band of BTNO. The initial concentration of the substrate was in the 0.5− to −4 × 10−2 M range, so to enable a pseudo-first-order treatment of the kinetic data. Plots of (At-A∞) vs time were well fitted by a first-order exponential over more than three half-lives and pseudo-first-order rate constant (k′) reckoned. From a plot of three-to five k′vs [subst]° data pairs, the second-order rate constant of H-abstraction (kH) was obtained. Determination of H-abstraction rate constants was analogously carried out at different temperatures (in the 13–50 °C range) for PhCH2NHCOMe and 4-MeO-C6H4CH2NHCOMe (Table 2), for the calculation of the activation parameters from the Arrhenius plot.Semiempirical calculations
The structure of a few substrates has been optimized by a semi-empirical method (PM3 level) on using the Hyperchem program.Acknowledgements
This work was supported by grants from the Italian MIUR (COFIN and FIRB projects) and from the University of Roma ‘La Sapienza’.References
- C. Galli, P. Gentili and O. Lanzalunga, Angew. Chem. Int. Ed., 2008, 47, 4790–4796 CrossRef CAS.
- I. W. C. E. Arends, and R. A. Sheldon, Modern Oxidations of Alcohols using Environmentally Benign Oxidants, Chapter 4, p. 83 in Modern Oxidation Methods, J.-E. Bäckvall, Ed., Wiley-VCH, Weinheim, 2004 Search PubMed.
- W. Adam, C. R. Saha-Möller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499–3538 CrossRef CAS.
- F. Minisci, C. Punta and F. Recupero, J. Mol. Catal. A:Chem., 2006, 251, 129–149 CrossRef CAS.
- Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef CAS.
- (a) K. Nobuyoshi, B. Saha and J. H. Espenson, J. Org. Chem., 2003, 68, 9364–9370 CrossRef CAS; (b) P. J. Figiel and J. M. Sobczak, New J. Chem., 2007, 31, 1668–1673 RSC.
- C. Ueda, M. Noyama, H. Ohmori and M. Masui, Chem. Pharm. Bull., 1987, 35, 1372–1377 CAS.
- (a) N. Koshino, Y. Cai and J. H. Espenson, J. Phys. Chem. A, 2003, 107, 4262–4267 CrossRef CAS; (b) C. Annunziatini, M. F. Gerini, O. Lanzalunga and M. Lucarini, J. Org. Chem., 2004, 69, 3431–3438 CrossRef CAS; (c) R. Amorati, M. Lucarini, V. Mugnaini, G. F. Pedulli, F. Minisci, F. Recupero, F. Fontana, P. Astolfi and L. Greci, J. Org. Chem., 2003, 68, 1747–1754 CrossRef CAS; (d) E. Baciocchi, O. Lanzalunga and M. F. Gerini, J. Org. Chem., 2004, 69, 8963–8966 CrossRef CAS.
- (a) C. Galli, P. Gentili, O. Lanzalunga, M. Lucarini and G. F. Pedulli, Chem. Commun., 2004, 2356–2357 RSC; (b) P. Brandi, C. Galli and P. Gentili, J. Org. Chem., 2005, 70, 9521–9528 CrossRef CAS.
- F. Minisci, C. Punta, F. Recupero, F. Fontana and G. F. Pedulli, J. Org. Chem., 2002, 67, 2671–2676 CrossRef CAS.
- D. Griller, J. A. Howard, P. R. Marriott and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 619–623 CrossRef CAS.
- V. Malatesta and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 609–614 CrossRef CAS.
- G. Prabhakar Rao and A. R. Vasudeva Murthy, J. Phys. Chem., 1964, 68, 1573–1576 CrossRef CAS.
- P. Astolfi, P. Brandi, C. Galli, P. Gentili, M. F. Gerini, L. Greci and O. Lanzalunga, New J. Chem., 2005, 29, 1308–1317 RSC.
- A. Coniglio, C. Galli, P. Gentili and R. Vadalà, J. Mol. Catal. B:Enzym., 2008, 50, 40–49 CrossRef CAS.
- T. H. Lowry, and K. S. Richardson, Mechanism and Theory in Organic Chemistry, 2nd ed., Harper & Row, New York, 1981 Search PubMed.
- For recent and detailed considerations on substituent effects on radical stabilization energy, see: D. A. Pratt, G. A. DiLabio, P. Mulder and K. U. Ingold, Acc. Chem. Res., 2004, 37, 334–340 Search PubMed.
- T. Sueda, D. Kajishima and S. Goto, J. Org. Chem., 2003, 68, 3307–3310 CrossRef CAS.
- P. Baiocco, A. M. Barreca, M. Fabbrini, C. Galli and P. Gentili, Org. Biomol Chem., 2003, 1, 191–197 RSC.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, Florida, USA, 2007 Search PubMed.
- E. Baciocchi, M. Bietti, M. F. Gerini and O. Lanzalunga, J. Org. Chem., 2005, 70, 5144–5149 CrossRef CAS.
- A. Cuppoletti, C. Dagostin, C. Florea, C. Galli, P. Gentili, O. Lanzalunga, A. Petride and H. Petride, Chem. Eur. J., 1999, 5, 2993–2999 CrossRef CAS.
- M. Aweke Tadesse, A. D'Annibale, C. Galli, P. Gentili and F. Sergi, Org. Biomol. Chem., 2008, 6, 868–878 RSC.
- Vogel's Textbook of Practical Organic Chemistry, IV edn, Longman, London, chapter VII, 1978 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |