Spectroscopic analysis of the pyrimidine(6–4)pyrimidone photoproduct: insights into the (6–4) photolyase reaction†
Junpei
Yamamoto
a,
Yoshiyuki
Tanaka
b and
Shigenori
Iwai
*a
aDivision of Chemistry, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: iwai@chem.es.osaka-u.ac.jp; Fax: +81 6 6850 6240; Tel: +81 6 6850 6250
bGraduate School of Pharmaceutical Sciences, Tohoku University, Aobayama, Aoba-ku, Sendai, Miyagi 980-8578, Japan
First published on 10th November 2008
Abstract
We synthesized a dinucleoside monophosphate of the 15N-labeled (6–4) photoproduct, which is one of the major UV-induced lesions in DNA, to investigate the (6–4) photolyase repair mechanism, and characterized its protonation state by measuring 15N NMR spectra as a function of pH. We expected that chemical-shift changes of the pyrimidone15N3, due to protonation, would be observed at pH 3, as observed for the 15N-labeled 5-methylpyrimidin-2-one nucleoside. Interestingly, however, the changes were observed only in alkaline solutions. In UV absorption spectroscopy and HPLC analyses under acidic conditions, a change in the maximum absorption wavelength, due to the protonation-induced hydrolysis, was observed at and below pH 1, but not at pH 2, whereas the protonation of 5-methylpyrimidin-2-one occurred at pH values between 2 and 3. These results indicated that the pKa value for this N3 is remarkably lower than that of a normal pyrimidone ring, and strongly suggest that an intramolecular hydrogen bond is formed between the N3 of the 3′ base and the 5-OH of the 5′ base under physiological conditions. The results of this study have implications not only for the recognition and reaction mechanisms of (6–4) photolyase, but also for the chemical nature of the (6–4) photoproduct.
Introduction
DNA in organisms is subjected to photochemical reactions between adjacent pyrimidine bases by exposure to the ultraviolet (UV) component in sunlight, leading to the formation of cis–syn cyclobutane pyrimidine dimers (CPDs) and pyrimidine(6–4)pyrimidone photoproducts ((6–4) photoproducts).1 These photoproducts prevent DNA replication by replicative polymerases, and induce mutations in DNA that can lead to carcinogenesis and cell death. In particular, the (6–4) photoproduct, formed at the TT site, causes a T→C transition mutation at the 3′ pyrimidone with a high frequency.2To maintain genetic integrity, living organisms have enzymes that can restore lesions to their intact form by excision repair,3translesion synthesis,4 and photoreactivation.5 Among them, photoreactivation is the simplest repair, in which a single enzyme, photolyase, uses blue to near-UV light (300–500 nm) to break the dimeric lesion. Although the CPD photolyase repair mechanism is well-established,5 (6–4) photolyase, which is responsible for (6–4) photoproduct repair, has been less studied. The (6–4) photolyase reportedly binds oligonucleotides containing the (6–4) photoproduct with high affinity. Kd values of ∼5 × 10−10 and 2 × 10−8 M have been reported for the Drosophila and Xenopus enzymes, respectively.6 Upon binding, (6–4) photolyase flips the (6–4) photoproduct out of the helix at a dinucleotide level to bring the lesion closer to the flavin adenine dinucleotide (FAD) at the catalytic site,7 as CPD photolyase does.8 The following (6–4) photolyase repair pathway consists of two distinct processes, the intramolecular rearrangement of the substrate to form the oxetane/azetidine intermediate and the light-driven electron transfer,9 while CPD photolyase restores CPD by a simple electron donation. The final step of the (6–4) photolyase reaction, namely the electron transfer-induced splitting of the oxetane intermediate, has been studied extensively by using various synthetic compounds10 and by quantum chemical calculations,11 although the latter method has been used predominantly for the study of the CPD photolyase.12
In contrast, the details of the oxetane intermediate formation are still poorly understood. This process is essential for the electron transfer-dependent repair of the (6–4) photoproduct, because the formation of the (6–4) photoproduct involves the migration of the functional group from the C4 position of the 3′ base to the C5 of the 5′ base.5 It was proposed that two conserved histidine residues work as acid and base catalysts,13 and an electron-nuclear double resonance (ENDOR) analysis of this enzyme suggested that one of these histidines that would act as an acid was protonated at pH 9.5.14 This protonated histidine initiates the formation of the four-membered ring intermediate by protonation of the N3 of the 3′ pyrimidone of the (6–4) photoproduct to form an iminium ion, followed by the intramolecular nucleophilic attack of the activated C5 functional group on the protonated pyrimidone ring (Fig. 1a).14 This proposed mechanism depends on the ability of the 3′ base to be protonated; in other words, it depends on the pKa for the conjugate acid of the N3 of the 3′ pyrimidone ring of the (6–4) photoproduct. However, no experimental data on the acidity/basicity of this protonation site have been reported until now, even though it is a fundamental parameter that restricts the enzyme reaction.
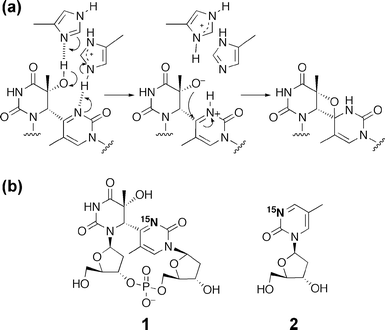 | ||
| Fig. 1 (a) Proposed mechanism for the first step of the (6–4) photolyase reaction. (b) The compounds used in this study, the dinucleoside monophosphate containing the 15N-labeled (6–4) photoproduct (1) and the 15N-labeled 5-methylpyrimidin-2-one 2′-deoxyribonucleoside (2). | ||
To gain new insights into the chemical nature of the (6–4) photoproduct and the repair mechanisms, we prepared a (6–4) photoproduct with the N3 of the pyrimidone ring 15N-labeled (1), and an analog of its 3′ component, 5-methylpyrimidin-2-one 2′-deoxyribonucleoside (2), for comparison (Fig. 1b). Using these compounds, we measured 15N NMR, UV absorption, and fluorescence spectra as a function of pH, to analyze their protonation states. We found that the 15N chemical shift and the fluorescence intensity of 1 changed at pH values higher than 10, and that the protonation-induced hydrolysis of 1 occurred under strongly acidic conditions. Based on these unexpected results, we discuss the chemical properties of the (6–4) photoproduct. This study not only has implications for the recognition and reaction mechanisms of (6–4) photolyase, but also will contribute to the development of molecules that recognize this lesion.
Results and discussion
Synthesis and characterization of the compounds
We synthesized [3-15N]-labeled thymidine according to the reported method,15 and then derived the 15N-labeled (6–4) photoproduct (6), following the procedure described previously (Scheme S1†),16 on a scale sufficient for its incorporation into oligonucleotides. Subsequently, an aliquot of the protected dinucleoside monophosphate containing the (6–4) photoproduct (6) was treated with ammonia water, and the deprotected product (1) was characterized by 1H NMR. Although most of the proton resonances were equivalent to those of the non-labeled dinucleoside monophosphate,17 the resonance of the H6 of the 5′ component was observed as a doublet signal with a coupling constant of about 3.7 Hz, at a chemical shift identical to that of the corresponding non-labeled sample. From a 1H{15N} NMR experiment, this coupling was determined to be a three-bond 1H-15N J-coupling through the (6–4) bond (3JNH: 15N3-C4-C6-H6, Fig. S1†). In the measurement of the 15N insensitive nuclei enhanced by polarization transfer (INEPT) utilizing the 3JNH, the 15N signal was observed at 270 ppm, which was low-field shifted by about 150 ppm from the thymine15N signal, due to the formation of the pyrimidone ring.To compare the spectroscopic behavior of the (6–4) photoproduct, we also synthesized 15N-labeled 5-methylpyrimidin-2-one 2′-deoxyribonucleoside (2) as an analog of the 3′ component of the (6–4) photoproduct. A possible problem in using this compound for comparison was that 1 contained a phosphodiester linkage, which had a negative charge. However, its effect was estimated to be small because the difference in the pKa values of thymidine and thymidine 5′-phosphate is only 0.2,18 and this phosphate does not dissociate at low pH. Compound 2 was obtained from [3-15N]-labeled 3′,5′-O-diacetylthymidine, a derivative of the [3-15N]-labeled thymidine, by a modification of the reported procedure19 (Scheme S2†). After purification, 2 was characterized by 1H and 15N NMR. In a manner similar to that for 1, the H4 of 2 was coupled with 15N3 (2JNH∼ 11 Hz) at the identical chemical shift.20 In the 15N INEPT measurement utilizing 2JNH, the 15N signal of 2 was observed at ∼ 267 ppm.
Analysis of the (6–4) photoproduct by 15N NMR and fluorescence spectroscopy
We examined the pH-dependency of the 15N chemical shift of 1, which would provide information on the equilibrium between the neutral and protonated states of the 3′ pyrimidone ring. For the 15N INEPT measurement, 1 was dissolved at a concentration of 2 mM in 20 mM phosphate buffer, consisting of 90% H2O and 10% D2O at various pH values. The lower-field shift of the 15N resonance was observed at pH values higher than 10, although the chemical shift did not change significantly in the acidic and neutral solutions (Fig. 2a). In the same way, the fluorescence emission derived from the pyrimidone ring of 1 was drastically decreased, and was slightly blueshifted in alkaline solutions (Fig. 2b). To confirm the 15N NMR spectroscopic behavior of the N3 of pyrimidone, we performed a 15N NMR measurement using 2. In the case of 2, no shift of the 15N resonance was observed in the alkaline and neutral solutions, but the signal was shifted upfield by about 7 ppm at pH 3 (Fig. S2†). We tried measurements in more acidic solutions, but the 15N signal was broadened out at pH 2. The fluorescence emission of 2 excited at 313 nm was measured, but no significant spectral change was observed in the alkaline and neutral solutions (data not shown).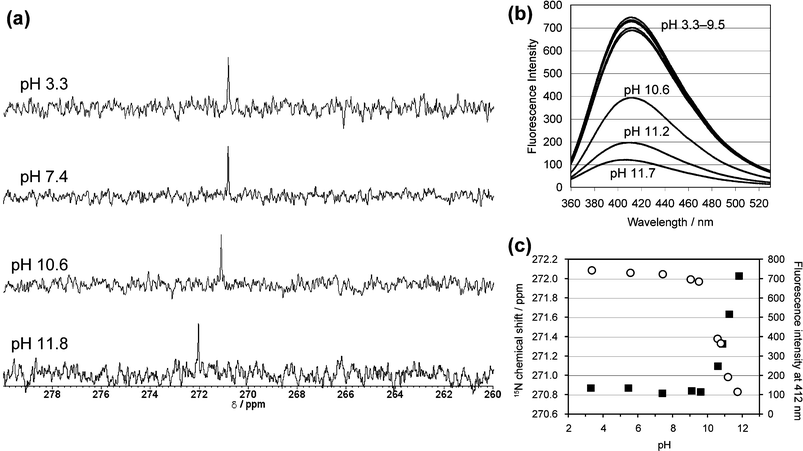 | ||
| Fig. 2 (a) Selected 15N NMR spectra of 2 mM dinucleoside monophosphate containing the 15N-labeled (6–4) photoproduct (1), measured at 303 K in 20 mM phosphate buffers composed of 90% H2O and 10% D2O. (b) Fluorescence emission spectra of 0.1 mM 1 in 20 mM phosphate buffers at various pH values. The excitation wavelength was set to 313 nm, and the spectra were measured at 298 K. At each pH, three independently measured spectra were averaged. (c) pH profiles of the 15N chemical shift (black squares) and the fluorescence intensity (open circles) obtained for 1. | ||
Since the changes in the alkaline solutions were observed only for 1, they probably occurred with the involvement of the 5′ component of the (6–4) photoproduct. The pKa value for the N3 imino function in the 5′ component of 1 is presumed to be similar to that reported for 5,6-dihydrothymidine (pKa = 11.6),21 and thus the change at higher pH can be attributed to the dissociation at the N3 position of the 5′ component. Furthermore, we converted 1 to its Dewar valence isomer by near-UV irradiation, as described previously,7 and analyzed the pH-dependency of the 15N chemical shift. The 15N resonance of the 15N-labeled Dewar valence isomer was also shifted to lower field in the alkaline solutions (Fig. S3†), supporting the idea that the 5′ component, which is common to the (6–4) photoproduct and its Dewar isomer, is important for the change observed at higher pH.
At pH 3, the 15N resonance of 2 was shifted upfield by about 7 ppm (Fig. S2†). The pKa value for the N3 of pyrimidin-2-one 2′-deoxyribonucleoside, which has almost the same structure as 2, was reportedly 3.13,22 suggesting that the shift of the 15N resonance represents the protonation of the N3 of 2. Moreover, the 15N resonance is reportedly shifted upfield upon protonation,23 and the direction of the shift observed in our present work is consistent with this report.
Analysis of the (6–4) photoproduct in acidic solutions by UV absorption spectroscopy
Since the 15N signal of 2 disappeared at pH 2, probably due to the hydrolysis during the time scale of the NMR measurement,24 we performed further analysis by UV absorption spectroscopy to characterize the (6–4) photoproduct in acidic solutions. The maximum absorption wavelengths of these compounds were plotted against the pH (Fig. 3). Compound 2 exhibited an absorption maximum at 314.5 nm at a neutral pH, but the absorption spectra were redshifted at pH values between 2 and 3 (Fig. 3b). On the other hand, the absorption spectra of the (6–4) photoproduct (1) showed a slight blueshift at pH values lower than 2 and higher than 10 (Fig. 3a). The change at alkaline pH observed for 1 corresponded with the results of the 15N NMR and fluorescence experiments, and the shift in acidic solutions occurred at pH values significantly lower than those in the case of 2.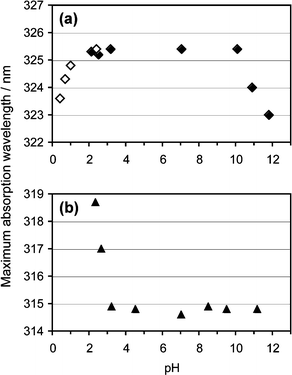 | ||
| Fig. 3 pH profiles of the maximum absorption wavelengths of 1 (a) and 2 (b). Four independently measured spectra of 0.1 mM 1, in 20 mM phosphate buffers at various pH values, were averaged, and then the maximum absorption wavelengths were obtained. The data showing the hydrolysis (open diamonds) were obtained at 5 min after the addition of hydrochloric acid. | ||
Since glycosidic bond cleavage in 2 reportedly occurs under acidic conditions,24 it is important to determine whether the spectral change at low pH was caused by this type of degradation or by protonation of the base moiety with the intact glycosidic bond. For this purpose, 1 and 2 were analyzed by HPLC after acid treatment (Fig. 4). Compounds without the 15N labeling were used in this experiment. The pH range was chosen from the results shown in Fig. 3, and the actual pH values of the solutions of 1 and 2 were 0.55 and 2.36, respectively. When 1 was treated with hydrochloric acid, a new peak emerged at a longer retention time (Rt 9.0 min, peak ii) than that of the intact material (Rt 8.5 min, peak i) after the incubation for 30 min at room temperature, and 1 was mostly converted to this product after 60 min (Fig. 4a). A comparison of the absorption spectra of these detected peaks revealed that the product obtained from 1 had an absorption maximum shorter than that of the intact material by about 10 nm (Fig. 4c), and the magnitude of the change depended on the reaction time (data not shown). In the case of 2, a new peak emerged at a short retention time (Rt 2.7 min, peak iv), and this product had an absorption maximum at 311 nm, blueshifted by about 4 nm from that of 2 (Fig. 4b and d). It should be noted that the spectral change observed for 2 in the experiment shown in Fig. 3b was a redshift. The products obtained from 1 and 2 (peaks ii and iv, respectively) were characterized by mass spectrometry. The product from 2 yielded a molecular ion peak at m/z 110.1, which indicated the hydrolysis of the glycosidic bond.24 In the case of 1, the m/z values obtained for [M – H]− (563.1) and the fragment ions (545.2, 447.1) revealed that the glycosidic bond of the 3′ base was hydrolyzed although the base and sugar moieties were linked at the 5′ component. From these results, we concluded that the blueshift observed for 1 at low pH (Fig. 3a) was caused by the glycosidic bond cleavage immediately after protonation of the base moiety, whereas the redshift for 2 could be attributed to simple protonation. The acid hydrolysis of the glycosidic bond in the 3′ component of 1 is caused by the protonation of the N3 that significantly decreases the electron density of the base moiety. Accordingly, the pH profile of the UV absorption maximum of 1 (Fig. 3a) revealed that the pKa for its N3 is much lower than 2, and smaller by at least 1 pKa unit than that of 2. The substitution at the C4 position of 2 may have affected the basicity of the N3. However, an alkyl group at this position, like the dihydrothymin-6-yl group in 1, would increase the pKa of the conjugate acid, whereas the pKa value obtained for the N3 of 1 was much lower than expected. Therefore, the effect of the C4 substitution is marginal.
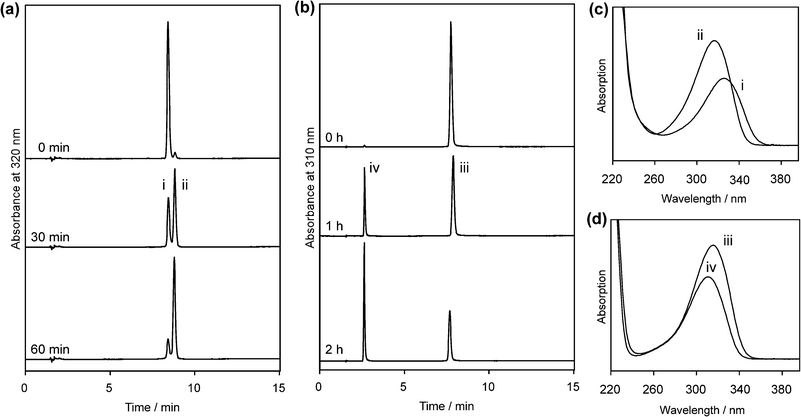 | ||
| Fig. 4 HPLC analysis of the acid hydrolysis of 1 and 2 (a and b, respectively) and absorption spectra of the detected peaks (c and d). The pH values of the solutions of 1 and 2 were 0.55 and 2.36, respectively. The absorption spectra were extracted from the 30 min (a) and 1 h (b) data sets with the data processing software for the photodiode array detector. | ||
Since the difference between compounds 1 and 2 is the presence of the 5′ pyrimidine base in the (6–4) photoproduct, a plausible explanation for the different pKa values between these two compounds is the formation of an intramolecular hydrogen bond between the hydroxyl group at the C5 position of the 5′ component and the N3 atom (15N in this study) of the 3′ pyrimidone ring within the (6–4) photoproduct (Fig. 5). This hydrogen bond can block the protonation of the N3 of the 3′ base, and will result in a low pKa value at this position. Although detection of the J-coupling between 15N and the 5-OH was not successful, the 5-OH signal of the protected dinucleoside monophosphate containing the (6–4) photoproduct (compound 3 in ref. 16) was observed at 3.59 ppm in CDCl3, whereas the chemical shift of the 5-OH of protected 5R-5,6-dihydro-5-hydroxythymidine, which lacks the 3′ pyrimidone ring, is reportedly 3.20 ppm in the same solvent.25 Since the signal of the hydrogen-bonding proton is observed at lower field,26 this result is consistent with the hydrogen-bond formation within the (6–4) photoproduct. On the other hand, the dissociation at the N3 position of the 5′ component in alkaline solutions increases the electron density at the O4 of the 5′ component by delocalization, to form an enolate (Fig. 5). In this case, a structural alteration occurs, as shown in Fig. 5, which can cause the spectral change because the hydrogen bond to the N3 is lost. We constructed a model structure by the density functional theory (DFT) calculation on Gaussian03W,27 and the results showed that the hydroxyl proton can be located at a distance of 1.84 Å from the N3 of the pyrimidone ring (Fig. S4†), indicating the feasibility of intramolecular hydrogen bond formation.
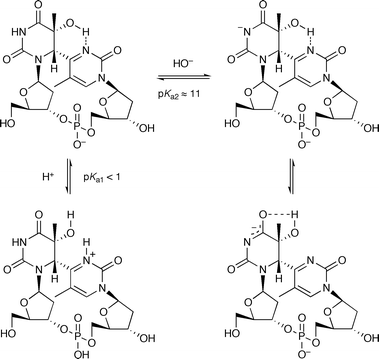 | ||
| Fig. 5 The protonation state of the (6–4) photoproduct and the hydrogen bond proposed in this study. | ||
The remarkably low pKa for the N3 of the 3′ pyrimidone ring of the (6–4) photoproduct has implications for the repair process of the (6–4) photolyase. Schleicher et al. suggested that one of the conserved histidines, which would play a role for the protonation of the (6–4) photoproduct, was protonated even at pH 9.5.14 Since the pKa value of the imidazole side chain of histidine is ordinarily between 6 and 7, this histidine residue is probably protonated by a nearby acidic residue, aspartic or glutamic acid, interacting with the histidine.28 When proton transfer occurs via the protonated histidine, it depends on the pKa value of the carboxyl group, which is between 3 and 4. However, our study suggested that it is not acidic enough to protonate the (6–4) photoproduct. For the protonation of the 3′ pyrimidone ring of the (6–4) photoproduct to initiate the repair process, it is necessary to cleave the intramolecular hydrogen bond discussed in this study. The other conserved histidine, acting as a base in the repair mechanism shown in Fig. 1a, must interact with the hydroxyl group at the C5 position, thus altering the orientation of the hydroxyl group, prior to or in a concerted manner with the protonation of the N3 of the pyrimidone ring of the (6–4) photoproduct.
Experimental
Materials
15N-labeled ammonium chloride and 4-phenyl-1,2,4-triazoline-3,5-dione were obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA) and Sigma-Aldrich Japan (Tokyo, Japan), respectively. All of the other solvents and reagents were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). TLC analyses were carried out on Merck Silica gel 60 F254 plates, which were visualized by UV illumination at 254 nm. For column chromatography, Wakogel C-200 was used. For reversed-phase chromatography, Preparative C18 125 Å 55–105 μm resin (Waters Corporation, Milford, MA) was used on a Bio-Rad Econo System.Spectroscopy
UV/VIS and fluorescence spectra were recorded on a Shimadzu UV-1700 Pharmaspec spectrophotometer and a JASCO FP-6500 spectrofluorometer, respectively. For the measurement of fluorescence spectra of 1, the excitation wavelength was set to 313 nm, and three independently measured spectra at 298 K were averaged. 1H NMR spectra were measured on a JEOL AL-400 or Varian INOVA 500 spectrometer. 31P NMR and 15N NMR spectra were recorded with a 5 mm switchable probe on a Varian INOVA 500 MHz NMR spectrometer. Proton chemical shifts were calibrated with tetramethylsilane (TMS) as an internal standard, and 31P chemical shifts were calibrated with trimethyl phosphate (TMP) as an external standard. 15N chemical shifts were calibrated with nitromethane as an external standard with a conversion factor of 375.8 ppm (the offset value from the 15N resonance of liquid ammonia), as described in ref. 23. High-resolution mass spectrometry was performed on a JEOL JMS-DX303 spectrometer with electron impact (EI) ionization and on a JEOL JMS-700 spectrometer with fast atom bombardment (FAB) ionization.HPLC analysis
HPLC analyses were carried out on a Gilson gradient-type analytical system equipped with a Waters 2996 photodiode array detector, and a Waters μBondasphere C18 5 μm 300 Å column (3.9 mm × 150 mm) was used, at a flow rate of 1.0 mL min−1, with a linear gradient of acetonitrile in 0.1 M triethylammonium acetate (pH 7.0). For the analysis of the acid hydrolysis, 1 and 2 without the 15N labeling were dissolved in diluted hydrochloric acid, and the actual pH values were measured on a Mettler Toledo SevenEasy S20 pH meter calibrated at pH 6.86, 4.01, and 1.68. Aliquots of these samples were injected after the reaction times shown in Fig. 4. The acetonitrile concentration was 0–7.5% for 15 min. The detected products were purified by HPLC using the same conditions, and were analyzed by mass spectrometry on a JEOL JMS-700 spectrometer with FAB (the product from 1) and EI (the product from 2) ionization.DFT calculation
The calculations based on the DFT were carried out with the Gaussian03W at the B3LYP/6-31G(d) level without considering the solvent effects. The initial structural models of dinucleoside monophosphates containing the (6–4) photoproduct and its Dewar valence isomer were constructed with Chem3D (CambridgeSoft, Cambridge, MA, USA). Although three different states of phosphate (an anion form, a neutral form, and a tetramethylammonium salt) were calculated, it was found that the hydrogen-bonding interaction between the N3 of the 3′ pyrimidone and the 5-OH of the 5′ pyrimidine was conserved among them, and the N3–H distances were 1.84, 1.88, and 1.87 Å for the anionic, neutral, and tetramethylammonium forms, respectively. The optimized structures of these compounds are listed as Appendix in the ESI.† We confirmed that all of the converged structures lacked an imaginary frequency at the B3LYP/6-31G(d) level. The structures of the anionic form of the (6–4) photoproduct and its Dewar valence isomer in Fig. S4† were produced with PyMOL (DeLano Scientific LLC, San Carlos, CA, USA). The structures obtained by the DFT calculation in the appendix were produced with Discovery Studio 2.0 (Accelrys Software Inc., San Diego, CA, USA).Conclusion
We performed comprehensive analyses on the protonation states of the pyrimidone N3 in the (6–4) photoproduct and its related compound. The results indicated that the pKa value for the N3 of the (6–4) photoproduct is remarkably lower than that of a normal pyrimidone ring, and strongly suggest that an intramolecular hydrogen bond is formed between the N3 of the 3′ base and the 5-OH of the 5′ base under physiological conditions. This interaction should be taken into account to elucidate the repair mechanism of the (6–4) photolyase. The histidine residue acting as a base in Fig. 1a is also required for the protonation of the 3′ pyrimidone base by the other histidine, at the first step of the (6–4) photolyase reaction.Acknowledgements
We thank Prof. Hiroshi Miyasaka (Osaka University, Japan) for discussions and Dr David Loakes (Medical Research Council, U.K.) for comments. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and a grant from Asahi Glass Foundation. J.Y. was supported by a Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists and expresses his special thanks to The Global COE Program of Osaka University.References
- S. Iwai, in Modified Nucleosides in Biochemistry, Biotechnology and Medicine, ed. P. Herdewijn, Wiley-VCH, Weinheim, 2008, ch. 5, pp. 97–131 Search PubMed.
- (a) J. E. LeClerc, A. Borden and C. W. Lawrence, Proc. Natl. Acad. Sci. USA, 1991, 88, 9685–9689 CAS; (b) C. A. Smith, M. Wang, N. Jiang, L. Che, X. Zhao and J.-S. Taylor, Biochemistry, 1996, 35, 4146–4154 CrossRef CAS; (c) H. Kamiya, S. Iwai and H. Kasai, Nucleic Acids Res., 1998, 26, 2611–2617 CrossRef CAS.
- L. C. J. Gillet and O. D. Schärer, Chem. Rev., 2006, 106, 253–276 CrossRef CAS.
- W. Yang and R. Woodgate, Proc. Natl. Acad. Sci. USA, 2007, 104, 15591–15598 CrossRef CAS.
- (a) A. Sancar, Chem. Rev., 2003, 103, 2203–2237 CrossRef CAS; (b) S. Weber, Biochim. Biophys. Acta, 2005, 1707, 1–23 CAS.
- (a) X. Zhao, J. Liu, D. S. Hsu, S. Zhao, J.-S. Taylor and A. Sancar, J. Biol. Chem., 1997, 272, 32580–32590 CrossRef CAS; (b) K. Hitomi, S.-T. Kim, S. Iwai, N. Harima, E. Otoshi, M. Ikenaga and T. Todo, J. Biol. Chem., 1997, 272, 32591–32598 CrossRef CAS.
- J. Yamamoto, K. Hitomi, T. Todo and S. Iwai, Nucleic Acids Res., 2006, 34, 4406–4415 CrossRef CAS.
- (a) K. S. Christine, A. W. MacFarlane, K. Yang and R. J. Stanley, J. Biol. Chem., 2002, 277, 38339–38344 CrossRef CAS; (b) A. Mees, T. Klar, P. Gnau, U. Hennecke, A. P. M. Eker, T. Carell and L.-O. Essen, Science, 2004, 306, 1789–1793 CrossRef CAS.
- S.-T. Kim, K. Malhotra, C. A. Smith, J.-S. Taylor and A. Sancar, J. Biol. Chem., 1994, 269, 8535–8540 CAS.
- (a) P. Clivio and J.-L. Fourrey, Chem. Commun., 1996, 2203–2204 RSC; (b) M. K. Cichon, S. Arnold and T. Carell, Angew. Chem., Int. Ed., 2002, 41, 767–770 CrossRef CAS; (c) T. Stafforst and U. Diederichsen, Chem. Commun., 2005, 3430–3432 RSC; (d) F. Boussicault and M. Robert, J. Phys. Chem. B, 2006, 110, 21987–21993 CrossRef CAS; (e) Q.-H. Song, H.-B. Wang, W.-J. Tang, Q.-X. Guo and S.-Q. Yu, Org. Biomol. Chem., 2006, 4, 291–298 RSC; (f) J. Trzcionka, V. Lhiaubet-Vallet, C. Paris, N. Belmadoui, M. J. Climent and M. A. Miranda, ChemBioChem, 2007, 8, 402–407 CrossRef CAS.
- O. A. Borg, L. A. Eriksson and B. Durbeej, J. Phys. Chem. A, 2007, 111, 2351–2361 CrossRef CAS.
- (a) C. B. Harrison, L. L. O'Neil and O. Wiest, J. Phys. Chem. A, 2005, 109, 7001–7012 CrossRef CAS; (b) H. Bohr, K. J. Jalkanen and F. B. Malik, Mod. Phys. Lett. B, 2005, 19, 473–487 CrossRef CAS; (c) T. R. Prytkova, D. N. Beratan and S. S. Skourtis, Proc. Natl. Acad. Sci. USA, 2007, 104, 802–807 CrossRef CAS; (d) X. Zheng, N. M. Ly and A. A. Stuchebrukhov, Int. J. Quant. Chem., 2007, 107, 3126–3131 CrossRef CAS; (e) X. Zheng, J. Garcia and A. A. Stuchebrukhov, J. Phys. Chem. B, 2008, 112, 8724–8729 CrossRef CAS.
- K. Hitomi, H. Nakamura, S.-T. Kim, T. Mizukoshi, T. Ishikawa, S. Iwai and T. Todo, J. Biol. Chem., 2001, 276, 10103–10109 CrossRef CAS.
- E. Schleicher, K. Hitomi, C. W. M. Kay, E. D. Getzoff, T. Todo and S. Weber, J. Biol. Chem., 2007, 282, 4738–4747 CAS.
- H. M. Bdour, J. L.-F. Kao and J.-S. Taylor, J. Org. Chem., 2006, 71, 1640–1646 CrossRef CAS.
- S. Iwai, M. Shimizu, H. Kamiya and E. Ohtsuka, J. Am. Chem. Soc., 1996, 118, 7642–7643 CrossRef CAS.
- J.-S. Taylor, D. S. Garrett and M. J. Wang, Biopolymers, 1988, 27, 1571–1593 CrossRef CAS.
- (a) J. J. Fox and D. Shugar, Biochim. Biophys. Acta, 1952, 9, 369–384 CAS; (b) R. O. Hurst, A. M. Marko and G. C. Butler, J. Biol. Chem., 1953, 204, 847–856 CAS.
- S. F. Singleton, F. Shan, M. W. Kanan, C. M. McIntosh, C. J. Stearman, J. S. Helm and K. J. Webb, Org. Lett., 2001, 3, 3919–3922 CrossRef CAS.
- B. Gildea and L. W. McLaughlin, Nucleic Acids Res., 1989, 17, 2261–2281 CAS.
- T. C. Butler, D. Johnson and K. H. Dudley, J. Heterocycl. Chem., 1982, 19, 657–658 CAS.
- M. Vives, R. Eritja, R. Tauler, V. E. Marquez and R. Gargallo, Biopolymers, 2004, 73, 27–43 CrossRef CAS.
- G. C. Levy and R. L. Lichiter, in Nitrogen-15 Nuclear Magnetic Resonance Spectroscopy, John Wiley & Sons, Inc., New York, 1979 Search PubMed.
- J. A. Iocono, B. Gildea and L. W. McLaughlin, Tetrahedron Lett., 1990, 31, 175–178 CrossRef CAS.
- T. J. Matray and M. M. Greenberg, J. Am. Chem. Soc., 1994, 116, 6931–6932 CrossRef CAS.
- G. Wagner, A. Pardi and K. Wüthrich, J. Am. Chem. Soc., 1983, 105, 5948–5949 CrossRef CAS.
- Gaussian 03, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- M. Schubert, D. K. Y. Poon, J. Wicki, C. A. Taling, E. M. Kwan, J. E. Nielsen, S. G. Withers and L. P. McIntosh, Biochemistry, 2007, 46, 7383–7395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, supplementary figures, and calculation results. See DOI: 10.1039/b815458a |
| This journal is © The Royal Society of Chemistry 2009 |