A facile synthesis of pyrrolo-(di)-benzazocinones via an intramolecular N-acyliminium ion cyclisation†
Received
1st September 2008
, Accepted 10th October 2008
First published on 12th November 2008
Abstract
A facile, moderate to high yielding synthesis of hexahydro-(di)-benzazocinones is described via an intramolecular N-acyliminium ion cyclisation. The iminium ion intermediates are formed from the readily available 4,4-diethoxybutyl amides with an excess of triflic acid. For electron-withdrawing substituents, better yields were obtained from the pre-formed 2-hydroxypyrrolidine amides. From NMR studies, at ambient temperatures the pyrrolo-benzazocin-3-ones exist as a slowly equilibrating mixture of two conformations.
Introduction
We have an on-going programme of work directed towards the development of new chemistry to allow the synthesis of privileged structures for the assembly of high quality screening libraries. We are interested in the synthesis of conformationally restricted aryl-substituted alkylamines, privileged structures which are found in CNS drugs,1–3 and particularly interested in 1,2,3,4-tetrahydro-isoquinoline and its homologues, for which there have been numerous reports on their biological activity.4–13 As an example of a new synthesis of the 1,2,3,4-tetrahydro-isoquinoline core, we recently described a synthesis of (±)-crispine A via an intramolecular cyclisation of a readily synthesized N-acyliminium ion.14 In this paper, we describe the unexpected isolation of a pyrrolo-dibenzazocin-3-one (3) from a pyrrolo-isoquinolone synthesis via an immimium ion cyclisation and the application of this methodology to related systems.
Results and discussion
Whilst investigating the scope of this tetrahydro-isoquinoline synthesis, we attempted the triflic acid-mediated cyclisation of compound 1, which was readily prepared from biphenyl-2-acetic acid.15 On treatment of 1 with triflic acid (10 equivs.) in CHCl3 we isolated the 6-membered cyclisation product 2 (64% yield) as expected (Scheme 1), but in addition a second, isomeric product. 1H and 13C NMR spectra provided evidence that the product was the dibenzazocine 3 (29% yield). The key difference between the 13C NMR spectra of 2 and 3 were the number of aromatic carbon signals, 10 signals for 2, 6 of which were C–H, and 12 signals for 3, 8 of which were C–H. The present observation is, as far as we can ascertain, the first example of an acyliminium ion cyclisation onto an unactivated aromatic to form an 8-membered ring.16,17
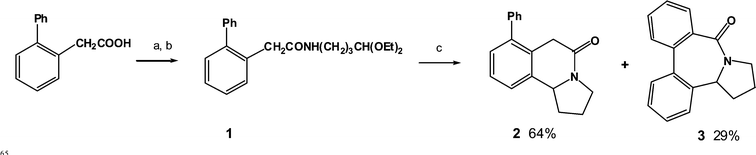 |
| | Scheme 1 Synthesis of the tetrahydro-dibenzazocine 3. Reagents and conditions: a) (COCl)2, CH2Cl2, DMF, b) H2N(CH2)3CH(OEt)2, EtNMe2, c) CF3SO3H, CHCl3, heat. | |
Although benzo-fused 7-membered rings have been extensively exploited in drug discovery, for example tricyclic antidepressants,18 benzodiazepines19 and as enzyme inhibitors,20 there have only been a few examples of pharmacologically active benzo-fused 8-membered rings described in the literature.21–24 To date, hexahydro-benzazocines have been prepared either by ring expansion,21,25–27 by ring-closing metathesis,28,29 and by intramolecular cyclisationvia either a Staudinger–aza-Wittig procedure22 or iminium ion cyclisations of sulfonamides.30–33 We therefore believed that this finding deserved further investigation.
Following the isolation of 3, we decided to investigate the synthesis of the simpler hexahydro-benzazocinone 5 from 4-phenylbutyramide 4 (Scheme 2), prepared in a similar manner to 1 from 4-phenylbutyric acid. Cyclisation with triflic acid gave a good yield (70%) of a colourless oil, tentatively assigned as the hexahydrobenzazocinone 5 together with a small amount of 1-tetralone (8% yield). The reaction was carried out at between 0.05–0.1 M concentration demonstrating that no special procedures (e.g. high dilution) were required to mediate this ring formation. Under these strongly acidic conditions, it is likely that the acyliminium ion is protonated on the carbonyl to form the dicationic superelectrophile, which undergoes the cyclisation.34
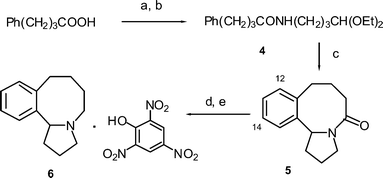 |
| | Scheme 2 Synthesis of benzazocines 5 and 6. Reagents and conditions: a) (COCl)2, CH2Cl2, DMF, b) H2N(CH2)3CH(OEt)2, EtNMe2 (100%), c) CF3SO3H, CHCl3, heat (70%), d) LiAlH4, THF, heat (88%), e) picric acid. | |
At ambient temperatures, the 1H and 13C NMR spectra of 5 in CDCl3 showed broadened signals for the majority of the peaks, which was attributed to an intramolecular dynamic process with a rate intermediate in the NMR timescale at temperatures near 298 K. Relatively sharp 1H and 13C NMR spectra were measured at 333 K, which were used for the structural confirmation of 5. Further variable-temperature 1H and 13C measurements, including two-dimensional exchange-correlated spectroscopy,35 revealed the presence of two conformations in the dynamic process. In particular, two sets of peaks were observed at 213 K in the 1H and 13C NMR spectra of 5 with the integral intensity ratio of 2.7:1. Proton H-2, the bridgehead proton of the major conformer, appeared as a doublet of doublets (J = 6.4 and 9.3 Hz), whereas that of the minor conformer showed only a doublet (J = 7.0 Hz). Based upon these values and the boundary values of the corresponding 3JHH couplings predicted for L-prolines,36 the five-membered ring of the major conformer is in the C4-exo conformation, whereas that of the minor comformer is in the C4-endo conformation. Here we use the previously suggested notation for L-proline conformations,36,37 where the endo-/exo-orientation of the C4 atom is determined relative to the C2–C1 bond. Overall, the 3JHH couplings of both the five- and eight-membered rings were in favour of a two-site conformational equilibrium shown in Fig. 1, where the C4-exo-C8-endo form is the preferred major conformer. The free energy barrier for the conformational change from the C4-exo-C8-endo conformer into the C4-endo-C8-exo conformer for 5 was estimated to be 55 ± 1 kJ mol−1 at the coalescence temperature of 266 K. For the reverse interconversion from the C4-endo-C8-exo conformer into the C4-exo-C8-endo conformer the free energy barrier was estimated to be 53 ± 1 kJ mol−1 at 266 K.
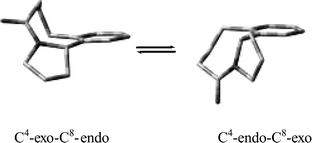 |
| | Fig. 1 A representation of the two conformers of 5 from NMR analysis. | |
In order to obtain a solid derivative to unambiguously confirm the molecular structure, 5 was reduced to the amine with LAH and a solid picrate salt 6 formed. Unfortunately good quality crystals of the picrate could not be obtained. However, the structure of this salt was confirmed by a low resolution X-ray crystal structure analysis. In addition, the structure of the corresponding free base was also confirmed by NMR measurements in CDCl3 (Fig. 2).
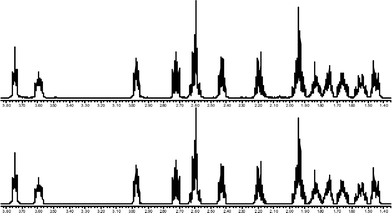 |
| | Fig. 2 Experimental (top, in CDCl3, 333 K) and calculated (bottom) 600 MHz 1H NMR spectra of the free base of 6. Fifteen protons of the five- and eight-membered aliphatic rings were included in the iterative fittings of the experimental spectrum. | |
The scope of this method was then investigated with respect to aromatic substituents, and the results are shown in Table 1.
Table 1 Synthesis of substitued hexahydrobenzazocin-3-ones 5a–f
|
Cpd. no. |
R |
T (h)a |
Cpd. no. |
R |
Isolated yield (%) |
|
Time of reaction in CHCl3 heated under reflux.
At ambient temperatures.
|
|
4a
|
4-Me
|
4 |
5a
|
14-Me
|
75 |
|
4b
|
3,4-diMeO |
1 |
5b
|
13,14-diMeO |
80 |
|
4c
|
2,5-diMeO |
21b |
5c
|
12,15-diMeO |
26 |
|
4d
|
4-Cl
|
18 |
5d
|
14-Cl
|
0 |
|
4e
|
4-Br
|
18 |
5e
|
14-Br
|
17 |
|
4f
|
4-Ph
|
0.5 |
5f
|
14-Ph
|
25 |
As expected of an electrophilic cyclisation, electron-donating substituents increased the rate of reaction and gave higher yields, as exemplified by the p-tolyl and 3,4-dimethoxy analogues (5a and 5b respectively). The 2,5-dimethoxy 5c gave a much lower yield, though the reaction was still fast and could be run at ambient temperatures. Electron-withdrawing substituents both slowed the reaction and gave lower yields. Thus the 4-Br 4e and 4-phenyl 4f gave only poor yields of the benzazocinones 5e and 5f respectively. None of the desired product could be detected from the 4-Cl 4dcyclisation. In all cases, where a poor yield was obtained a significant quantity of insoluble, presumably polymeric material, was produced. For the very electron-rich 4b, the cyclisation could be achieved using 10 equivs. of TFA (1 h reflux, 90% yield).
In the 1H and 13C NMR spectra of 5b and 5e at 213 K, again two sets of peaks were observed with the integral intensity ratios of 4.3:1 and 3.4:1 respectively. Similar to 5, the free energy barrier for the conformational change from the C4-exo-C8-endo conformer into the C4-endo-C8-exo conformer for 5e was estimated to be 56 ± 1 kJ mol−1 at the coalescence temperature of 266 K. For the reverse interconversion from the C4-endo-C8-exo conformer into the C4-exo-C8-endo conformer the free energy barrier was estimated to be 53 ± 1 kJ mol−1 at 266 K (Fig. 3).
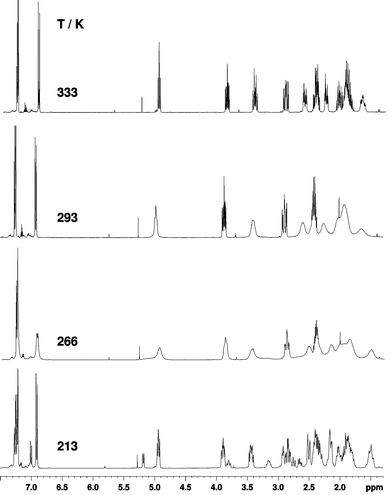 |
| | Fig. 3 The temperature dependence of the 1H NMR spectra of 5e (CDCl3, 400 MHz). | |
A sample of 5c was submitted for X-ray analysis, which confirmed the structure as the benzazocin-3-one (Fig. 4). Unlike the other benzazocinones, 5c showed sharp 1H and 13C NMR spectra in CDCl3 at 298 K with the 3JHH-couplings indicating of a single C4-exo-C8-endo conformer, in agreement with the solid-state structure. Presumably the steric constraints of the 12- and 15-substituents restricts the conformational freedom of the 8,5 system.
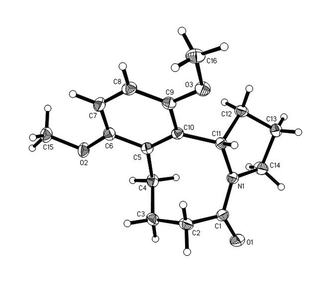 |
| | Fig. 4 Structure of hexahydrobenzazocinone 5c as determined by X-ray crystallography. ORTEP diagram (50% probability ellipsoids) showing the crystallographic atom-numbering scheme. | |
In our previous paper,14 a higher yield of tetrahydro-isoquinolinone was obtained from the N-acyl-2-hydroxy-pyrrolidine, readily prepared by mild acid hydrolysis of the amidoacetal. Thus treatement of an acetone solution 4d–h with 1 M HCl solution for 15 min gave the intermediate N-acyl- 2-hydroxypyrrolidines 7d–h in essentially quantitative yields (Scheme 3).
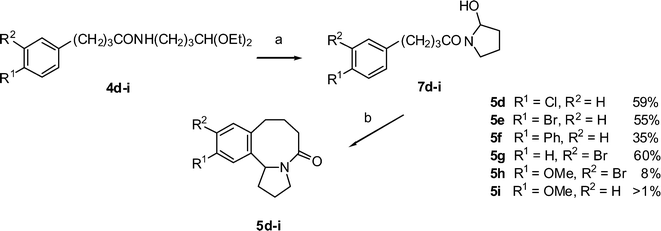 |
| | Scheme 3 Synthesis of the hexahydrobenzazocines 5d–ivia the 2-hydroxypyrolidines 7d–i. Reagents and conditions: a) HCl, acetone, b) CF3SO3H, CHCl3, heat. | |
Previous experience had shown that N-acyl-2-hydroxy- pyrrolidines were too unstable to purify by column chromatography and so 7d–i were used without further purification. Triflic acid cyclisation of 7d–g gave higher yields of benzazocines 5d–g, than previously obtained from the amides (yields quoted are the overall yields from 4d–g). Cyclisation of the m-Br congener 7g gave almost exclusively the 13-Br product 5g, with only a trace of a less polar isomer, presumably the 15-Br, detectable by MS. Cyclisation of the 4-methoxy lactam 7i gave only a trace (TLC, MS) of the hexahydrobenzazocine, but an improved yield was obtained from the 3-Br, 4-MeO lactam 7h as a single isomer. A similar low yield was reported for the TiCl4-mediated cyclisation of 1-(4-methoxyphenylacetyl)-2-methoxypyrrolidine to the pyrrolidino-tetrahydroisoquinoline.38
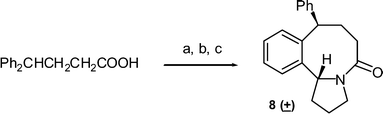
This methodology also works well for phenyl butyramides substituted in the alkyl chain. Cyclisation of the 4,4-diphenyl-butyric acid amide gave a good yield of the phenyl-substituted benzazocinone, which from NMR, the isolated product was a mixture of isomers in a ratio of 20:1 (Scheme 4). The major isomer 8 was isolated as a solid and its structure confirmed by X-ray analysis (Fig. 5), which shows that the phenyl group is in a pseudo-equatorial orientation ‘cis’ to the bridgehead C4 proton.
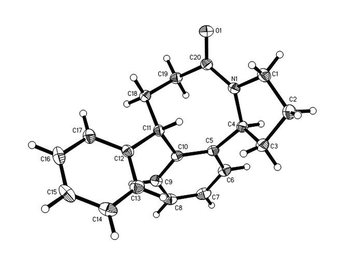 |
| | Fig. 5 Structure of hexahydro-benzazocinone 8 as determined by X-ray crystallography. ORTEP diagram (50% probability ellipsoids) showing the crystallographic atom-numbering scheme. | |
 |
| | Scheme 4 Synthesis of benzazocine 8; Reagents and conditions: a) (COCl)2, CH2Cl2, DMF, b) H2N(CH2)3CH(OEt)2, EtNMe2 (100%), c) CF3SO3H, CHCl3, heat (73%). | |
Both the attempted application of this methodology to the synthesis of the 9-membered ring homologue from the 5-phenylpentanoic acid amide 9 and the attempted cyclisation of the 4-phenylbutanoic acid amide of 5-aminopentananal diethylacetal 10 failed to give any cyclised products.
| Ph(CH2)4CONH(CH2)3CH(OEt)2 9 |
| Ph(CH2)3CONH(CH2)4CH(OEt)2 10 |
An alternative system was also evaluated where there is no possibility for the formation of a 6-membered ring (Scheme 5). The commercial α-phenyl-o-toluic acid was converted into the 4-aminobutyraldehyde diethylacetal amide derivative via the acid chloride. Cyclisation with triflic acid afforded the tetracycle 12a in an excellent yield. Both the 1H and 13C NMR spectra were consistent with the proposed structure. The structure of 12a was confirmed by an X-ray structural analysis (Fig. 6). Cyclisation of the ortho-phenoxyamide derived from 11b also gave the cyclic product 12b, though in a slightly lower yield.
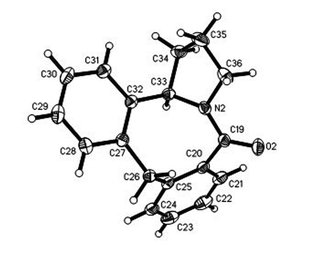 |
| | Fig. 6 Structure of hexahydro-dibenzazocinone 12a as determined by X-ray crystallography. ORTEP diagram (50% probability ellipsoids) showing the crystallographic atom-numbering scheme. There are two independent molecules in the asymmetric unit and only one of these is shown in the figure. The asymmetric unit was selected so that both molecules have the same R-stereochemistry. | |
 |
| | Scheme 5 Synthesis of dibenzoazocines 12a and 12b: Reagents and conditions: a) (COCl)2, CH2Cl2, DMF, b) H2N(CH2)3CH(OEt)2, EtNMe2 (100%), c) CF3SO3H, CHCl3, heat. | |
Although it is well known that acyliminium ions are more electrophilic than iminium ions,39 for comparison the Pictet–Spengler type cyclisation of the amine 13, prepared from 4 by reduction with LAH, to 6 was investigated.
| Ph(CH2)4NH(CH2)3CH(OEt)2 13 |
No formation of 6 was observed under the acidic conditions of TFA or triflic acid 2 h in refluxing CHCl3, or 2 M aqueous HCl at reflux for 24 h.
Conclusion
In conclusion, we have shown that pyrrolo-benzazocin-3-ones can be readily prepared from the commercially available 4-aminobutyraldehyde diethyl acetal and the appropriate carboxylic acids in moderate to high yield via an intramolecular N-acyl iminium ion cyclisation. For electron-withdrawing substituents, pre-formation of the 2-hydroxy-pyrrolidine amides gave better yields. Two series of pyrrolo-dibenzazocines were also prepared. This method is not applicable for the synthesis of the 3-aza-benzocyclononanes or the piperidino-benzazocines, nor was the Pictet–Spengler cyclisation successful. Our future work will concentrate on further investigation of the scope of this methodology and on the synthesis of 8-membered analogues of biologically active tetrahydroisoquinolines and benzazepines, and it is our belief that this chemistry will further open up the possibility of 8-membered rings for exploitation in, for example, chemical library synthesis and drug discovery.
Experimental
All reagents were commercially available, unless specified, and used without purification. The chloroform used was stabilised with amylene. All non-crystalline final compounds were found to be >95% pure by HPLC, and all crystalline compounds >98% pure. Solution 1H and 13C NMR spectra (Tables 2, 3 and 4) were recorded on Bruker NMR spectrometers AMX300, Avance III 400, DRX500 and Avance III 600 equipped with z-gradient facilities. 1H and 13C chemical shifts are given relative to TMS. Unless otherwise specified, spectra were recorded at 298 K. The 1H spectra of 5b, 5c and 6 were analysed using full lineshape analysis.40 Low temperature NMR measurements were carried out for hexahydro-benzazocinones 5, 5b and 5e. The value of the free energy of activation for 5 and 5e was calculated using the procedure described previously.41
Table 2
1H NMR chemical shifts (δH, ppm) of hexahydrobenzazocinones in CDCl3. The spectrum of 5c was measured at 298 K, all other spectra were measured at 333 K. The structure shows the proton and carbon numbering used in the NMR assignments in Tables 2–4. The relative orientations of the protons are defined relative to proton H-2c: “c” and “t” are used to denote protons with the cis and trans orientations relative to proton H-2c
|
Proton
|
5
|
5a
|
5b
|
5c
|
5d
|
5e
|
5f
|
6
a
|
|
2.590 ppm (H-7c) and 2.611 ppm (H-7t).
|
|
2c
|
5.03 |
4.98 |
4.967 |
5.182 |
5.00 |
4.98 |
5.13 |
3.746 |
|
3c
|
2.40 |
2.40 |
2.412 |
2.520 |
2.44 |
2.42 |
2.49 |
2.192 |
|
3t
|
2.07 |
2.06 |
2.081 |
1.719 |
2.07 |
2.06 |
2.17 |
1.949 |
|
4c
|
1.87 |
1.86 |
1.888 |
1.813 |
1.90 |
1.89 |
1.92 |
1.832 |
|
4t
|
1.91 |
1.91 |
1.960 |
1.853 |
1.96 |
1.95 |
1.98 |
1.933 |
|
5c
|
3.87 |
3.87 |
3.863 |
3.838 |
3.90 |
3.88 |
3.94 |
2.430 |
|
5t
|
3.43 |
3.44 |
3.447 |
3.549 |
3.44 |
3.42 |
3.50 |
2.974 |
|
8c
|
2.49 |
2.48 |
2.508 |
2.371 |
2.47 |
2.44 |
2.57 |
1.546 |
|
8t
|
2.26 |
2.24 |
2.265 |
2.106 |
2.29 |
2.27 |
2.32 |
1.453 |
|
9c
|
1.70 |
1.66 |
1.713 |
1.447 |
1.70 |
1.68 |
1.77 |
1.662 |
|
9t
|
1.95 |
1.95 |
1.935 |
1.927 |
1.96 |
1.95 |
2.04 |
1.754 |
|
10c
|
2.66 |
2.48 |
2.584 |
3.207 |
2.64 |
2.62 |
2.73 |
2.722 |
|
10t
|
2.96 |
2.92 |
2.912 |
2.529 |
2.95 |
2.92 |
3.03 |
3.593 |
| 12 |
7.05 |
6.93 |
6.553 |
3.747 (OMe) |
7.00 |
6.93 |
7.15 |
7.042 |
| 13 |
7.16 |
6.97 |
3.833 (OMe) |
6.738 |
7.14 |
7.27 |
7.41 |
7.15 |
| 14 |
7.14 |
2.29 (Me) |
3.836 (OMe) |
6.689 |
— |
— |
— |
7.14 |
| 15 |
7.12 |
6.92 |
6.638 |
3.747 (OMe) |
7.13 |
7.26 |
7.36 |
7.13 |
Table 3
Proton
J-couplings (in Hz) of hexahydrobenzazocinones 5–5f and 6 in CDCl3. The spectrum of 5c was measured at 298 K, all other spectra were measured at 333 K
|
Protons
|
5
|
5a
|
5b
|
5c
|
5d
|
5e
|
5f
|
6
|
| 2c–3c |
7.1 |
7.1 |
6.99 |
6.15 |
7.1 |
7.1 |
7.1 |
7.55 |
| 2c–3t |
7.1 |
7.1 |
6.98 |
9.69 |
7.1 |
7.1 |
7.1 |
7.74 |
|
3c–3t |
−12.7 |
−12.7 |
−12.80 |
−12.57 |
−12.7 |
−12.9 |
−12.7 |
−12.87 |
|
3c–4t |
5.1 |
5.0 |
5.29 |
3.29 |
5.2 |
5.1 |
5.0 |
5.29 |
| 3c–4c |
7.6 |
7.5 |
7.58 |
6.51 |
7.5 |
7.4 |
7.5 |
10.06 |
|
3t–4c |
8.7 |
8.7 |
8.41 |
10.81 |
8.5 |
8.7 |
8.6 |
5.83 |
|
3t–4t |
7.3 |
7.2 |
7.25 |
6.68 |
7.0 |
7.1 |
7.0 |
10.72 |
|
4c–4t |
−12.6 |
−12.5 |
−12.63 |
−12.41 |
−12.7 |
−12.6 |
−12.6 |
−12.54 |
|
4c–5c |
8.6 |
8.5 |
8.42 |
8.65 |
8.4 |
8.4 |
8.5 |
8.63 |
|
4c–5t |
8.1 |
8.1 |
8.12 |
9.06 |
7.9 |
8.2 |
8.3 |
3.74 |
|
4t–5c |
4.1 |
4.2 |
4.41 |
3.44 |
4.3 |
4.2 |
4.3 |
7.68 |
|
4t–5t |
8.1 |
8.1 |
7.85 |
7.45 |
7.9 |
8.0 |
8.1 |
8.38 |
|
5c–5t |
−12.2 |
−12.2 |
−12.17 |
−12.21 |
−12.2 |
−12.1 |
−12.2 |
−9.14 |
|
8c–8t |
−12.4 |
−12.3 |
−12.32 |
−11.88 |
−12.5 |
−12.5 |
−12.4 |
−15.00 |
|
8c–9c |
4.8 |
4.9 |
4.79 |
5.38 |
4.8 |
4.7 |
4.9 |
11.97 |
|
8c–9t |
12.8 |
12.8 |
12.72 |
13.27 |
12.7 |
12.8 |
12.8 |
3.34 |
|
8t–9c |
4.2 |
4.1 |
4.03 |
2.61 |
4.1 |
4.0 |
4.1 |
3.35 |
|
8t–9t |
4.2 |
4.0 |
4.18 |
4.47 |
4.1 |
3.9 |
4.1 |
5.37 |
|
9c–9t |
−13.0 |
−12.8 |
−13.41 |
−13.06 |
−12.8 |
−12.8 |
−12.8 |
−13.62 |
|
9c–10c |
4.0 |
4.9 |
3.71 |
3.90 |
3.8 |
3.9 |
3.9 |
6.71 |
|
9c–10t |
11.7 |
11.9 |
11.82 |
13.55 |
11.8 |
11.9 |
11.9 |
5.31 |
|
9t–10c |
5.0 |
4.9 |
5.04 |
3.04 |
5.0 |
5.0 |
4.7 |
5.32 |
|
9t–10t |
3.9 |
3.9 |
3.80 |
4.09 |
3.9 |
3.9 |
3.9 |
8.85 |
|
10c–10t |
−13.8 |
−13.8 |
−13.96 |
−13.29 |
−13.9 |
−13.9 |
−13.8 |
−12.82 |
| Other |
— |
7.8 (12–13), 1.7 (13,15) |
— |
8.94 (13–14) |
∼8 (12–13), 2.2 (13,15) |
∼8 (12–13), 2.1 (13,15) |
7.9 (12–13), 2.0 (13,15) |
−12.57 (7c–7t), 3.09 (7c–8c), 5.27 (7c–8t), 0.85 (7c–9t), 11.28 (7t–8c), 3.13 (7t–8t) |
Table 4
13C NMR chemical shifts (δC, ppm) of hexahydrobenzazocinones 5–5f and 6 in CDCl3. The spectrum of 5c was measured at 298 K, all other spectra were measured at 333 K. The values of 1JCH couplings (in Hz) are also included in brackets for 5c
|
Carbon
|
5
|
5a
|
5b
|
5c
|
5d
|
5e
|
5f
|
6
|
|
1-Cq |
140.54 |
140.35 |
132.75 |
130.09 |
142.38 |
142.85 |
140.90 |
141.08 |
|
2-CH
|
62.80 |
62.93 |
62.53 |
59.44 (145) |
62.35 |
62.40 |
63.11 |
65.65 |
| 3-CH2 |
36.83 |
37.12 |
36.91 |
36.09 (134) |
36.78 |
36.99 |
37.33 |
34.16 |
| 4-CH2 |
22.75 |
22.77 |
22.80 |
22.50 (131) |
22.66 |
22.77 |
22.92 |
22.71 |
| 5-CH2 |
45.94 |
46.01 |
46.04 |
46.81 (143) |
45.93 |
46.08 |
46.15 |
55.30 |
| 7 |
172.48 |
172.50 |
172.61 |
172.71 |
172.22 |
172.32 |
172.65 |
54.01 |
| 8-CH2 |
33.81 |
33.67 |
33.91 |
32.26 (131) |
33.59 |
33.63 |
33.80 |
24.67 |
| 9-CH2 |
27.37 |
27.60 |
27.61 |
25.74 (132) |
27.22 |
27.25 |
27.52 |
30.17 |
| 10-CH2 |
32.62 |
32.03 |
32.22 |
21.01 (131) |
31.93 |
32.06 |
32.32 |
32.98 |
|
11-Cq |
138.16 |
135.01 |
130.96 |
127.48 |
136.60 |
137.18 |
137.25 |
140.19 |
| 12 |
131.49 |
131.46 |
115.20 |
151.02 |
132.81 |
133.22 |
132.17 |
130.50 |
| 13 |
126.57 |
128.31 |
148.66 |
109.47 (159) |
127.58 |
130.67 |
126.38 |
125.92 |
| 14 |
127.61 |
135.97 |
147.89 |
108.31 (159) |
132.29 |
120.29 |
141.04 |
126.86 |
| 15 |
126.90 |
127.63 |
111.61 |
150.19 |
126.91 |
129.95 |
125.84 |
127.13 |
| Other |
— |
20.97 (14-Me) |
56.54 (13-OMe), 56.24 (14-OMe) |
56.21(143) (12-OMe), 55.75 (143) (15-OMe) |
— |
— |
139.78 (i-Ph), 127.13 (o-Ph), 128.96 (m-Ph), 127.50 (p-Ph) |
|
4-(3-Bromophenyl)-butyric acid
4-(3-Bromophenyl)-4-oxo-butyric acid42 (4.8 g) and KOH (3.6 g) was dissolved in 35 ml ethylene glycol and hydrazine hydrate (2.4 ml) added. The reaction mixture was heated to reflux (140 °C) for 2 h, then heated to 220 °C (heating block temperature) to distill out the H2O and excess hydrazine hydrate. After all the distillation had ceased, the reaction mixture was heated for 1 h, the cooled to room temperature, H2O added (100 ml) and conc. HCl to acidic, then ice (50 g). On scratching with a glass rod, a solid had formed which was collected, dissolved in Et2O (150 ml) and the product extracted into 1 M NaOH (2 × 50 ml). The aqueous solution was acidified with 2 M HCl and the product extracted into CH2Cl2 (3 × 50 ml). The organic extracts were dried (MgSO4) and filter through a SiO2 bed (∼20 g), washing with Et2O until no more non-polar material was extracted. The combined washings were evaporated in vacuo to give 3.1 g of pale yellow oil. 1H NMR (300 MHz, CDCl3): δ 1.95 (quintet, 2H, J = 7 Hz), 2.39 (t, 2H, J = 7 Hz), 2.65 (t, 2H, J = 7 Hz), 7.10–7.21 (m, 2H), 7.31–7.40 (m, 2H including 7.35, d, 1H, J = 1.5 Hz). 13C NMR and DEPT (75 MHz, CDCl3) δ = 25.6 (CH2), 33.3 (CH2), 34.6 (CH2), 122.5 (C), 127.2 (CH), 129.3 (CH), 130.0 (CH), 133.6 (CH), 143.6 (C), 180.0 (C)
Synthesis of 4-(3-bromo-4-methoxyphenyl)-butyric acid
A solution bromine (0.52 ml, 10 mmol) in CHCl3 (10 ml) was added, drop-wise to a stirred solution of 4-(4-methoxyphenyl)-butyric acid (1.94 g, 10 mmol) in CHCl3 (30 ml) at ambient temperature and the stirring was continued for 1 h. The solvent was removed in vacuo and the residue triturated purified by column chromatography on silica, eluting with CH2Cl2–10% Et2O/CH2Cl2. and triturated with petrol to give 1.4 g of white solid (50%). Mpt = 78–80 °C; 1H NMR (300 MHz, CDCl3): δ 1.95 (quintet, 2H, J = 7 Hz), 2.39 (t, 2H, J = 7 Hz), 2.65 (t, 2H, J = 7 Hz), 7.10–7.21 (m, 2H), 7.31–7.40 (m, 2H including 7.35, d, 1H, J = 1.5 Hz). 13C NMR and DEPT (75 MHz, CDCl3) δ = 6.2 (CH2), 33.2 (CH2), 33.7 (CH2), 56.3 (CH3), 111.6 (C), 112.0 (CH), 128.4 (CH), 133.3 (CH), 134.9 (C), 154.3 (C), 180.0 (C).
General procedure for the synthesis of the amides
Oxalyl chloride (10 mmol) was added to a stirred solution of the appropriate acid (10 mmol) in DCM (30 mL) and 1–2 drops of DMF were added. After stirring at ambient temperatures for 2 h, by which time gas evolution had ceased, the solvent was removed by rotary evaporationin vacuo. The crude acid chloride was re-dissolved in DCM (20 mL) and was added, dropwise, to a stirred, cooled (0 °C) solution of 4-aminobutyraldehyde diethyl acetal (10 mmol) and Et2NMe (12 mmol) in Et2O (50 mL) over 5 min. The stirred reaction mixture was warmed to room temperature over 1 h. A 1 M aqueous solution of NaHCO3 (30 mL) was then added and the mixture transferred to s separating funnel, shaken then allowed to settle. The lower aqueous layer was separated and the organic layer washed with brine (30 mL). The organic layer was collected, dried (K2CO3), filtered and the filtrate concentrated in vacuo, then dried under high vacuum to give the essentially pure amides (>98% by HPLC) which were used without further purification.
2-Biphenyl-2-yl-N-(4,4-diethoxybutyl)acetamide (1).
Isolated as an oil (95% yield). 1H NMR (300 MHz, CDCl3): δ 1.17 (t, 6H, J = 7 Hz), 1.40–1.56 (m, 4H), 3.16 (dt, 2H, J = 7, 13 Hz), 3,37–3.66 (m, 4H), 3.52 (s, 2H), 4.42 (t, 1H, J = 5 Hz), 5.35 (brs, 1H), 7.24–7.48 (m, 9H): 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 30.8 (CH2), 39.3 (CH2), 41.4 (CH2), 61.3 (CH2), 102.5 (CH), 127.3 (CH), 127.4 (CH), 128.0 (CH), 129.1 (CH), 130.5 (CH), 130.7 (CH), 132.5 (C), 140.9 (C), 142.6 (C), 170.9 (C)
N-(4,4-Diethoxybutyl)-4-phenyl-butyramide (4).
Isolated as an oil (100% yield). 1H NMR (300 MHz, CDCl3): δ 1.17 (t, 6H, J = 6 Hz), 1.48–1.66 (m, 4H), 1.94 (quintet, 2H, J = 7 Hz), 2.13 (t, 2H, J = 7 Hz), 2.62 (t, 2H, J = 7 Hz), 3.22 (quartet, 2H, J = 6 Hz), 3.39–3.52 (m, 2H), 3.56–3.68 (m, 2H), 4.45 (t, 1H, J = 5.5 Hz), 5.82 (brs, 1H), 7.10–7.20 (m, 3H), 7.21–7.29 (m, 2H): 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 27.2 (CH2), 31.1 (CH2), 35.2 (CH2), 36.0 (CH2), 39.2 (CH2),61.5 (CH2), 102.7 (CH), 125.9 (CH), 128.4 (CH), 128.5 (CH), 141.5 (C), 172.7 (C).
N-(4,4-Diethoxybutyl)-4-(4-methylphenyl)-butyramide (4a).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3): δ = 1.19 (t, 6H, J = 7 Hz), 1.53–1.70 (m, 4H), 1.93 (quin., 2H, J = 8 Hz), 2.14 (t, 2H, J = 8 Hz), 2.31 (s, 3H), 2.60 (t, 2H, J = 8 Hz), 3.24 (quartet, 2H, J = 6 Hz), 3.43–3.55 (m, 2H), 3.57–3.70 (m, 2H), 4.47 (t, 1H, J = 5.5 Hz), 5.62 (brs, 1H), 7.04 (d, 2H, J = 5.5 Hz), 7.08 (d, 2H, J = 5.5 Hz): 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 21.0 (CH3), 24.6 (CH2), 27.3 (CH2), 31.1 (CH2), 34.8 (CH2), 36.0 (CH2), 39.2 (CH2), 61.5 (CH2), 102.7 (CH), 128.4 (CH), 129.1 (CH), 135.4 (C), 138.4 (C), 172.7 (C).
N-(4,4-Diethoxybutyl)-4-(3,4-dimethoxy)phenyl-butyramide (4b).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3): δ = 1.05–1.22 (m, 6H), 1.45–1.66 (m, 4H), 1.81–1.99 (m, 2H), 2.06–2.19 (m, 2H), 2.46–2.62 (m, 2H), 3.15–3.30 (m, 2H), 3.35–3.50 (m, 2H), 3.51–3.66 (m, 2H), 3.77–3.88 (m, 6H), 4.36–3.49 (m, 1H), 5.63–5.91 (brs, 1H), 6.59–6.80 (m, 3H). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 27.4 (CH2), 31.1 (CH2), 34.8 (CH2), 35.9 (CH2), 39.2 (CH2), 55.8 (CH3), 55.9 (CH3), 61.5 (CH2), 102.7 (CH), 111.2 (CH), 111.8 (CH), 120.3 (CH), 134.2 (C), 147.2 (C), 148.8 (C), 172.7 (C).
N-(4,4-Diethoxybutyl)-4-(2,5-dimethoxy)phenyl-butyramide (4c).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3): δ = 1.17 (t, 6H, J = 7 Hz), 1.52–1.66 (m, 4H), 1.89 (quintet, 2H, J = 7.5 Hz), 2.16 (t, 2H, J = 7.5 Hz), 2.60 (t, 2H, J = 7.5 Hz), 3.24 (q, 2H, J = 6 Hz), 3.43–3.50 (m, 2H), 3.58–3.75 (m, 2H), 3.73 (s, 3H), 3.75 (s, 3H), 4.45 (t, 1H, J = 5.5 Hz), 5.75 (brs, 1H). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.38 (CH3), 24.75 (CH2), 25.96 (CH2), 29.82 (CH2), 31.14 (CH2), 36.29 (CH2), 39.27 (CH2), 55.72 (CH3), 56.00 (CH3), 61.48 (CH2), 102.74 (CH), 111.26 (CH), 111.38 (CH), 116.42 (CH), 131.26 (C), 151.80 (C), 153.57 (C), 172.93 (C)
N-(4,4-Diethoxybutyl)-4-(4-chlorophenyl)-butyramide (4d).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3): δ = 1.18 (t, 6H, J = 7 Hz), 1.45–1.66 (m, 4H), 1.81–1.99 (m, 2H), 2.12 (t, 2H J = 7 Hz), 2.59 (t, 2H, J = 7 Hz), 3.25 (qt, 2H, J = 6.5 Hz), 3.38–3.53 (m, 2H), 3.56–3.68 (m, 2H), 4.44 (t, 1H, J = 7.5 Hz), 5.68 (brs, 1H), 7.08 (d, 2H, J = 8 Hz), 7.22 (d, 2H, J = 8 Hz). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 27.0 (CH2), 31.1 (CH2), 34.5 (CH2), 35.7 (CH2), 39.2 (CH2), 61.5 (CH2), 102.7 (CH), 128.4 (CH), 129.8 (CH), 131.6 (C), 140.0 (C), 172.4 (C).
N-(4,4-Diethoxybutyl)-4-(4-bromophenyl)-butyramide (4e).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3): δ = 1.17 (t, 6H, J = 7 Hz), 1.49–1.67 (m, 4H), 1.90 (quin, 2H, J = 7 Hz), 2.11 (t, 2H, J = 7 Hz), 2.57 (t, 2H, J = 7.5 Hz), 3.22 (quartet, 2H, J = 6.5 Hz), 3.39–3.52 (m, 2H), 3.56–3.69 (m, 2H), 4.45 (t, 1H, J = 5 Hz), 5.91 (brs, 1H), 7.02 (d, 2H, J = 8 Hz), 7.36 (d, 2H, J = 8 Hz). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 26.9 (CH2), 31.1 (CH2), 34.6 (CH2), 35.7 (CH2), 39.2 (CH2), 61.5 (CH2), 102.7 (CH), 119.7 (C), 130.2 (CH), 131.4 (CH), 140.5 (C), 172.4 (C).
N-(4,4-Diethoxybutyl)-4-(4-biphenyl)-butyramide (4f).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3): δ = 1.12 (t, 6H, J = 7 Hz), 1.45–1.66 (m, 4H), 1.99 (quintet, 2H, J = 7 Hz), 2.18 (t, 2H, J = 7 Hz), 2.68 (t, 2H J = 7 Hz), 3.25 (quartet, 2H, J = 6.5 Hz), 3.38–3.53 (m, 2H), 3.56–3.68 (m, 2H), 4.49 (t, 1H, J = 7.5 Hz), 5.83 (brs, 1H), 7.19–7.59 (m, 9H); 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.4 (CH3), 24.6 (CH2), 27.1 (CH2), 31.1 (CH2), 34.9 (CH2), 36.0 (CH2), 39.3 (CH2), 61.5 (CH2), 102.7 (CH), 127.0 (CH), 127.1 (CH), 128.7 (CH), 128.9 (CH), 138.9 (C), 140.7 (C), 141.0 (C), 172.7 (C).
N-(4,4-Diethoxybutyl)-4-(3-bromophenyl)-butyramide (4g).
Isolated as an oil (100%). 1H NMR (300 MHz, CDCl3) δ = 1.18 (t, 6H, J = 7 Hz), 1.49–1.67 (m, 4H), 1.92 (quin, 2H, J = 7 Hz), 2.13 (t, 2H, J = 7 Hz), 2.60 (t, 2H, J = 7.5 Hz), 3.24 (quartet, 2H, J = 6.5 Hz), 3.39–3.52 (m, 2H), 3.56–3.69 (m, 2H), 4.46 (t, 1H, J = 5 Hz), 5.69 (brs, 1H), 7.08–7.16 (m, 2H), 7.26–7.33 (m, 2H): 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 26.9 (CH2), 31.1 (CH2), 34.9 (CH2), 35.7 (CH2), 39.2 (CH2), 61.5 (CH2), 102.7 (CH), 122.4 (C), 127.2 (CH), 129.1 (CH), 130.0 (CH), 131.5 (CH), 143.9 (C), 172.3 (C).
N-(4,4-Diethoxybutyl)-4-(3-bromo-4-methoxyphenyl)-butyramide (4h).
Isolated as an oil (100%). 1H NMR (500 MHz, CDCl3): δ = 1.16 (t, 6H, J = 7 Hz), 1.51–1.65 (m, 4H), 1.88 (quin, 2H, J = 7 Hz), 2.11 (t, 2H, J = 7 Hz), 2.53 (t, 2H, J = 7.5 Hz), 3.24 (quartet, 2H, J = 6.5 Hz), 3.40–3.49 (m, 2H), 3.55–3.64 (m, 2H), 3.83 (s, 3H), 4.45 (t, 1H, J = 5 Hz), 5.76 (brs, 1H), 6.78 (d, 1H, J = 8.5 Hz), 7.04 (dd, 1H, J = 8.5, 2.0 Hz), 7.32 (d, 1H, J = 2.0 Hz).: 13C NMR and DEPT (125 MHz, CDCl3) δ = 15.38 (CH3), 24.62 (CH2), 27.23 (CH2), 31.13 (CH2), 34.00 (CH2), 35.80 (CH2), 39.28 (CH2), 56.33 (CH3), 61.56 (CH2), 102.73 (CH), 111.45 (C), 112.02 (CH), 128.46 (CH), 135.31 (CH), 135.31 (C), 154.17 (C), 172.58 (C).
N-(4,4-Diethoxybutyl)-4-(4-methoxyphenyl)-butyramide (4i).
Isolated as an oil (100%) 1H NMR (300 MHz, CDCl3): δ = 1.17 (t, 6H, J = 7 Hz), 1.45–1.66 (m, 4H), 1.89 (quintet, 2H, J = 7 Hz), 2.12 (t, 2H J = 7 Hz), 2.55 (t, 2H, J = 7 Hz), 3.22 (quartet, 2H, J = 6 Hz), 3.38–3.53 (m, 2H), 3.56–3.68 (m, 2H), 3.75 (s, 3H), 4.45 (t, 1H, J = 5 Hz), 5.79 (brs, 1H), 6.79 (d, 2H, J = 8 Hz), 7.06 (d, 2H, J = 8 Hz); 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 27.4 (CH2), 31.1 (CH2), 34.3 (CH2), 35.9 (CH2), 39.2 (CH2), 55.3 (CH3), 61.5 (CH2), 102.7 (CH), 113.8 (CH), 129.4 (CH), 133.6 (C), 157.8 (C), 172.8 (C).
N-(4,4-Diethoxybutyl)-4,4-diphenyl-butyramide
.
Isolated as an oil (88%) 1H NMR (300 MHz, CDCl3): δ = 1.18 (t, 6H, J = 7 Hz), 1.45–1.66 (m, 4H), 2.07 (t, 2H, J = 7 Hz_), 2.39 (qu, 2H, J = 7 Hz), 3.21 (quartet, 2H, J = 6.5 Hz), 3.38–3.53 (m, 2H), 3.56–3.68 (m, 2H), 3.91 (t, 1H, J = 8 Hz), 4.46 (t, 1H, J = 7.5 Hz), 5.57 (brs, 1H), 7.10–7.32 (m, 10H). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH), 31.1 (CH2), 31.2 (CH2), 35.0 (CH2), 39.2 (CH2), 50.6 (CH), 61.4 (CH2), 102.7 (CH), 126.3 (CH), 127.9 (CH), 128.5 (CH), 144.3 (C), 172.4 (C).
5-Phenyl-pentanoic acid (3,3-diethoxybutyl)-amide (9).
Isolated as an oil (95%): 1H NMR (300 MHz, CDCl3): δ = 1.18 (t, 6H, J = 7 Hz), 1.50–1.73 (m, 8 H), 2.15 (t, 2H, J = 7 Hz), 2.61 (t, 2H, J = 7 Hz), 3.23 (q, 2H, J = 6 Hz), 3.40–3.53 (m, 2H), 3.55–3.69 (m, 2H), 4.46 (t, 1H, J = 5 Hz), 5.71 (brs, 1H), 7.12–7.19 (m, 3H), 7.22–7.27 (m, 2H). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.6 (CH2), 25.4 (CH2), 31.1 (CH2), 35.7 (CH2), 36.7 (CH2), 39.2 (CH2), 61.5 (CH2), 102.7 (CH), 125.7 (CH), 128.3 (CH), 128.4 (CH), 142.2 (C), 172.8 (C)
N-(5,5-Diethoxypentyl)-4-phenyl-butyramide (10).
Isolated as an oil (100%) 1H NMR (300 MHz, CDCl3): δ = 1.18 (t, 6H, J = 7 Hz), 1.32–1.67 (m, 6H), 1.95 (quintet, 2H, J = 7.4 Hz), 2.15 (t, 2H, J = 7.4 Hz), 2.64 (t, 2H, J = 7.4 Hz), 3.22 (q, 2H, J = 6 Hz), 3.40–3.55 (m, 2H), 3.56–3.71 (m, 2H), 4.45 (t, 1H, J = 5.6 Hz), 5.50 (brs, 1H), 7.13–7.23 (m, 3H), 7.24–7.31 (m, 2H). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 22.1 (CH2), 27.2 (CH2), 29.5 (CH2), 33.3 (CH2), 35.2 (CH2), 36.0 (CH2), 39.4 (CH2), 61.1 (CH2), 102.8 (CH), 125.9 (CH), 128.4 (CH), 128.5 (CH), 141.5 (C), 172.6 (C).
2-Benzyl-N-(4,4-diethoxybutyl)benzamide (11a).
Isolated as an oil (100%) 1H NMR (300 MHz, CDCl3): δ = 1.16 (t, 6H, J = 7 Hz), 1.45–1.65 (m, 4H), 3.30 (q, 2H, J = 6 Hz), 3.35–3.50 (m, 2H), 3.53–3.68 (m, 2H), 4.17 (s, 2H), 4.43 (t, 1H, J = 5.3 Hz), 5.90 (brs, 1H), 7.07–7.37 (m, 9H). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.4 (CH2), 31.1 (CH2), 38.9 (CH2), 39.6 (CH2), 61.4 (CH2), 102.6 (CH), 126.0 (CH), 126.3 (CH), 127.1 (CH), 128.4 (CH), 129.0 (CH), 130.9 (CH), 137.0 (C), 138.8 (C), 140.9 (C), 170.0 (C).
N-(4,4-Diethoxybutyl)-2-phenoxybenzamide (11b).
Isolated as an oil (95%). 1H NMR (300 MHz, CDCl3): δ = 1.13 (t, 3H, J = 7 Hz), 1.14 (t, 3H, J = 7 Hz), 1.53–1.67 (m, 4H), 3.30–3.61 (m, 6H), 4.41–4.40 (m, 1H), 6.80 (dd, 1H, J = 8.2, 1.7 Hz), 7.00–7.04 (m, 2H), 7.13–7.20 (m, 2H), 7.29–7.41 (m, 3H), 7.69 (brs, 1H), 8.20 (dt, 1H, J = 7.8, 1.5 Hz). 13C NMR and DEPT (75 MHz, CDCl3) δ = 15.3 (CH3), 24.7 (CH2), 30.9 (CH2), 39.5 (CH2), 61.1 (CH2), 102.5 (CH), 118.4 (CH), 119.4 (CH), 123.7 (CH), 124.3 (C), 124.6 (CH), 130.1 (CH), 132.2 (CH), 132.5 (CH), 155.3 (C), 155.6 (C), 164.7 (C)
General procedure for the synthesis of the 2-hydroxypyrrolidino-lactams 7d–h
A solution of the 4,4-diethoxybutyl butyramide (3 mmol) in acetone (50 ml) and 1 M HCl (12 ml) was allowed to stand at room temperature for 30 min., by which time TLC showed no starting material remaining. The reaction mixture was basified with 1 M NaHCO3 solution (30 ml) and the acetone removed under reduced pressure on a rotary evaporator. The product was extracted from the aqueous residue with CH2Cl2 (3 × 50 ml). The combined organic extracts were dried (K2CO3), filtered and evaporated to give the 2-hydroxypyrrolidino-lactam, used without further purification. NMR analysis showed them to be a complex mixture of ∼20% aldehyde and amide rotomers.
General procedure for the triflic acid-mediated cyclisation
A solution of the acetal (5 mmol) in chloroform (10 mL) was added over 10 min to a heated (65 °C), stirred mixture of triflic acid (50 mmol) in chloroform (40 mL). The reaction was heated under gentle reflux for a given period. On cooling to ambient temperatures, water (20 ml) was added. The reaction mixture was transferred to a separating funnel and the lower layer separated. The aqueous layer was extracted with DCM (50 ml) and the combined organic extracts dried (K2CO3). Filtration and evaporationin vacuo gave the crude products that were separated by column chromatography on silica.
7-Phenyl-2,3,6,10b-tetrahydro-1H-pyrrolo[2,1-a]isoquinolin-5-one (2).
Reaction heated to reflux for 3 h, purified by elution with 1:1 CH2Cl2/Et2O to 2%MeOH/Et2O and fraction 2 isolated as a solid from trituration with Et2O (64%): mpt 157–9 °C. HRMS Theoretical Mass: 263.12551, found: 263.12589. 1H NMR (300 MHz, CDCl3): δ = 1.91–2.25 (m, 3H), 2.61–2.79 (m, 1H), 3.41–3.55 (m, 2H), 3.63 (d, 1H, J = 18 Hz), 3.66–3.77 (m, 1H), 4.63–4.77 (m, 1H), 7.15–7.45 (m, 8H). 13C NMR and DEPT (75 MHz, CDCl3): δ = 23.4 (CH2), 31.7 (CH2), 37.0 (CH2), 44.8 (CH2), 59.7 (CH), 123.2 (CH), 126.6 (CH), 127.4 (CH), 128.4 (CH), 129.1 (CH), 129.2 (CH), 130.8 (C), 136.8 (C), 140.2 (C), 140.9 (C), 167.7 (C).
1,2,3,6,10b-Pentahydro-1H-pyrrolo[2,1-a]-dibenzo[c,e]azocin-5-one (3).
Reaction heated to reflux for 3 h, purified by elution with 1:1 CH2Cl2/Et2O to 2%MeOH/Et2O and as fraction 1 isolated as an oil (29%). HRMS Theoretical Mass: 263.12551: found: 263.12567. 1H NMR (300 MHz, CDCl3): δ = 1.66–1.86 (m, 1H), 1.98–2.20 (m, 2H), 2.35–2.50 (m, 1H), 3.12 (d, 1H, J = 15 Hz), 3.57 (d, 1H, J = 15 Hz), 3.60–3.80 (m, 2H), 4.27 (t, 1H, J = 8 Hz), 7.25–7.55 (m, 8H). 13C NMR and DEPT (75 MHz, CDCl3): δ = 23.4 (CH2), 33.6 (CH2), 43.3 (CH2), 48.4 (CH2), 53.5 (CH2), 56.7 (CH), 126.0 (CH), 127.5 (CH), 127.7 (CH), 127.9 (CH), 128.3 (CH), 128.5 (CH), 128.6 (CH), 130.0 (CH), 132.6 (C), 137.1 (C), 141.2 (C), 142.6 (C), 168.7 (C).
6-Aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5).
Reaction heated to reflux for 4 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as an oil (70% yield). HRMS theoretical 215.13047, found 215.13000: FTIR (film) 2947, 2873, 1624, 1455, 1420, 754 cm−1.
14-Methyl-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5a).
Reaction heated to reflux for 4 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as an oil (70% yield). HRMS Theoretical Mass: 229.14611, found 229. 14545
13,14-Dimethoxy-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5b).
From the acetal at reflux for 1 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as a white solid, recrystallised from EtOAc/petrol (80% yield), m. pt. 116–7 °C. HRMS Theoretical Mass: 275.15160, found 275.15154
12,15-Dimethoxy-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11), 12,14-trien-7-one (5c).
Isolated from the acetal at room temperature for 21 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as a white solid, recrystallized from EtOAc/petrol (26% yield), m. pt. 126–7 °C. HRMS Theoretical Mass: 275.15160, found 275.15102. FTIR (solid) 1634, 1471, 1417, 1252, 1141, 1090, 784, 718 cm−1.
14-Chloro-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5d).
Prepared from the 2-hydroxypyrrolidine, heated to reflux for 18 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as an oil (59% yield). HRMS: Theoretical Mass: 249.09149, found 249.09081
14-Bromo-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5e).
Prepared from the 2-hydroxypyrrolidine, heated to reflux for 18 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as an oil (55% yield). HRMS Theoretical Mass: 293.04098, found 293.03969.
14-Phenyl-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5f).
Prepared from the 2-hydroxypyrrolidine, heated to reflux for 18 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as an oil (45% yield). HRMS Theoretical Mass: 291.16177; found: 291.16191
13-Bromo-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5g).
From the 2-hydroxypyrrolidine, heated to reflux for 18 h, purified by elution with Et2O-4% MeOH/Et2O, isolated as white solid (Et2O/petrol) (60% yield). HRMS Theoretical Mass: 293.04098: found 293.04037. 1H NMR (500 MHz, CDCl3) at 332 K: δ = 1.68–1.80 (brm, H-9), 1.87–2.11 (m, H-3,4,4,9), 2.26–2.35 (brm, H-8), 2.40–2.52 (m, H-3, 8), 2.61–2.70 (brm, H-10), 2.96 (ddd, H-10, J = 3.8, 11.9, 13.8 Hz), 3.40–3.50 (brm, H-5), 3.90 (ddd, H-5, J = 4.1, 8.6, 12.4 Hz), 5.01 (t, H-2, J = 7 Hz), 7.02 (d, H-15, J = 8.3 Hz), 7.25 (d, H-12, J = 2.2 Hz), 7.32 (dd, H-14, J = 2.2, 8.3 Hz). 13C NMR and DEPT (150 MHz, CDCl3) at 332 K: δ = 22.60, 26.99, 32.22, 33.48, 36.79, 45.88, 62.34 (2-C), 120.99 (C-13), 128.54 (C-15), 129.50 (C-14), 134.01 (C-12), 139.54 (C-1), 140.27 (C-11), 172.14 (C-7)
13-Bromo-14-methoxy-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5h).
Isolated from the acetal at reflux for 1 h, purified by elution with Et2O-2% MeOH/Et2O, isolated as a white solid, recrystallized from EtOAc/petrol (80% yield), m. pt. 116–7 °C. HRMS Theoretical Mass: 323.05154, found 323.05138. 1H NMR (600 MHz, CDCl3) at 331 K: δ = 1.64–1.77 (brm, 1H), 1.87–2.03 (m, 3H), 2.04–2.13 (m, 1H), 2.26–2.34 (brm, 1H), 2.41–2.52 (m, 2H), 2.54–2.2 (brm, 1H), 2.89 (ddd, 1H, J = 3.8, 12.1, 14 Hz), 3.46 (dt, 1H, J = 8.0, 12.0 Hz), 3.86 (s, 3H), 3.89 (ddd, 1H-5, J = 12.1, 8.6, 4.2 Hz), 5.00 (t, 1H, J = 7.1 Hz), 6.66 (s, 1H), 7.26 (s, 1H). 13C NMR and DEPT (150 MHz, CDCl3) at 333 K: δ = 22.86 (CH2), 27.62 (CH2), 31.24 (CH2), 33.43 (CH2), 46.30 (CH2), 56.61 (CH3), 63.01 (CH), 110.68 (C), 111.19 (CH), 131.89 (C), 136.06 (CH), 141.04 (C), 154.65 (C), 172.69 (C).
6-Aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(15),11,13-triene (6).
A solution of 6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (0.9 g, 4.2 mmol) in dry THF (5 ml) was added, drop-wise, to a stirred solution of 1 M LiAlH4 (4.2 ml, 4.2 mmol) in dry THF (20 ml) and the reaction mixture heated to reflux for 2 h. On cooling to 0 °C, 2 M NaOH (0.6 ml) was carefully added, drop-wise, the reaction mixture stirred for 30 min. and then Et2O (50 ml) was added. The white solid was removed, washed with Et2O (2 × 50 ml) and the combined organics dried (K2CO3), filtered and concentrated in vacuo to give the title compound as an oil (0.8 g, 88% yield). HRMS Theoretical Mass: 201.15120, found 201.15131. Attempts to form an HCl salt resulted in a sticky, hygroscopic gum. A sample was treated with 1 equivalent of picric acid (35% water) in ethanol to give the picrate, (m pt 162–4 °C)
(2R,10R)(2S,10S)-10-Phenyl-6-aza-tricyclo[9.4.0.0*2,6*] pentadeca-1(15),11,13-triene-7-one (8).
From the acetal at reflux for 3 h, purified by elution with Et2O and the solid triturated with petrol (73%), a small sample was re-crystallized from CH2Cl2/petrol, m. pt. 162–4 °C. 1H NMR (500 MHz, CDCl3) at 332 K: δ = 1.75–2.60 (m, 7H), 3.60–3.79 (m, 1H), 3.87 (dt, 1H, J = 9.5, 2.4 Hz), 4.53 (dd, 1H, J = 12.3, 3.2 Hz), 4.93 (dd, 1H, J = 10.9, 5.1 Hz), 6.71 (d, 1H, J = 6.8 Hz), 7.05–7.45 (m, 8H). 13C NMR and DEPT (125 MHz, CDCl3) at 332 K: δ = 22.4 (CH2), 31.2 (CH2), 32.7 (CH2), 39.5 (CH2), 43.3 (CH), 46.9 (CH2), 65.8 (CH), 126.5 (CH), 126.6 (CH), 127.7 (CH), 127.9 (CH), 128.5 (CH), 128.7 (CH), 129.2 (CH), 138.9 (C), 141.0 (C), 142.9 (C), 172.3 (C)
4b,5,6,7-Tetrahydro-14H-dibenzo[c,f]pyrrolo[1,2-a]azocin-9-one (12a).
From the acetal at reflux for 3 h, purified by elution with CH2Cl2–20% Et2O/CH2Cl2, isolated as a white solid, recrystallised from EtOAc/petrol (95% yield), m. pt. 127–9 °C. HRMS Theoretical Mass: 263.13046, found 263. 12983. 1H NMR (300 MHz, CDCl3): δ = 1.80–2.17 (m, 3H), 2.55–2.71 (m, 1H), 3.23–3.30 (m, 1H), 3.79 (d, 1H J = 14.5 Hz), 3.99–4.13 (m, 1H), 4.3 (d, 1H, J = 14.5 Hz), 4.67 (d, 1H, J = 5.5 Hz), 7.12–7.41 (m, 7H), 7.52 (dd, 1H, J = 7, 1 Hz). 13C NMR and DEPT (75 MHz, CDCl3): δ = 23.0 (CH2), 29.0 (CH2), 41.0 (CH2), 44.4 (CH2), 60.6 (CH), 125.2 (CH), 125.6 (CH), 127.1 (CH), 127.2 (CH), 127.9 (CH), 129.6 (CH), 130.6 (CH), 131.2 (CH), 134.9 (C), 136.4 (C), 138.0 (C), 139.2 (C), 171.0 (C).
4b,5,6,7-Tetrahydro-14-oxa-dibenzo[c,f]pyrrolo[1,2-a]azocin-9-one (12b).
From the acetal at reflux for 2 h, purified by elution with CH2Cl2–10% Et2O/CH2Cl2, (66% yield), m. pt. 144–6 °C. HRMS Theoretical Mass: 265.10973, found 265.10973. 1H NMR (600 MHz, CDCl3): δ = 1.85–1.96 (m, 6–1H), 1.96–2.04 (m, 5–1H), 2.04–2.10 (m, 6–1H), 2.62 (dd, 5–1H, J = 5.3, 12.8 Hz), 3.40–3.48 (m, 7–1H), 3.87–3.95 (m, 7–1H), 4.72 (d, 4d-1H, J = 6.8 Hz), 7.10–7.16 (m, 2H), 7.22 (d, 1H, J = 7.9 Hz), 7.27 (t, 1H, J = 7.9 Hz), 7.32–7.48 (m, 2H), 7.42 (dt, 1H, J = 7.9, 1.2 Hz), 7.58 (dd, 1H, J = 7.6, 1.2 Hz). 13C NMR and DEPT (150 MHz, CDCl3): δ = 23.18 (6-CH2), 28.75 (5-CH2), 45.17 (7-CH2), 58.79 (4b-CH), 122.57 (CH), 122.74 (CH), 124.71 (CH), 125.99 (CH), 126.29 (CH), 127.52 (CH), 129.22 (C), 129.50 (CH), 132.23 (CH), 132.61 (C), 152.82 (C), 157.63 (C), 168.08 (C).
(4,4-Diethoxy-butyl)-(4-phenyl-butyl)-amine (13).
A solution of the amide 4 (2.0 g, 6.5 mmol) in dry THF (20 ml) was added to a stirred solution of 1 M LAH (30 ml, 30 mmol) in THF (30 ml) under argon at 0 °C and the reaction was heat under gentle reflux for 24 h. The reaction mixture was cooled to 0 °C and H2O (1 ml), then 2 M NaOH (2 ml) were carefully add and the reaction mixture stirred until a white solid formed. Et2O (50 ml) was then addded, the solid collected which was thoroughly washed with Et2O (4 × 25 ml). The combined organic filtrate and washings were dried (K2CO3), filter and concentrate in vacuo to give a colourless oil: 1.9 g (∼100%). 1H NMR (500 MHz, CDCl3): δ = 1.19 (t, 6H, J = 7 Hz), 1.50–1.57 (m, 4H), 1.61–1.70 (m, 4H), 2.58–2.65 (m, 6H), 3.45–3.54 (m, 2H), 3.59–3.67 (m, 2H), 4.48 (t, 1H, J = 5.7 Hz), 7.14–7.18 (m, 3H), 7.24–7.28 (m, 2H); 13C NMR and DEPT (125 MHz, CDCl3): δ = 15.41 (CH3), 25.51 (CH2), 29.32 (CH2), 29.83 (CH2), 31.54 (CH2), 35.92 (CH2), 49.89 (CH2), 49.94 (CH2), 61.06 (CH2), 61.16 (CH2), 102.90 (CH), 125.74 (CH), 128.40 (CH), 128.53 (CH), 142.56 (CH).
Crystal data and structure refinement
12,15-Dimethoxy-6-aza-tricyclo[9.4.0.0*2,6*]pentadeca-1(11),12,14-trien-7-one (5c).
Chemical formula C16H21NO3; Formula weight 275.34; Temperature 150(2) K; Radiation, wavelength MoKα, 0.71073 Å; Crystal system, space group monoclinic, P21/c; Unit cell parameters a = 8.3263(9) Å; α = 90°; b = 18.599(2) Å; β = 107.795(2)°; c = 9.2541(10) Å; γ = 90°; Cell volume 1364.5(3) Å3 Z 4; Calculated density 1.340 g/cm3; Absorption coefficient μ 0.092 mm−1; F(000) 592; Crystal colour and size colourless, 0.40 × 0.12 × 0.02 mm3; Data collection method Bruker SMART APEX CCD diffractometer ω rotation with narrow frames; θ range for data collection 2.56 to 28.27°; Index ranges h −10 to 11, k −24 to 24, l −12 to 12; Completeness to θ = 26.00° 99.4%; Reflections collected 11463; Independent reflections 3261 (Rint = 0.0276); Reflections with F2>2σ 2760; Absorption correction semi-empirical from equivalents; Min. and max. transmission 0.9641 and 0.9982; Structure solution direct methods; Refinement method Full-matrix least-squares on F2; Weighting parameters a, b 0.0681, 0.3291; Data/restraints/parameters 3261/0/181; Final R indices [F2>2σ] R1 = 0.0439, wR2 = 0.1168; R indices (all data) R1 = 0.0519, wR2 = 0.1221; Goodness-of-fit on F2 1.066; Largest and mean shift/su 0.000 and 0.000; Largest diff. peak and hole 0.343 and −0.260 e Å−3.
2R,10R)(2S,10S)-10-Phenyl-6-aza-tricyclo[9.4.0.0*2,6*]-pentadeca-1(15),11,13-triene-7-one (8).
Chemical formula C20H21NO; Formula weight 291.38; Temperature 150(2) K; Radiation, wavelength MoKα, 0.71073 Å; Crystal system, space group monoclinic, P21/c; Unit cell parameters a = 7.6569(8) Åα = 90°, b = 9.6582(11) Åβ = 90.423(2)°, c = 20.404(2) Åγ = 90°; Cell volume 1508.9(3) Å3; Z 4; Calculated density 1.283 g/cm3; Absorption coefficient μ 0.078 mm−1; F(000) 624; Crystal colour and size colourless, 0.48 × 0.46 × 0.43 mm3; Data collection method Bruker SMART APEX CCD diffractometer ω rotation with narrow frames; θ range for data collection 3.34 to 28.29°; Index ranges h −10 to 10, k −12 to 12, l −26 to 26; Completeness to θ = 26.00° 99.3%; Reflections collected 12268; Independent reflections 3580 (Rint = 0.0327); Reflections with F2>2σ 3271; Absorption correction semi-empirical from equivalents; Min. and max. transmission 0.9634 and 0.9671;Structure solution direct methods; Refinement method Full-matrix least-squares on F2; Weighting parameters a, b 0.0720, 0.4836; Data/restraints/parameters 3580/0/200; Final R indices [F2>2σ] R1 = 0.0440, wR2 = 0.1201; R indices (all data) R1 = 0.0473, wR2 = 0.1231; Goodness-of-fit on F2 1.041; Extinction coefficient 0.027(4); Largest and mean shift/su 0.000 and 0.000; Largest diff. peak and hole 0.402 and −0.248 e Å−3.
4b,5,6,7-Tetrahydro-14H-dibenzo[c,f]pyrrolo[1,2-a]azocin-9-one (12a).
Chemical formula C18H17NO; Formula weight 263.33; Temperature 150(2) K; Radiation, wavelength MoKα, 0.71073 Å; Crystal system, space group triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; Unit cell parameters a = 9.3106(8) Åα = 75.9490(10)° b = 10.4150(9) Åβ = 88.8050(10)° c = 16.0747(14) Åγ = 63.5860(10)°; Cell volume 1347.4(2) Å3 Z 4; Calculated density 1.298 g/cm3; Absorption coefficient μ 0.080 mm−1; F(000) 560; Crystal colour and size colourless, 0.32 × 0.28 × 0.18 mm3; Data collection method Bruker SMART APEX CCD diffractometer ω rotation with narrow frames θ range for data collection 2.63 to 28.30°; Index rangesh −12 to 12, k −13 to 13, l −20 to 21; Completeness to θ = 26.00° 97.7%; Reflections collected 11524; Independent reflections 6146 (Rint = 0.0221); Reflections with F2>2σ 5332; Absorption correction semi-empirical from equivalents; Min. and max. transmission 0.9748 and 0.9857; Structure solution direct methods; Refinement method Full-matrix least-squares on F2; Weighting parameters a, b 0.1039, 0.3159; Data/restraints/parameters 6146/0/361; Final R indices [F2>2σ] R1 = 0.0520, wR2 = 0.1554; R indices (all data) R1 = 0.0583, wR2 = 0.1641. Goodness-of-fit on F2 1.079; Largest and mean shift/su 0.000 and 0.000; Largest diff. peak and hole 0.476 and −0.433 e Å−3.
; Unit cell parameters a = 9.3106(8) Åα = 75.9490(10)° b = 10.4150(9) Åβ = 88.8050(10)° c = 16.0747(14) Åγ = 63.5860(10)°; Cell volume 1347.4(2) Å3 Z 4; Calculated density 1.298 g/cm3; Absorption coefficient μ 0.080 mm−1; F(000) 560; Crystal colour and size colourless, 0.32 × 0.28 × 0.18 mm3; Data collection method Bruker SMART APEX CCD diffractometer ω rotation with narrow frames θ range for data collection 2.63 to 28.30°; Index rangesh −12 to 12, k −13 to 13, l −20 to 21; Completeness to θ = 26.00° 97.7%; Reflections collected 11524; Independent reflections 6146 (Rint = 0.0221); Reflections with F2>2σ 5332; Absorption correction semi-empirical from equivalents; Min. and max. transmission 0.9748 and 0.9857; Structure solution direct methods; Refinement method Full-matrix least-squares on F2; Weighting parameters a, b 0.1039, 0.3159; Data/restraints/parameters 6146/0/361; Final R indices [F2>2σ] R1 = 0.0520, wR2 = 0.1554; R indices (all data) R1 = 0.0583, wR2 = 0.1641. Goodness-of-fit on F2 1.079; Largest and mean shift/su 0.000 and 0.000; Largest diff. peak and hole 0.476 and −0.433 e Å−3.
Acknowledgements
The authors would like to thank GSK for funding and L. Harris for the mass spectra.
References
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS , and references quoted therein.
-
E. J. Ariens, A. J. Beld, J. F. Rodrigues de Miranda and A. M. Simonis, in The Receptors A Comprehensive Treatise, R. D. O'Brien, Ed.; Plenum Press, New York, 1979, p 33 Search PubMed.
- P. R. Andrews and E. J. Lloyd, Med. Res. Rev., 1982, 2, 355 CrossRef CAS.
- E. S. Vermeulen, M. van Smeden, A. W. Schmidt, J. S. Sprouse, H. V. Wikström and C. J. Grol, J. Med. Chem., 2004, 47, 5451 CrossRef CAS.
- M. Dukat, M. Taroua, A. Dahdouh, U. Siripurapu, D. Imad, B. R. Martin and R. A. Glennon, Bioorg. Med. Chem. Lett., 2004, 14, 3651 CrossRef CAS.
- J. M. Keith, L. A. Gomez, R. L. Wolin, A. J. Barbier, S. J. Wilson, J. D. Boggs, C. Mazur, I. C. Fraser, B. Lord, L. Aluisio, T. W. Lovenberg and N. I. Carruthers, Bioorg. Med. Chem. Lett., 2007, 17, 702 CrossRef CAS.
- C. Ma, S.-J. Liun, L. Xin, Q. Zhang, K. Ding, J. R. Falck and D.-S. Shin, Chem. Letts., 2006, 35, 1010 Search PubMed.
- J. W. Lane, A. Estevezc, K. Mortarac, O. Callanc, J. R. Spencer and R. M. Williams, Bioorg. Med. Chem. Lett., 2006, 16, 3180 CrossRef CAS.
- N. E. Austin, K. Y. Avenell, I. Boyfield, C. L. Branch, M. C. Coldwell, M. S. Hadley, P. Jeffrey, A. Johns, C. N. Johnson, D. J. Nash, G. J. Riley, S. A. Smith, R. C. Stacey, G. Stemp, K. M. Thewlis and A. K. Vong, Bioorg. Med. Chem. Lett., 1999, 9, 179 CrossRef CAS.
- W. L. Huang, X. Q. Song, S. X. Peng and Z. Y. Huang, Yaoxue Xuebao, 1990, 25, 815 CAS.
- A. J. Bojarski, M. J. Mokrosz, S. C. Mino, A. Koziol, A. Wesolowska, E. Tatarczynska, A. Klodzinska and E. Chojnacka-Wojcik, Bioorg. Med. Chem., 2002, 10, 87 CrossRef CAS.
- B. E. Maryanoff, D. F. McComsey, J. F. Gardocki, R. P. Shank, M. J. Costanzo, S. O. Nortey, C. R. Schneider and P. E. Setler, J. Med. Chem., 1987, 30, 1433 CrossRef CAS.
- G. Stemp, T. Ashmeade, C. L. Branch, M. S. Hadley, A. J. Hunter, C. N. Johnson, D. J. Nash, K. M. Thewlis, A. K. Vong, N. E. Austin, P. Jeffrey, K. Y. Avenell, I. Boyfield, J. J. Hagan, D. N. Middlemiss, C. Reavill, G. J. Riley, C. Routledge and M. Wood, J. Med. Chem., 2000, 43, 1878 CrossRef CAS.
- F. D. King, Tetrahedron, 2007, 63, 2053 CrossRef CAS.
- L. Gang, R. Franzen, Q. Zhang and Y. Xu, Tetrahedron Lett., 2005, 46, 4255 CrossRef.
- B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431 CrossRef CAS.
- G. Hilt, F. Galbiati and K. Harms, Synthesis, 2006, 3575 CrossRef CAS.
-
Antidepressants: Past, Present and Future, in Handbook of Experimental Pharmacology, S. Preskorn, C. Y. Stanga, J. P. Feighner, and R. Ross, (eds.) 2004, 157, Springer Search PubMed.
- Z. F. Solomko and A. N. Kost, Chem. Het. Cpds., 1975, 11, 1231 Search PubMed.
- G. Flynn, E. Giroux and R. Dage, J. Am. Chem. Soc., 1987, 109, 7914 CrossRef CAS.
- G. L. Grunwald, V. H. Dahanukar, P. Ching and K. R. Criscione, J. Med. Chem., 1996, 39, 3539 CrossRef CAS.
- H. Fuwa, Y. Okamura, Y. Morohashi, T. Tomita, T. Iwatsubo, T. Kan, T. Fukuyama and H. Natsugari, Tetrahedron Lett., 2004, 45, 2323 CrossRef CAS.
- S. Bergemann, R. Brecht, F. Büttner, D. Guénard, R. Gust, G. Seitz, M. T. Stubbs and S. Thoret, Bioorg. Med. Chem., 2003, 11, 1269 CrossRef CAS.
- S. Seto, A. Tanioka, M. Ikeda and S. Izawa, Bioorg. Med. Chem., 2005, 13, 5717 CrossRef CAS.
- D. H. Jones, G. F. Stephenson, G. W. Spray and W. R. Wragg, J. Chem. Soc. C, 1969, 2176 RSC.
- L. A. Paquette, L. B. Anderson, J. F. Hansen, S. A. Lang, Jr. and H. Berk, J. Am. Chem. Soc., 1972, 94, 4907 CrossRef CAS.
- A. M. Monro, R. M. Quinton and T. I. Wrigley, J. Org. Chem., 1963, 28, 255.
- M. Qadir, J. Cobb, P. W. Sheldrake, N. Whittall, A. J. P. White, K. K. Hii, P. N. Horton and M. B. Hursthouse, J. Org. Chem., 2005, 70, 1552 CrossRef CAS.
- J.-L. Panayides, R. Pathak, H. Panagiotopoulos, H. Davids, M. A. Fernandes, C. B. de Koning and W. A. L. van Otterlo, Tetrahedron, 2007, 63, 4737 CrossRef CAS.
- O. O. Orazi, R. A. Corral and H. Giacco, J. Chem. Soc., Perkin Trans. 1, 1986, 1977 RSC.
- R. A. Corral, Monograf. Acad, Nac. Cien. Exact. Fis. Nat., 1988, 3, 37–42 Search PubMed.
- J. Zinczuk, I. H. Sorokin, O. Orfeo and R. A. Corral, J. Het. Chem., 1992, 29, 859 Search PubMed.
- M. Mizukami, H. Saito, T. Higuchi, M. Imai, H. Bando, N. Kawahara and S. Nagumo, Tetrahedron Lett., 2007, 48, 7228 CrossRef CAS.
- Y. Zhang, D. J. DeSchepper, T. M. Gilbert, K. K. S. Sai and D. A. Klumpp, Chem. Commun., 2007, 4032 RSC.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546 CrossRef CAS.
- A. E. Aliev and D. Courtier-Murias, J. Phys. Chem. B, 2007, 111, 14034 CrossRef CAS.
- C. A. G. Haasnoot, F. A. A. M. DeLeeuw, H. P. M. DeLeew and C. Altona, Biopolymers, 1981, 20, 1211 CAS.
- K. D. Moeller, P. W. Wang, S. Tarazi, M. R. Marzabadi and P. L. Wong, J. Org. Chem., 1991, 56, 1058 CrossRef CAS.
- H. E. Zaugg, Synthesis, 1984, 85 Search PubMed.
- D. Stephenson and G. J. Binsch, Magn. Reson., 1980, 37, 395 Search PubMed;
G. Hägele, M. Engelhardt, and M. Boenigk, Simulation und automatisierte Analyse von Kernresonanzspektren, VCH, Weinheim, 1987 Search PubMed;
gNMR, Version 5.0.6, NMR Simulation Program, Budzelaar PHM, 2006 Search PubMed.
- H. Shanan-Atidi and K. H. Bar-Eli, J. Phys. Chem., 1970, 74, 961 CrossRef CAS.
-
M. J. Chapdeleine, and D. McLaren, GB 2251616A (1992) CA 117, 212302.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 