Antimicrobial activity, biocompatibility and hydrogelation ability of dipeptide-based amphiphiles†
Rajendra Narayan Mitra, Anshupriya Shome, Pritha Paul and Prasanta Kumar Das‡*
Department of Biological Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata, 700 032, India. E-mail: bcpkd@iacs.res.in; Fax: +(91)-33-24732805
First published on 5th November 2008
Abstract
The development of new antibiotics is of increasing importance due to the growing resistance power of microbes against conventional drugs. To this end, cationic peptides are emerging as clinically potent antimicrobial agents. In the present study, we have synthesized six dipeptide-based cationic amphiphiles with different head group structures by varying combinations of L-amino acid residues. These amphiphiles showed remarkable growth inhibiting activity on several Gram-positive (minimum inhibitory concentration (MIC) = 0.1–10 µg/mL) and Gram-negative (MIC = 5–150 µg/mL) bacteria as well as on fungus (MIC = 1–50 µg/mL). The inherent antimicrobial efficacies of these cationic dipeptides were influenced by the head group architecture of the amphiphiles. Encouragingly, these amphiphiles selectively attacked microbial cells, while showing biocompatibility toward mammalian cells. The results show that the rational designing of short peptide-based cationic amphiphiles might serve as a promising strategy in the development of antimicrobial agents with greater cell specificities. In addition, the amphiphiles showed water gelation ability at room temperature. The formation of non-covalent supramolecular networks in gelation was established by microscopic and spectroscopic studies.
Introduction
The design and synthesis of new antimicrobial agents is getting exponentially important in medicinal chemistry due to the increasing resistance power of microbes to conventional antibiotics.1–3 This resistance arises from newly acquired genes, which encode a variety of resistance mediating proteins.4 However, microbes with disrupted cell membranes are not expected to reform and hence lose their resistance ability.1 In this regard, cationic antimicrobial peptides (AMPs) are emerging as potent antimicrobial agents owing to their important role in the innate defense mechanism against microbes.5,6 The naturally derived AMPs are generally composed of ∼ 12–50 amino acid based peptides with a net positive charge due to which it can attack the negatively charged cell membrane.1 In addition, the AMPs also require an optimum hydrophobicity known as ‘threshold hydrophobicity’ to exhibit their effectiveness at killing microbes.7,8 The optimal hydrophobicity may be attained by the presence of a critical number of hydrophobic amino acids in peptide chains or by the incorporation of long alkyl chains in cationic peptide amphiphiles. Hence, a perfect amphipathicity resulting from the optimal combination of cationic charge and hydrophobic residue was found to be crucial for the antimicrobial activity of AMPs.9 The AMPs permeate and disintegrate the cell membrane probably because of their successful incorporation into the hydrophobic lipid bilayer leading to cell death.1,8 In spite of the excellent antimicrobial activity of AMPs there are some hurdles in their use as effective drugs. The AMPs are not generally cell-specific i.e. they are also toxic to mammalian cells along with the microbes.5,8 In addition, synthetic mimics of AMPs should contain short peptide chains to avoid low metabolic stabilitys and high production costs.10,11 To date, there are few reports on the antimicrobial activity of short peptides.1,10,12 The cell specificity of these AMPs is determined by the sequence of the short peptide moiety. Most of these peptides are comprised of amino acids like lysine and/or arginine that get protonated easily at biological pH and thus can interact with the negatively charged microbial cell membrane. However, if we can design a short peptide that already contains cationic charges, variation in the peptide structure can be substantially increased using combinations of different categories (polar, non-polar) of amino acids as the presence of arginine or lysine is no longer mandatory. Thus the design of cationic amphiphiles based on short peptide sequences with diversified cell specificities is the prime requirement for the development of biocompatible antimicrobial agents. In this context, cationic amphiphilic compounds are quite well known to exhibit antimicrobial activity13 but most of them are also cytotoxic to eukaryotic cells. In a recent report, we showed the selective antibacterial activity of L-tryptophan based cationic biocompatible amphiphiles.14 However for the diversified cell specificities, biomedicinal chemistry and the allied research front is demanding a greater number of structurally varied short peptides as antimicrobial agents.In our present work, we report the synthesis of six dipeptide-based amphiphiles with varying head group architecture (1–6, Fig. 1). These amphiphiles showed inherent growth-inhibiting activity on several Gram-positive and Gram-negative bacteria, as well as on fungus. The antimicrobial efficiency was dependent on the head-group structure particularly with regard to Gram-negative bacteria and fungus. The most important feature of these amphiphiles is their biocompatibility with different mammalian cell lines like HepG2, HeLa and SiHa. Thus, rational design of small peptide-based cationic amphiphiles by head-group modulation led to the development of inherently antimicrobial amphiphiles with low toxicity to mammalian cells.
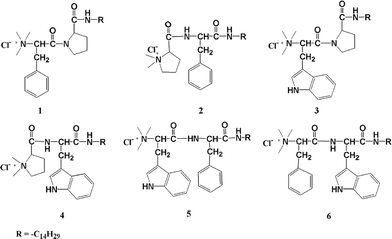 | ||
| Fig. 1 Structure of different amphiphiles 1–6. | ||
Interestingly, these excellent antimicrobial peptide-based amphiphiles also exhibit water gelation ability at room temperature with varying efficiency (minimum gelation concentration; MGC = 1.2–22%, w/v), similar to their C-16 analogues.15 The hydrogelation ability of the amphiphiles and their supramolecular self-assembly was investigated by microscopic and spectroscopic techniques (ESI†).
Results and discussion
The development of new antimicrobial agents having broad-spectrum activity against bacteria and fungi with considerable biocompatibility is finding tremendous importance in medicinal chemistry. AMP is an important class of antimicrobial agent with a net positive charge and optimum hydrophobicity that destroys the cell membrane of microbes resulting in cell-death.1,7,8 A synthetic mimic of these AMPs and/or lipopeptide, such as peptide based cationic amphiphiles, may also act as antimicrobial agents. To this end, our recent study on the alkyl chain length dependent antimicrobial efficacy of the single amino acid-based cationic amphiphile showed optimum antibacterial activity with C-14 chain length.14 Thus, the development of C-14 aliphatic chains containing short peptide-based antimicrobial cationic amphiphiles with greater cell specificities is the key aim of the present study. Hence, we synthesized six dipeptide-based cationic amphiphiles that showed strong antimicrobial activity against several Gram-positive and Gram-negative bacteria, as well as fungus (1–6, Fig. 1) but low toxicity to mammalian cells. A very simple synthetic route was used to prepare these amphiphiles as discussed in our previous report.15,16 We modulated the head group structure of the amphiphiles by changing the amino acids of the dipeptide moiety. An attempt has been made to correlate the antimicrobial activity with the varying head group structure of the dipeptide amphiphiles.The antibacterial activity of the dipeptide amphiphiles was investigated against three strains of Gram-positive bacteria (Bacillus subtilis, Micrococcus luteus, and Staphylococcus aureus) and four Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa, Klebsiella aerogenes and Klebsiella pneumoniae). The minimum inhibitory concentrations (MICs), the lowest concentration of amphiphile at which no viable cells are present, are reported in Table 1. The observed MIC values by both broth dilution and the spread plate method are comparable. The killing efficiency of amphiphiles 1–5 against Gram-positive bacteria, B. subtilis, M. luteus, and S. aureus was very high as the observed MIC values were very low, 0.1–1 µg/mL. The amphiphile 6 showed slightly higher MIC values (5–10 µg/mL) against three Gram-positive bacteria. In the case of Gram-negative bacteria, E. coli, P. aeruginosa, K. aerogenes, and K. pneumoniae, 1 showed better killing efficiency (MIC = 5–10 µg/mL, Table 1) than the other amphiphiles (2–6), which showed higher MIC values (35–150 µg/mL). Again, among the others (2–6), 2 showed comparatively better killing ability (35–75 µg/mL).
| Amphiphile | Bacteria | Fungus | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Gram-positive | Gram-negative | ||||||||
| B. subtilis | M. luteus | S. aureus | E. coli | P. aeruginosa | K. aerogenes | K. pneumoniae | C. albicans | S. cerevisiae | |
| a The MIC values are confirmed by both broth dilution and the spread plate method. | |||||||||
| 1 | 0.5 | 0.5 | 0.1 | 10 | 10 | 5 | 10 | 1 | 5 |
| 2 | 1 | 1 | 1 | 60 | 75 | 35 | 45 | 5 | 15 |
| 3 | 0.1 | 0.1 | 0.1 | 80 | 120 | 80 | 75 | 15 | 1 |
| 4 | 1 | 1 | 1 | 100 | 85 | 100 | 80 | 50 | 45 |
| 5 | 1 | 1 | 1 | 80 | 110 | 150 | 90 | — | 15 |
| 6 | 5 | 10 | 5 | 100 | 125 | 90 | 80 | — | 50 |
We have also tested the antifungal activity of these amphiphiles against yeast and fungi, Candida albicans and Saccharomyces cerevisiae, respectively. The summarized results (Table 1) revealed that in all cases the amphiphiles showed significant antifungal activity up to 50 µg/mL. In concurrence with the observed antibacterial activity, amphiphile 1 showed potent antifungal activity against both the yeast and fungus. MIC values were 1 and 5 µg/mL for C. albicans and S. cerevisiae, respectively. Amphiphile 2 also showed significant killing ability with C. albicans (MIC = 5 µg/mL) and S. cerevisiae (MIC = 15 µg/mL), although the killing efficiency of 2 against the fungus is 3–5 times lower than that was observed for 1. The other amphiphiles (3–6) showed comparatively less activity towards the yeast and fungi, except for 3 against S. cerevisiae (MIC = 1 µg/mL, Table 1). However, both amphiphiles 5 and 6 were ineffective against C. albicans even up to a concentration of 200 µg/mL. The antimicrobial activities of the dipeptide amphiphiles were compared with the well known standard AMP, magainin 2, consisting of 23 amino acids.9,17,18 The dipeptide amphiphiles 1–6, showed better killing efficiency for S. aureus and C. albicans (Table 1) compared to magainin 2 whose MIC values for the above mentioned organisms were 50 and 80 µg/mL respectively.18 Also, for E. coli, the amphiphile 1 showed better activity than magainin 2 (MIC = 50 µg/mL) while the MIC values of amphiphiles 2–6 are little higher (Table 1) than magainin 2.18 Interestingly it was also observed that the amphiphiles showed better activity against Gram-positive bacteria compared to the widely used traditional antibiotics streptomycin, gentamycin, and kanamycin (5–10 µg/mL), tested in our recent work.14,19 However, the amphiphiles demonstrated less potency against Gram-negative bacteria compared to these antibiotics.14,19 Interestingly, the antifungal activity of the synthesized amphiphiles against C. albicans was found to be better than the traditional antibiotic, amphotericin B (25 µg/mL).19
Until now we have measured the thermodynamic activity20 of peptides in equilibrium against several microorganisms. We were also interested in measuring the time dependent killing ability of the cationic dipeptide amphiphiles by exposing the microorganisms to the liquid medium (Luria Brutani (LB) medium for bacteria and yeast extract, peptone, and dextrose (YEPD) medium for fungi and yeast) for a wide range of times. The concentration of the amphiphiles was kept at 10 µg/mL except that of 6, where 40 µg/mL was used against all the tested Gram-positive bacteria. Fig. 2a–c shows the plot of log(survivors) versus exposure time for all the amphiphiles against B. subtilis, M. luteus, and S. aureus, respectively. All the Gram-positive bacteria were killed within 45–90 min of exposure. The killing effect was more prominent in the cases of 1 and 3, having phenylalanine-proline and tryptophan-proline moieties at the head group. The killing kinetics of Gram-negative bacteria, E. coli, P. aeruginosa, K. aerogenes, and K. pneumoniae (Fig. 2d–g, respectively) were studied using all the peptide amphiphiles at 200 µg/mL. Here also 1 and 3 showed better killing efficiency within 120–240 min compared to the other amphiphiles (2 and 4–6) which took 180–300 min contact time to show the bactericidal effect. The cationic peptide amphiphiles are more efficient at killing Gram-positive bacteria than the Gram-negative ones. Similar to the killing kinetics trend with Gram-negative bacteria, the amphiphiles 2 and 4–6 killed the yeast and fungi within 300 min (Fig. 2h–i), probably with slower permeabilization kinetics compared to 1 and 3, which took 240 min to kill them.
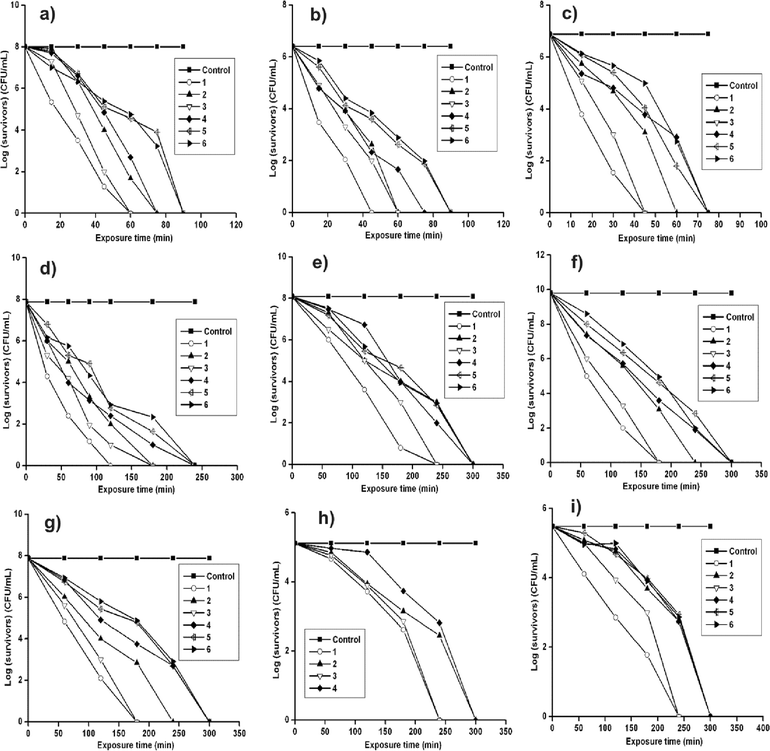 | ||
| Fig. 2 Log(survivors) versus exposure time (min) plots for amphiphiles 1–6 against (a) B. subtilis (b) M. luteus (c) S. aureus (d) E. coli (e) P. aeruginosa (f) K. aerogenes (g) K. pneumoniae (h) C. albicans and (i) S. cerevisiae. Each value represents the mean ± SD of six determinations. | ||
On the whole, from the time dependent killing assay it was observed that the amphiphiles 1 and 3 kill microbes at a faster rate compared with the other amphiphiles. Amphiphiles 1–6 are structurally varied at the polar head group with dipeptides of different L-amino acids but comprising the same C-14 long chain moiety as the hydrophobic tail. Hence, the observed antimicrobial activity of the amphiphiles may be regulated by the molecular architecture at the head group. So, the antimicrobial efficiency of the cationic dipeptide amphiphiles is because of their molecular properties. 1 and 3 possess the rigid proline moiety in the interior part of the head group. Thus, the positive charge on the quaternary ammonium segment presumably did not delocalize on the entire head group of 1 and 3, rather it was concentrated on a smaller area comprised of either phenylalanine (for 1) or tryptophan (for 3) residues. While in the case of the other amphiphiles 2 and 4–6, due to the presence of π-electron containing aromatic rings either at the inside or both at the interior and exterior regions of the head group, the positive charge can easily delocalize over the larger head group area. Thus, the charge densities at the head groups of 1 and 3 were higher compared with those in other amphiphiles, resulting in greater electrostatic interactions with the negatively charged cytoplasmic cell membranes of microbes and enhanced killing efficiency. Probably for a similar reason amphiphiles 5 and 6 showed comparatively lower antimicrobial activity (higher MICs) and could not even disrupt the C. albicans cell membrane at ≤200 µg/mL.
Mechanistically, there are two major steps that contribute to the antimicrobial activity of cationic amphiphiles. At first the cationic head group attacks the negatively charged cell membrane of microbes because of the electrostatic interaction.21 This electrostatic interaction is also entropically favored due to the release of a large number of counterions.22 Next, the hydrophobic tail of the amphiphile undergoes self-promoted transport across the cell membrane resulting in disruption in the bilayer and the release of the intracellular constituents leading to cell death.23–26 In this step, an optimum hydrophobicity of the amphiphile possibly allows it to enter and diffuse in the non-polar environment created by the lipid bilayers of the cell membrane. Thus the composition of the cell membrane of microbes plays a crucial role in determining the efficiency of antimicrobial agents. In the present study, all the amphiphiles showed higher antimicrobial activity against Gram-positive bacteria than Gram-negative bacteria (Table 1, Fig. 2). The cell membrane of Gram-positive bacteria is composed of a peptidoglycan layer, a class of polysaccharides, whereas Gram-negative bacteria contain an outer layer consisting of lipopolysaccharide and phospholipids in addition to the peptidoglycan layer.27,28 This additional protection in Gram-negative bacteria was presumably the main reason behind the higher MIC values of the amphiphiles against them (Table 1). The better killing efficiency of the peptide amphiphiles against Gram-positive bacteria can also be attributed to the presence of high amounts of negatively charged lipids in their membranes resulting in stronger electrostatic interactions.29 In the case of fungus, the negative charge density at the cell membrane is not as high as in a bacterial one, so most of the antibacterial compounds do not show antifungal activity.5 However, interestingly in our case, dipeptide-based cationic amphiphiles showed significant antifungal activity (Table 1).
To get an insight into the effect of amphiphiles on the morphology of bacterial cells,30,31 scanning electron microscopic (SEM) images of E. coli and S. aureus were taken before and after treatment with amphiphile 1 at above the MIC at 50 and 1.0 µg/mL (Fig. 3a–d). The incubation of E. coli and S. aureus with amphiphile 1 for around 30–40 min causes a significant change in their cell morphology. The untreated bacteria E. coli (Fig. 3a) and S. aureus (Fig. 3c) exhibited cell membrane integrity. However, after treatment with the 1, the SEM images showed collapse in the membrane integrity of lysed E. coli (Fig. 3b) and S. aureus (Fig. 3d).
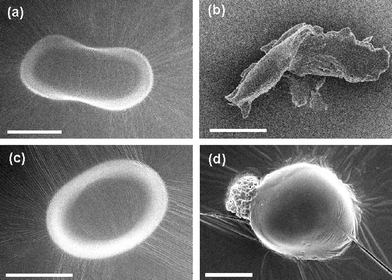 | ||
| Fig. 3 Scanning electron microscopic (SEM) images of (a) control bacterium E. coli; (b) E. coli after treatment with 50 µg/mL of amphiphile 1; (c) control bacterium S. aureus; and (d) S. aureus after treatment with 1.0 µg/mL of amphiphile 1. Scale bar = 0.5 µm. | ||
We have also studied the antibacterial activity of the representative amphiphile 1 by fluorescence microscopy using commercially available LIVE/DEAD® BacLight™ bacterial viability kit.14,22,32,33 The kit is composed of two nucleic acid binding strains, known as SYTO 9 and propidium iodide. These dyes have different cell penetration properties as well as different spectral characteristics. SYTO 9 can binds to the nucleic acid of both living and dead cells and fluoresce green after excitation with BP460–495 nm filter.14 Propidium iodide can only bind to the nucleic acid of dead cells and shows red fluorescence. Fig. S1a and S1c (ESI†) show the control image of E. coli and S. aureus, respectively while Fig. S1b and S1d (ESI†) show the fluorescence images of the E. coli and S. aureus cells treated with 50 and 1 µg/mL of 1, respectively. All the untreated bacterial cells exhibited green fluorescence (Fig. S1a and S1c, ESI†) indicating that they were alive. After treating with the amphiphile 1 above the corresponding MIC values, only red fluorescence (Fig. S1b and S1d, ESI†) was observed which confirmed the killing efficiency of the amphiphile.
The major problem with using cationic amphiphiles as antimicrobial agents is their potential toxicity to mammalian cells. So, we were quite eager to know how the peptide-based amphiphiles (1–6, Fig. 1) affect eukaryotic cells. The cytotoxicity of all the amphiphiles in the concentration range of 10 to 200 µg/mL was followed using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) based cell viability assay in human hepatic cancer derived HepG2 cells, human cervical cancer derived SiHa and HeLa cells (Fig. 4).14,34 The dipeptide-based cationic amphiphiles, irrespective of their molecular structure, showed low toxicity to all the mammalian cells. Only a maximum of 30% cytotoxicity was detected for amphiphile 1 at a concentration of 200 µg/mL on SiHa cells while others had a maximum toxicity up to 26% at the same concentration in HepG2 (Fig. 4a) and SiHa (Fig. 4b) cell lines. A maximum toxicity of 36% was observed at a concentration of 200 µg/mL of amphiphile 3 on HeLa cell line (Fig. 4c). At lower concentrations, all the amphiphiles were mostly non-toxic to HepG2, SiHa and HeLa cells after an exposure of 3 h. In the same time duration most of the bacterial cells were killed by the amphiphiles even at a much lower concentration. Thus, all the dipeptide based cationic amphiphiles are lethal to bacterial cells, but encouragingly safe with mammalian cells. The differences in the lipid compositions and the membrane potential gradient between the microbial and the mammalian cell membranes is very crucial for such cell selectivity of the amphiphiles.34,35 Eukaryotic cell membranes are primarily comprised of zwitterionic lipids such as phosphatidylcholine, sterols and sphingomyelin.36 On the other hand, bacterial cell membranes are made up of negatively charged phospholipids. Therefore the positively charged amphiphile preferentially binds to the more negatively charged bacterial membrane. While in the case of fungus, despite its eukaryotic nature, the amphiphiles selectively kill the fungi over the mammalian cells probably due to the presence of cholesterol in the latter membrane which is known to prevent the membrane disruption.37 The lower membrane potential across the mammalian cell membrane and its larger size may also be another reason for such cell selectivity.34,35
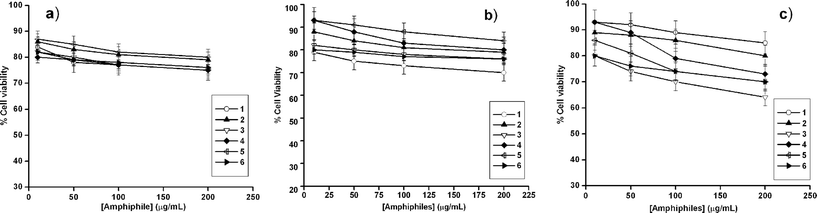 | ||
| Fig. 4 MTT assay based percent (a) HepG2, (b) SiHa and (c) HeLa cell viability as a function of concentration of 1–6. Each value represents the mean ± SD of six determinations. | ||
As mentioned earlier, these antimicrobial amphiphiles (1–6, Fig. 1) also showed water gelation capacity. The gelation behavior of these amphiphiles was investigated by different microscopic and spectroscopic studies and rheological studies. In this regard, we recently reported the hydrogelation ability of the C-16 alkyl chain analogues of these dipeptide cationic amphiphiles at room temperature.15 The present amphiphiles with C-14 alkyl chains exhibited notable hydrogelation efficiencies with a wide range of minimum gelation concentrations (MGC = 1.2–22%, w/v, Table S1, ESI†). The spectroscopic and microscopic investigations to understand the gelation mechanism as well as its characterization with related experimental details are discussed in the ESI†. The dried hydrogels at their MGC showed typical entangled fibrous networks of varying thickness in their FESEM images (Fig. S3, ESI†). The gel-to-sol transition, Tgel was determined by the tube inversion method (Fig. S2, ESI†), fluorescence study (Fig. S4, ESI†), and by rheological study (Fig. S5, ESI†). The thermoreversibility of the hydrogel was also confirmed by intrinsic fluorescence study (Fig. S4, ESI†).
Conclusion
This study demonstrates the important role of the head group structure of the cationic dipeptide-based amphiphiles toward influencing their antimicrobial activity as well as their physical properties. Interestingly, all the cationic amphiphiles are lethal to microbial cells while non-toxic to mammalian cells leading to their greater possibility in biomedical applications. This class of amphiphiles could be potent antimicrobial candidates in the future since there has been a continual increase in the demand for antimicrobial agents. Thus, we have been successful in developing the shortest sequence of cationic peptide amphiphile that has cell specificity toward a number of microbes over mammalian cells, leading to its promising future in biomedicines.Experimental
Materials
Silica gel of 60–120 mesh, L-tryptophan, L-phenylalanine, L-proline, n-tetradecylamine, N,N-dicyclohexylcarbodiimide (DCC), 4-N,N-(dimethylamino)pyridine (DMAP), 1-hydroxybenzotriazole (HOBT), iodomethane, solvents and all other reagents were procured from SRL, India. Water used throughout the study was Milli-Q water. Thin layer chromatography was performed on Merck precoated silica gel 60-F254 plates. CDCl3, Amberlite Ira-400 chloride ion exchange resins were obtained from Aldrich Chemical Company. Ethylene diaminetetraacetic acid (EDTA) and reagents required to prepare the Luria Burtani (LB) and YEPD culture medium like tryptone, peptone, yeast extract, and agar powder were purchased from Himedia Chemical Company, India. The LIVE/DEAD® BacLight™ bacterial viability kit was purchased from Molecular Probes, Invitrogen Chemical Company. All the materials used in the cell culture study such as Dulbecco's Modified Eagles’ Medium (DMEM), heat inactivated fetal bovine serum (FBS), trypsin from porcine pancreas and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were obtained from Sigma Aldrich Chemical Company. 1H NMR spectra were recorded on an AVANCE 300 MHz (BRUKER) spectrometer. Mass spectrometric (MS) data were acquired by the Electron Spray Ionization (ESI) technique on a Q-tof-Micro Quadruple mass spectrophotometer, Micromass.Synthetic procedure and characterization of amphiphiles 1–6
All the peptide amphiphiles (1–6, Fig. 1) were synthesized following the reaction procedure previously reported by us.15,16 Boc-protected L-amino acids (10 mmol) were coupled with the methyl ester (11 mmol) of the corresponding L-amino acids using dicyclohexylcarbodiimide (DCC, 11 mmol) and a catalytic amount of DMAP in the presence of HOBT (11 mmol) in dry DCM on an ice-bath. The reaction mixture was then allowed to come to room temperature and stirred for 12 h. Next, the resultant mixture was filtered and the filtrate was concentrated in a rotary evaporator. The whole residue was then extracted in ethyl acetate washed with 2 M HCl, brine, 1 M sodium carbonate and brine to neutrality. The organic part was dried over anhydrous sodium sulfate and concentrated. The residue obtained was then purified by a silica gel 60–120 mesh column using ethyl acetate/toluene as the eluent. The concentrated column purified white solid material (-OMe protected) was dissolved in the required amount of MeOH and subjected to saponification by the addition of 20 mL 1 M NaOH solution at room temperature. The stirring was carried out for 6 h, after which the MeOH was evaporated in a rotary evaporator. The remaining material was then added to water and washed with diethyl ether; the pH of the aqueous part was adjusted to 2 using 1 M HCl, and then extracted with ethyl acetate, which was further washed with brine to remove any traces of acid. The organic part was then dried over anhydrous sodium sulfate and concentrated on a rotary evaporator, which yielded a solid -OMe deprotected peptide. This free C-terminal peptide (8 mmol) was coupled with n-tetradecylamine (8.8 mmol), DCC (8.8 mmol) and a catalytic amount of DMAP in the presence of HOBT (8.8 mmol) in dry DCM on an ice-bath and then the reaction mixture was allowed to come to room temperature and stirred for 12 h. Work up, and extraction of this reaction were carried out similarly as mentioned above. The residue thus obtained was then purified by a silica gel 60–120 mesh column using ethyl acetate/toluene as the eluent. After that the purified BOC-protected long chain dipeptide was subjected to deprotection by trifluoroacetic acid (TFA, 32 mmol) in dry DCM at room temperature. After 2 h of stirring, the solvent was removed on a rotary evaporator and the residue was taken up in ethyl acetate, which was thoroughly washed with aqueous 10% sodium carbonate solution followed by brine to neutrality. The organic part was dried over anhydrous sodium sulfate and concentrated to get the corresponding amine (7 mmol). The free amine (7 mmol) was quaternized with excess iodomethane using anhydrous potassium carbonate (7.7 mmol for 2, 4 and 15.4 mmol for 1, 3, 5 and 6) and a catalytic amount of 18-crown-6-ether in dry DMF for 5–6 h at room temperature (25–27 °C). After concentrating the reaction mixture, the residue was taken up in ethyl acetate and washed with aqueous 5% thiosulfate solution and brine to neutrality. The concentrated ethyl acetate part was purified using a 60–120 mesh silica gel column using a mixture of chloroform/methanol as the eluent and crystallized from methanol/ether to obtain the solid quaternized iodide salt, which was subjected to ion exchange on an Amberlite Ira-400 chloride ion exchange resin column to get the colorless chloride salts of 1–6. The overall yields were 62, 69, 63, 70, 70, and 68% for 1–6, respectively.Microorganisms and culture conditions
The in vitro antimicrobial activity of all the cationic amphiphiles was investigated against pathogenic representatives of Gram-positive bacteria, Gram-negative bacteria and fungus. The Gram-positive bacteria used in the present study were Bacillus subtilis, Micrococcus luteus, and Staphylococcus aureus. The Gram-negative strains investigated included Escherichia coli, Pseudomonas aeruginosa, Klebsiella aerogenes and Klebsiella pneumoniae. Investigations of antibacterial activities were performed by both broth dilution and the spread plate method. The LB medium (tryptone (10 g), yeast extract (5 g) and NaCl (10 g) in 1 L sterile distilled water at pH 7.0) was used as a liquid medium and LB agar (tryptone (10 g), yeast extract (5 g), NaCl (10 g) and agar (15 g) in 1 L of sterile distilled water of at 7.0) was used as a solid medium in all antibacterial experiments. The antifungal activity on the two fungi Candida albicans and Saccharomyces cerevisiae was investigated by YEPD broth dilution and the spread plate method. The YEPD medium (yeast extract (3 g), peptone (10 g) and dextrose (20 g) in 1 L sterile distilled water at pH 7.0) was used as a liquid medium and YEPD agar (yeast extract (3 g), peptone (10 g), dextrose (20 g) and agar (15 g) in 1 L sterile distilled water at pH 7.0) was used as a solid medium for the antifungal studies. All the microbial strains were purchased from the Institute of Microbial Technology, Chandigarh, India. The stock solutions of all the amphiphiles as well as the required dilutions were made in autoclaved sterile water.The freeze-dried ampules of all microbial strains were opened and a loopful of culture was spread to give single colonies on the respective solid agar (LB agar for bacterial strains and YEPD agar for fungal strains) media and incubated for 24 h (for all the bacterial strains and for fungi S. cerevisiae) and 48 h (for fungi C. albicans) at 37 °C. For all the microbial strains, a representative single colony was picked up with a wire loop and was spread on an agar slant to give single colonies. The slants were incubated at 37 °C for the respective time. These incubated cultures of all the microorganisms were diluted as required to give a working concentration in the range of 106–109 and 104–105 colony forming units (cfu)/mL for bacteria and fungus, respectively before every experiment.
Antimicrobial studies
Minimum inhibitory concentrations (MICs) of 1–6 were estimated by both broth dilution and the spread plate method. The MIC was measured using a series of test tubes containing the amphiphiles (0.05–200 µg/mL) in 5 mL liquid medium. Diluted microbial culture was added to each test tube in identical concentration to obtain the working concentration of B. subtilis; 7.5 × 107–1 × 108 cfu/mL, for M. luteus; 1 × 106–2.5 × 106 cfu/mL, for S. aureus; 5 × 106–7.5 × 106 cfu/mL, for E. coli; 3.75 × 107–7.5 × 107 cfu/mL, for P. aeruginosa; 9 × 107–1.2 × 108 cfu/mL, for K. aerogenes; 1.5 × 109–6.25 × 109 cfu/mL, for K. pneumoniae; 1.4 × 107–7.5 × 107 cfu/mL, for C. albicans; 9.8 × 104–1.3 × 105 cfu/mL, for S. cerevisiae; 1.5 × 105–3 × 105 cfu/mL. All the test tubes were then incubated at 37 °C for 24 h. The optical density of all the solutions was measured before and after incubation at 650 nm. Liquid medium containing microorganisms was used as a positive control. All the experiments were performed in triplicate and repeated twice.We also tested the antimicrobial activity through the spread plate method. For these experiments, the same amount of bacterial culture was added to test tubes containing the desired concentration of amphiphile solution in liquid medium. The test tubes were then incubated at 37 °C for 5–7 h. Then, 50 µL from each tube was spread onto LB agar plates inside the laminar hood. Finally, plates were incubated for 24 h (for all the bacterial strains and for fungi S. cerevisiae) and 48 h (for fungi C. albicans) at 37 °C and the viable cells were counted. The experiments were performed in triplicate and were repeated twice.
Time dependent microbial killing assay
A serial dilution method was used for determining the kinetic effect, also called Contact Germicidal Efficiency (CGE), i.e., the time required for the antimicrobial effect. Overnight cultures on solid agar medium were diluted with liquid medium to give a bacterial concentration of 106–109 cfu/mL. Amphiphile (1–6) stock solutions were added at concentrations higher than MIC values, and were incubated at room temperature inside the laminar hood. In the case of Gram-positive bacteria the concentrations of 1–5 were 10 µg/mL and of 6 it was 40 µg/mL while in the case of Gram-negative bacteria it was 200 µg/mL for all amphiphiles. At regular intervals, the same amount of aliquot was withdrawn and kept at 4 °C. Immediately, the aliquots were diluted and 50 µL of diluted suspension was spread onto LB agar plates. Next, all the plates were incubated at 37 °C for 24 h and after incubation the viable cells were counted. The control experiment was performed in the absence of amphiphile. The same technique was followed for the two fungi. In this case 25 µg/mL (amphiphile 1) and 200 µg/mL (amphiphiles 2–6) of the amphiphiles were utilized for the corresponding kinetic study and for C. albicans and S. cerevisiae respectively. Here all the YEPD agar plates were incubated at 37 °C for 48 h (C. albicans) and 24 h (S. cerevisiae) and after incubation the viable cells were counted. With the same technique the control study was carried out without amphiphiles.FESEM of dipeptide amphiphile treated E. coli and S. aureus
E. coli (3.75 × 107–7.5 × 107 cfu/mL) and S. aureus (5 × 106–7.5 × 106 cfu/mL) cells (1 mL) were treated with compound 1 above the MIC at 50 and 1.0 µg/mL, respectively. The bacterial cells with and without (untreated cells as control) amphiphile were incubated for 30–40 min. After incubation the mixtures were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The media was removed completely and the cells were redispersed in 0.9 wt% saline. Finally, 5 µL of the redispersed samples was mounted on a glass slide and dried under vacuum for 4 h and the SEM images were taken on JEOL-6700F scanning electron microscope.
000 rpm for 10 min. The media was removed completely and the cells were redispersed in 0.9 wt% saline. Finally, 5 µL of the redispersed samples was mounted on a glass slide and dried under vacuum for 4 h and the SEM images were taken on JEOL-6700F scanning electron microscope.Fluorescence microscopic study
The LIVE/DEAD® BacLight ™ bacterial kit was used to examine bacterial cell viability under a fluorescence microscope. The kit contains a mixture of two nucleic-acid binding strains, specifically referred to as SYTO 9 and propidium iodide. The kit was stored at −20 °C in the dark, and was taken out and thawed at room temperature just prior to assay. E. coli (3.75 × 107–7.5 × 107 cfu/mL) and S. aureus (5 × 106–7.5 × 106 cfu/mL) cells (1 mL) were treated with 1 above the MIC (50 µg/mL for E. coli and 1 µg/mL for S. aureus, respectively) and also untreated cells were taken in a centrifuge tube as a control. The mixtures were centrifuged at 10000 rpm for 10 min. Then, media was removed completely and the cells were redispersed in 0.9 wt% saline. Finally, BacLight dye mixture (3 µL) was added and the cells incubated in the dark at room temperature for 15–20 min. After incubation, 5 µL of the solution mixture was mounted over microscope slides, which were then air-dried and viewed under the light microscope (BX61, Olympus) using an excitation filter of BP460–495 nm and a band absorbance filter covering wavelengths below 505 nm.Cell cultures
Human hepatic cancer derived HepG2 cells, human cervical cancer derived SiHa and HeLa cells were obtained from the National Center for Cell Science (NCCS), Pune (India), and maintained in DMEM medium supplemented with 10% FBS, 100 mg/L streptomycin and 100 IU/mL penicillin. Cells were grown in a 25 mL cell culture flask and incubated at 37 °C in a humidified atmosphere of 5% CO2 to approximately 70–80% confluence. A subculture was performed every 2–3 days. After 48–72 h, media was removed to eliminate the dead cells. Next, the adherent cells were detached from the surface of the culture flask by trypsinization. Cells were now in the exponential phase of growth for checking the viability of the cationic amphiphilic amphiphiles (1–6).Cytotoxicity assay
The cytotoxicities of amphiphiles 1–6 were assessed by the microculture MTT reduction assay as used in the literature. This assay is based on the reduction of a soluble tetrazolium salt by the mitochondrial dehydrogenase of the viable cells to form an insoluble colored product, formazan. The amount of formazan formed can be measured spectrophotometrically after dissolution of the dye in DMSO. The activity of the enzyme and the amount of the formazan produced is proportional to the number of cells alive. Reduction of the absorbance value can be attributed to the killing of cells or inhibition of cell proliferation by the amphiphiles. Cells were seeded at a density 15000 cells per well in a 96-well microtiter plate for 18–24 h before the assay. A stock solution of all the six compounds was prepared in DMSO. Sequential dilutions of these stock solutions were done during the experiment to vary the concentrations of the amphiphiles (10 to 200 µg/mL) in the microtiter plate. The amphiphiles were insoluble beyond this concentration in the DMEM medium. The cells were incubated with 1–6 for 3 h at 37 °C under 5% CO2. Then, 15 µL MTT stock solution (5 mg/mL) in phosphate buffer saline was added to the above mixture and the cells were further incubated for another 4 h. The precipitated formazan was dissolved thoroughly in DMSO and the absorbance at 570 nm was measured using BioTek® Elisa Reader. The number of surviving cells were expressed as percent viability = (A570(treated cells) − background/A570(untreated cells) − background) × 100.Statistics
Significant differences between two groups of data were evaluated by Student's t-test.Acknowledgements
P.K.D. is thankful to the Department of Science and Technology, India for financial assistance through a Ramanna Fellowship (No. SR/S1/RFPC-04/2006). A.S. acknowledges the Council of Scientific and Industrial Research, India for Research Fellowships. P.P. is thankful to the Indian Association for the Cultivation of Science. We are very much thankful to Sangita Roy for helpful discussions.References
- A. Makovitzki, D. Avrahami and Y. Shai, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 15997–16002 CrossRef CAS.
- S. Pritz, M. Pätzel, G. Szeimies, M. Dathea and M. Bienert, Org. Biomol. Chem., 2007, 5, 1789–1794 RSC; A. Zumbuhel, L. Ferreira, D. Kuhn, A. Astashkina, L. long, Y. Yeo, T. Iaconis, M. Ghannoum, G. R. Fink, R. Langer and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12994–12998 CrossRef; B. Xing, C.-W. Yu, K.-H. Chow, P.-L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS; Z. Yang, K. Xu, Z. Guo, Z. Guo and B. Xu, Adv. Mater., 2007, 19, 3152–3156 CrossRef CAS; J. C. Tiller, Angew. Chem., Int. Ed. Engl., 2003, 42, 3072–3075 CrossRef CAS; J. C. Tiller, S. B. Lee, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2002, 79, 465–471 CrossRef CAS.
- S. A. Sieber and M. A. Marahiel, Chem. Rev., 2005, 105, 715–738 CrossRef CAS; E. D. Brown and G. D. Wright, Chem. Rev., 2005, 105, 759–774 CrossRef CAS.
- J. C. B. DeNap and P. J. Hergenrother, Org. Biomol. Chem., 2005, 3, 959–966 RSC.
- A. Makovitzki and Y. Shai, Biochemistry, 2005, 44, 9775–9784 CrossRef CAS.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS.
- Y. Shai and Z. Oren, Peptides, 2001, 22, 1629–1641 CrossRef CAS.
- D. Avrahami and Y. Shai, J. Biol. Chem., 2004, 279, 12277–12285 CAS.
- M. Dathe, M. Schümann, T. Wieprecht, A. Winkler, M. Beyermann, E. Krause, K. Matsuzaki, O. Murase and M. Bienert, Biochemistry, 1996, 35, 12612–12622 CrossRef CAS; T. Wieprecht, M. Dathe, R. M. Epand, M. Beyermann, E. Krause, W. Lee Maloy, D. L. MacDonald and M. Bienert, Biochemistry, 1997, 36, 12869–12880 CrossRef CAS; Y. Wolf, S. Pritz, Saıd Abes, M. Bienert, B. Lebleu and J. Oehlke, Biochemistry, 2006, 45, 14944–14954 CrossRef CAS; N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 2794–2799 CrossRef CAS.
- M. Dathe, H. Nikolenko, J. Klose and M. Bienert, Biochemistry, 2004, 43, 9140–9150 CrossRef CAS.
- R. E. W. Hancock, Drugs, 1999, 57, 469–473 CrossRef CAS.
- M. B. Strom, B. E. Haug, M. L. Skar, W. Stensen, T. Stiberg and J. S. Svendsen, J. Med. Chem., 2003, 46, 1567–1570 CrossRef CAS.
- J. Haldar, P. Kondaiah and S. Bhattacharya, J. Med. Chem., 2005, 48, 3823–3831 CrossRef CAS.
- S. Roy and P. K. Das, Biotechnol. Bioeng., 2008, 100, 756–764 CrossRef CAS.
- R. N. Mitra, D. Das, S. Roy and P. K. Das, J. Phys. Chem. B, 2007, 111, 14107–14113 CrossRef CAS.
- A. Dasgupta, R. N. Mitra, S. Roy and P. K. Das, Chem. Asian J., 2006, 1, 780–788 CrossRef CAS; D. Das, A. Dasgupta, S. Roy, R. N. Mitra, S. Debnath and P. K. Das, Chem. Eur. J., 2006, 12, 5068–5074 CrossRef CAS; S. Roy, A. Dasgupta and P. K. Das, Langmuir, 2007, 23, 11769–11776 CrossRef CAS; A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280–4288 CrossRef CAS.
- D. Avrahami and Y. Shai, Biochemistry, 2002, 41, 2254–2263 CrossRef CAS; T. Wieprecht, M. Dathe, M. Beyermann, E. Krause, W. Lee Maloy, D. L. MacDonald and M. Bienert, Biochemistry, 1997, 36, 6124–6132 CrossRef CAS.
- M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5449–5453 CAS.
- C. Ramalingan, Y. T. Park and S. Kabilan, Eur. J. Med. Chem., 2006, 41, 683–696 CrossRef CAS; I. Oren, O. Temiz, I. Yalcin, E. Sener and N. Altanlar, Eur. J. Pharm. Sci., 1998, 7, 153–160.
- N. Bodor, J. J. Kaminsky and S. Selk, J. Med. Chem., 1980, 23, 469–474 CrossRef CAS; R. M. Baird, and S. F. Bloomfield, in Microbial quality assurance in cosmetics, toiletries and non-sterile pharmaceuticals, published by CRC Press, 1996, pp. 258 Search PubMed.
- K. M. Stewart, K. L. Hortonb and S. O. Kelley, Org. Biomol. Chem., 2008, 6, 2242–2255 RSC; D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS.
- R. Kügler, O. Bouloussa and F. Rondelez, Microbiology, 2005, 151, 1341–1348 CrossRef CAS.
- J. A. Castillo, P. Clapes, M. R. Infante, J. Comas and A. Manresa, J. Antimicrob. Chemother., 2006, 57, 691–698 CrossRef CAS.
- C. J. Ioannou, G. W. Hanlon and S. P. Denyer, Antimicob. Agents Chemother., 2007, 51, 296–306 Search PubMed.
- E. Jawetz, J. L. Melnick, E. A. Adelberg, G. F. Brooks, J. S. Butel, and L. N. Ornston, in Medical microbiology, Prentice-Hall, London, 1989, pp. 46 Search PubMed.
- T. J. Franklin, and G. A. Snow, in Antiseptics, antibiotics, and the cell membrane, Biochemistry of Antimicrobial Action, Chapman and Hall Ltd., London, 1981, pp. 58-78 Search PubMed.
- J. W. Costerton and K. J. Cheng, J. Antimicrob. Chemother., 1975, 1, 363–377 CrossRef CAS.
- S. P. Denyer and J. Y. Maillard, J. Appl. Micobiol., 2002, 92, 35S–45S Search PubMed.
- W. M. O'Leary and S. G. Wilkinson, in Gram-positive bacteria, in Microbial lipids (C. Ratledge, and S. G. Wilkinson, Eds.), Academic Press Ltd., London, 1988, pp. 117–201S. G. Wilkinson, in Gram-negative bacteria, in Microbial lipids (C. Ratledge, and S. G. Wilkinson, Eds.); Academic Press Ltd., London, 1988, pp. 299–488 Search PubMed.
- M. L. Mangoni, N. Papo, D. Barra, M. Simmaco, A. Bozzi, A. Di Giulio and A. C. Rinaldi, Biochem. J., 2004, 380, 859–865 CrossRef CAS.
- S. Sharma, P. Sen, S. N. Mukhopadhyay and S. K. Guha, Colloids Surf. B, 2003, 32, 43–50 CrossRef CAS.
- C. Qin, X. Zhong, X. Bu, N. L. J. Ng and Z. Guo, J. Med. Chem., 2003, 46, 4830–4833 CrossRef CAS.
- L. Boulos, M. Prevost, B. Barbeau, J. Coallier and R. Desjardins, J. Microbiol. Methods, 1999, 37, 77–86 CrossRef CAS.
- M. B. Hansen, S. E. Nielsen and K. Berg, J. Immunol. Methods, 1989, 119, 203–210 CrossRef CAS.
- K. Matsuzaki, M. Harada, T. Handa, S. Munakoshi, N. Fujii, H. Yajima and K. Miyajima, Biochim. Biophys. Acta, 1989, 981, 130–134 CAS.
- S. Munro, Cell, 2003, 115, 377–388 CrossRef CAS; F. J. Alvarez, L. M. Douglas and J. B. Konopka, Eukaryotic Cell, 2007, 6, 755–763 Search PubMed.
- J. Brajtburg and J. Bolard, Clin. Microbiol. Rev., 1996, 9, 512–531 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The hydrogelation study, related experimental details and the corresponding figures and tables, the fluorescence micrograph of E. coli and S. aureus before and after treating with compound 1 in the presence of LIVE/DEAD® BacLight™ Bacterial Viability Kit as well as the NMR and ESIMS spectra of the amphiphiles 1–6. See DOI: 10.1039/b815368j |
| ‡ Also at the Centre for Advanced Materials, Indian Association for the Cultivation of Science. |
| This journal is © The Royal Society of Chemistry 2009 |