Synthesis of highly substituted 2-perfluoroalkyl quinolines by electrophilic iodocyclization of perfluoroalkyl propargyl imines/amines†
Pravin R.
Likhar
*a,
Madavu Salian
Subhas
a,
Sarabindu
Roy
a,
Mannepalli Lakshmi
Kantam
a,
Balasubramanian
Sridhar
b,
Ratanesh Kumar
Seth
c and
Sukla
Biswas
c
aInorganic and Physical Chemistry Division, Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad, 500670, India. E-mail: plikhar@iict.res.in; Fax: +91-40-27160921
bX-Ray Crystallography Division, Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad, 500670, India
cNational Institute of Malaria Research (ICMR), 22 Shamnath Marg, Delhi, 110 054, India
First published on 10th November 2008
Abstract
A series of highly substituted 2-perfluoroalkyl-3-iodoquinolines are prepared by two different methods in good to excellent yields under mild reaction conditions. The first method involves iodocyclization of perfluoroalkyl propargyl imines with I2-CAN. The second method involves iodocyclization of perfluoroalkyl propargyl amines using I2 and ICl. The perfluoroalkyl propargyl amines are prepared in excellent yields viaSonogashira coupling of easily accessible imidoyl iodides with alkynes followed by reduction with NaBH3CN. The scope of this methodology is extended by using the resulting 2-perfluoroalkyl-3-iodo quinolines in Suzuki, annulation, dehalogenation and carboxylation reactions. Antimalarial activity of the 2-perfluoroalkyl-3-iodoquinolines is discussed.
Introduction
Recently, fluorinated compounds have attracted considerable attention from the synthetic and medicinal chemists due to the unique physical and biological properties imparted by fluorine. Substitution of an aromatic hydrogen atom by a perfluoroalkyl group has a substantial effect on these characteristics.1 In particular, trifluoromethylated quinolines have been found to possess special biological properties.2–5 For example, 2-trifluoromethylated quinolines are of significant pharmacological interest for their use as potent antimalarial agents (mefloquine),2 PDE4 inhibitors,3 DPP-IV inhibitors,4 and leishmanicidal agents.5In view of the importance of fluorine-containing quinoline compounds, considerable effort has been placed in developing an efficient method for the synthesis of trifluoromethylated quinoline derivatives. The usual synthetic methods for the introduction of a CF3group into aromatic systems have been fluorination of a suitable functional group6 (i.e.halogen exchange of –CCl3, –CBr3), fluorination of –CO2H7 upon treatment with a fluorinated Lewis acid (SbF3, SbF5, SF4, etc.) or by Ullmann-type reaction of perfluoroalkyl iodides and aryl halides using copper powder.8 Many other methods are known for the synthesis of 2-perfluoroalkyl-subsituted quinolines but many such methodologies afford the products in low yields.9
Baraznenok et al.10 have reported an elegant high yielding synthesis of 2-perfluroalkyl substituted quinolines from anilines and 4-dimethylamino-1,1,1-trifluoro-3-buten-2-one. However this protocol does not provide access to 3- or 4- substituted 2-perfluoroalkylquinolines. In 2001, Amii et al. reported one-pot synthesis of highly substituted fluorinated quinolines by Rh(I)-catalyzed cyclization of N-aryl trifluoroacetimidoyl chlorides using alkynes.11 Unfortunately this procedure suffered from poor regioselectivity when unsymmetrical internal alkynes were used. Consequently, the development of an efficient and selective synthesis of 2-perfluoroalkyl substituted quinolines using mild reaction conditions remains an active research area.
The synthesis of carbocycles and heterocycles can be achieved through cyclization of aryl-substituted alkynes via intramolecular hydroarylation. Recently, the electrophilic cyclization of functionalized alkenes and alkynes has received considerable attention by organic chemists for synthesizing furans, quinolines, benzo[b]furans, benzothiophenes, indoles, isoquinolines and furopyridines etc.12 Such work provides encouragement for the synthesis of 2-perfluoroalkyl quinolines via an electrophilic iodocyclization reaction.
Results and discussion
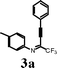
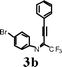
It is assumed that 2-perfluoroalkyl quinolines can be synthesized by electrophilic cyclization of 2-perfluoroalkynyl imines as shown in Scheme 1. Accordingly, the 2-perfluoroalkynyl imine (3b) was synthesized viaSonogashira coupling13,14 of imidoyl iodide (1b)15 and phenylacetylene (2b) (Scheme 2).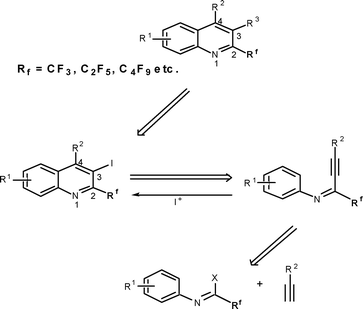 | ||
| Scheme 1 Retrosynthetic approach for synthesis of 2-perfluoroalkyl-substituted quinolines. | ||
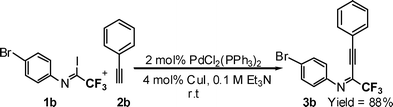 | ||
| Scheme 2 Synthesis of 2-perfluoroalkynyl imines using coupling of imidoyl iodide and phenylacetylene. | ||
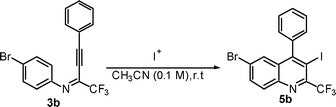
In an attempt to cyclize the imine product, 3b was treated with I2 (2.0 equiv) and NaHCO3 (2.0 equiv) in acetonitrile at room temperature. Only a trace amount of the product, 5b was obtained (Table 1, entry 1). When a strong eletrophile, ICl was employed, the product was isolated in 10% yield (entry 2).
| Entry | I+ (equiv) | CAN (equiv) | Time (h) | Yield (%)b |
|---|---|---|---|---|
| a Reaction condition: 0.5 mmol of 3b, I2 and CAN in 0.1M of CH3CN at room temp. b Yield of isolated product, 5b. c 2 equiv of NaHCO3 was used. d 2 equiv of NaHCO3 in 0.1M CH2Cl2 was used. | ||||
| 1 | I2 (2.0) | 0 | 24 | Trace c |
| 2 | ICl (2.0) | 0 | 24 | 10d |
| 3 | I2 (2.0) | 1.0 | 5 | 65 |
| 4 | I2 (2.0) | 2.0 | 2 | 85 |
| 5 | I2 (3.0) | 2.0 | 3 | 85 |
| 6 | I2 (1.0) | 2.0 | 5 | 53 |
After recent success in iodocyclization of the (2-methoxyaryl)- system,16 the iodocyclization of 3b (0.5 mmol) was examined, using I2 (2.0 mmol)-CAN (1.0 mmol) in 0.1 M CH3CN at room temperature. Product 5b was obtained in 65% isolated yield (entry 3). Subsequently, conditions for optimization of I2 and CAN to afford maximum product yield were tested (Table 1). The highest yield (85%) was obtained with 2 equiv of I2 and 2 equiv of CAN in 0.1 M CH3CN at room temperature (entry 4).
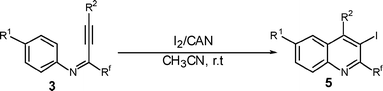
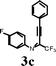
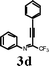
To investigate the scope of the reaction, the effect of various R1groups on the aniline moiety were examined. The electron-donating methyl group afforded corresponding quinoline in 86% yield, while the strongly electron-withdrawing fluorine failed to afford the product (Table 2, entries 1 and 3). The unsubstituted aniline and naphthylamine provided good product yields (entries 4 and 5). The effect of various substitutents on the alkyne moiety was then examined. The substituted aromatic alkynes underwent smooth iodocyclization to afford the corresponding quinolines in excellent yields (entries 6, 7 and 8). The olefin-substituted alkyne gave the desired product with 81% yield (entry 9). A good yield was observed when the perfluoroalkyl chain was changed from CF3 to C4F9 (entry 10).
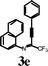
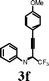
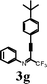
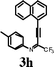
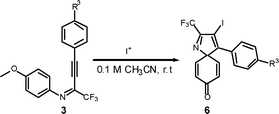
Surprisingly, when para-substituted methoxy perfluoroalkyl propargyl imine 3k was subjected to iodocyclization, only the azaspiro compound 6a was obtained in 92% and in 0.5h instead of the desired quinoline product, 5k (Table 3, entry 1).17 Similarly, 6b was obtained in 90% yield from 3l (entry 2). As an alternative, it was also found that I2/NaHCO3 was effective for the synthesis of azaspiro compound, however the reaction was more sluggish than the I2-CAN system (entries 3 and 4). The structure of the azaspiro compound 6a was confirmed by 1H NMR, 13C NMR, HRMS and by single-crystal X-ray diffraction study (Fig. 1).18
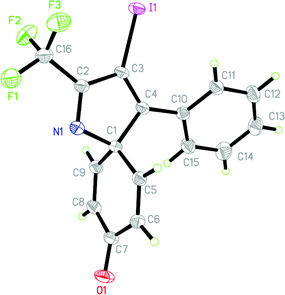 | ||
| Fig. 1 ORTEP diagram of compound 6a (see Table 3). | ||
The observed results can be plausibly explained by electrophilic attack of the iodine cation which is generated in situ by activating with CAN, on the carbon-carbon triple bond of the propargylic aniline to give the iodonium intermediate A. It is believed that CAN acts as an efficient and convenient activator for I2 and during the activation process, reduces Ce(IV) to Ce(III).19 The intramolecular orthocyclization of the aniline aromatic ring with the activated triple bond results in the formation of perfluoroalkyl quinolines 5 (Scheme 3).
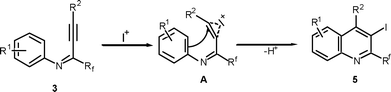 | ||
| Scheme 3 Plausible mechanism for the formation of compound 5 (Table 2). | ||
In the case of para-methoxy substituted aniline, the formation of the azaspiro compound can be explained similarly to above where the iodonium intermediate A is generated in the first step. The intermediate A can in turn undergo intramolecular ipso-cyclization with the electron rich aromatic ring. The removal of the methyl groupvia nucleophilic displacement results into formation of compound 617(Scheme 4).
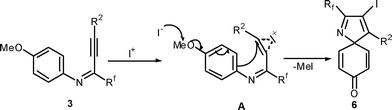 | ||
| Scheme 4 Plausible mechanism for the formation of compound 6 (Table 3). | ||
To potentially overcome the limitations of I2/CAN system in the above iodocyclization process, the iodocyclization of perfluoroalkyl propargyl amines, a reduction product of perfluoroalkyl propargyl imines was examined. Thus 2-trifluoromethyl-substituted propargyl imine 3k was treated with NaBH3CN in acetic acid at 0 °C to afford 2-trifluoromethyl-substituted propargyl amine, 4k in quantitative yields (Scheme 5).
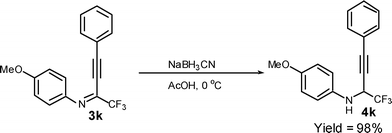 | ||
| Scheme 5 Reduction of 2-trifluoromethyl-substituted propargyl imine, 3k to 2-trifluoromethyl-substituted propargyl amine 4k | ||
The potentially useful trifluoro propargyl amines were prepared by the addition of acetylenes to trifluoro methyl iminium ions, generated from the corresponding oxazolidines20 or aminal21 in the presence of Lewis acid. Recently, another preparation has been reported which describes addition of acetylides to trifluoromethyl aldimines.22 However, to the best of the authors’ knowledge, the synthesis of perfluoroalkyl propargyl amines by the reduction of perfluoroalkyl propargyl imines prepared from Sonogashira coupling reaction of easily accessible corresponding imidoyl iodides and alkynes has not yet been reported.
Next, 4k was treated with 3 equiv. of iodine in the presence of 2 equiv. of NaHCO3 in 0.1 M acetonitrile and 2-trifluromethyl-substituted quinoline 5k obtained in 90% yield (Table 4, entry 9). It can be seen from these experiments that by changing the imine-amine substrates, selective ipso or orthocyclization may be realised.
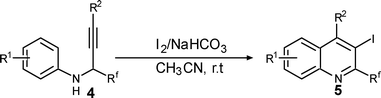
| Entry | 4 | I+ | Time (h) | 5 | Yield (%)b |
|---|---|---|---|---|---|
| a Reaction conditions: 0.5 mmol of 3, 2 equiv of I2 and 2 equiv of NaHCO3 in 5 mL of CH3CN at room temp. b Isolated yields. c Room temperature. d Yield of isolated product, 3,6-diiodo-substituted quinoline. | |||||
| 1 |
|
I2 | 0.5 | 5a | 90 |
| 2 |
|
I2 | 24 | 5b | 54 |
| 3 | ICl | 0.5 | 5b | 82 | |
| 4 |
|
I2 | 24 | 5c | 43 |
| 5 | ICl | 0.5 | 5c | 74 | |
| 6 |
|
I2 | 1 | 5d | 0 |
| 7 | ICl | 1 | 5d | 38 | |
| 8 |
|
I2 | 0.5 | 5e | 80 |
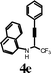
| 9 |
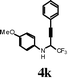
|
I2 | 0.5 | 5k | 90 |
| 10 | ICl | 0.5 | 5k | 92 (79) c | |
| 11 |
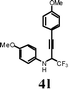
|
I2 | 0.5 | 5l | 57 |
| 12 | ICl | 0.5 | 5l | 84 | |
| 13 |
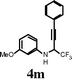
|
I2 | 2 | 5m | 0, 25 d |
| 14 | ICl | 0.5 | 5m | 0 | |
| 15 |
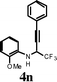
|
I2 | 2 | 5n | 72 |
| 16 | ICl | 0.5 | 5n | 74 | |
| 17 |
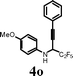
|
I2 | 0.5 | 5o | 80 |
| 18 |
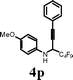
|
I2 | 0.5 | 5p | 76 |
| 19 |
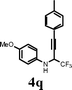
|
I2 | 0.5 | 5q | 81 |
| 20 |
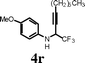
|
I2 | 1 | 5r | 64 |
| 21 |
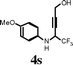
|
I2 | 1 | 5s | 58 |
| 22 | ICl | 1 | 5s | 64 | |
| 23 |
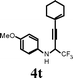
|
I2 | 0.5 | 5t | 78 |
The effect of different solvents and bases on the iodocyclization of 4k to improve the yield was then examined. It was found that the combination of 0.1 M of acetonitrile and 2 equiv. of NaHCO3 was favourable. (Table 4, entry 9). Encouraged by the results obtained with iodine, the efficacy and generality of the current protocol was studied by employing the stronger electrophile ICl on the cyclization of compound 4k. It was observed that ICl demonstrated improved results at 0 °C compared to the reaction at room temperature (Table 4, entry 10).
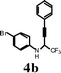
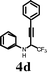
In an effort to understand the scope of the reaction, the effect of various substituents on the aniline ring was scrutinized. As expected, the electron-rich-substituted anilines promote the reaction and cyclize to give the corresponding quinolines in high yields (Table 4, entry 1). The 4-bromo and 4-fluoro-substituted anilines, in the presence of iodine, afforded moderate yields of the corresponding quinolines with incomplete conversion (entries 2 and 3). Yields were dramatically enhanced when the cyclization was carried out with ICl (entries 3 and 5). Unsubstituted aniline (4d) in the presence of iodine yielded iodo-substituted propargyl amine rather than the quinoline 5d (entry 6). Replacing the iodine with ICl afforded 5d in 38% isolated yield. The napthylamine 4h also provided the corresponding iodoquinoline 5e in high yield (entry 8). When 3-methoxy-substituted aniline 4m was treated with iodine, only 25% of the 3,6-diiodo-substituted quinoline was obtained along with uncyclized aromatic iodinated side products (entry 13). No desired quinoline product was observed in this case when ICl was used (entry 14). The iodocyclization of 2-methoxy-substituted aniline, 4n afforded the corresponding quinoline in good yield when both I2 and ICl electrophiles were used. (entries 15 and 16).
Attention was then focused on investigating the scope of this cyclization protocol in terms of the perfluoroalkyl chain Rf. It was observed that both the C2F5 and C4F9-substituted propargyl amines gave high yields of the corresponding quinolines (Table 4, entries 17 and 18). Further, the aryl substitution on the alkyne moiety of the 2-perfluoroalkyl propargyl amines was varied. The iodocyclization of 4-methoxyphenyl-substituted propargyl amines 4l gave moderate yield (57%) of the quinoline 5l in the presence of I2. Yield was further increased to 84% when I2 was replaced with ICl (Table 4, entry 12). The structure of the 2-trifluoromethylated quinoline 5l was confirmed by 1H NMR, 13C NMR, HRMS and by single crystal X-ray diffraction study (Fig. 2).18
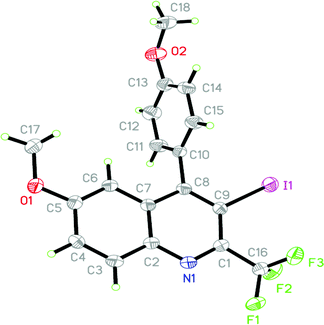 | ||
| Fig. 2 ORTEP diagram of compound 5l. | ||
Changing the substitution at the terminal position of the C–C triple bond to alkyl chain afforded a satisfactory yield of the corresponding quinolines (entries 19–23) in the presence of iodine. When ICl was employed in place of iodine in the cyclization of propargyl alcohol 4s, only a marginal increase in the yield was observed (entry 22). The substrate with vinylic substitution on the terminal alkyne 4t afforded the desired 2-trifluoromethyl-substituted quinoline 5t in 78% yield (entry 23).
The plausible mechanism for iodocyclization of perfluoroalkyl propargyl amine 4 presented herein is outlined in Scheme 6. Initially, I2 coordinates to the alkyne moiety to form the iodonium intermediate A. This is followed by intramolecular nucleophilic attack of the aromatic ring of aniline on the activated alkyne moiety leading to formation of intermediate 1,2-dihydroquinolineB. Subsequent oxidation of B by I2 produces the corresponding quinoline 5.23
 | ||
| Scheme 6 Plausible mechanism for the iodocyclization of perfluoroalkyl propargyl amine and subsequent dehydrogenation to quinoline. | ||
The presence of the iodide functional group at the 3-position of the quinoline ring provides an opportunity to explore the scope of the chemistry. Using 3-iodoquinoline 5k, some complex 2-perfluoroalkyl-substituted heterocycles have been synthesized in good yields.
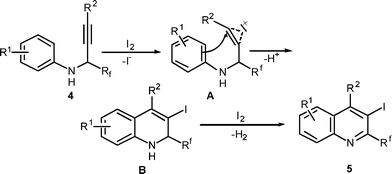
Iodoquinoline 5k underwent facile Suzuki24 coupling with para-tolylboronic acid to afford the highly substituted 6-methoxy-4-phenyl-3-p-tolyl-2-trifluoromethylquinoline 8 in good yield (75%).
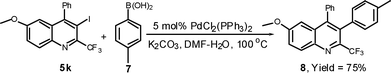
Next, 5k was further subjected to the palladium-catalyzed annulation reaction25 with the diphenyl acetylene. This yielded trifluoromethyl-substituted benzophenanthridine 10 in 70% yield.
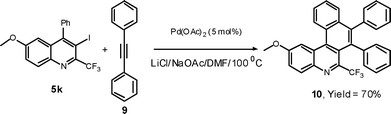
In the metal-halogen exchange reaction,26 the treatment of n-BuLi with 5k gave the 2-trifluoromethyl-substituted quinoline 11 in 95% yield, and subsequent treatment with CO227 afforded highly substituted 2-trifluoromethyl quinoline-3-carboxylic acid 12 in 62% yield.
Antimalarial activity of perfluoroalkyl quinolines
The antimalarial activity of some the compounds (5a, 5c, 5e, 5k, 5o, 5p, 5r, 8 and 10) against a Plasmodium falciparum strain (FDL-B) was examined at different doses, starting from 250 μg/ml with 5-fold serial dilutions up to 0.016 μg/ml.28 The doses were kept constant for all compounds in order to maintain the comparative profile of their activities. The compounds, 5a and 5o showed substantial in vitro activity for total parasite growth inhibition.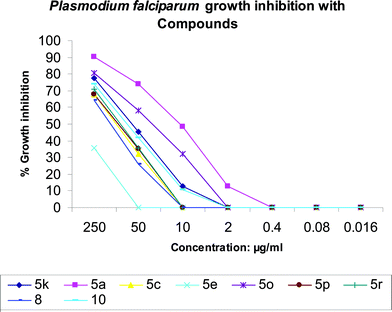
Conclusions
In conclusion, efficient and simple new methodologies for the synthesis of highly substituted perfluoroalkyl quinolines have been developed. These methodologies offer various substitutions at aniline ring or at the terminal alkyne using various perfluoroalkyl groups to allow a diverse range of quinolines to be synthesized. Therefore, such reactions may be distinctly important for pharmacological applications. The iodocyclization processes using I2/CAN, I2 and ICl showed considerable synthetic advantages in terms of product diversity, mild reaction conditions, the simplicity of the reaction process and good-to-excellent yields. A new route for the synthesis of 2-perfluoroalkylpropargyl amines, which are the building blocks of perfluoroalkyl heterocycles, have also been demonstrated. Further work is in progress for the synthesis of more complex molecular structures.Experimental
All reactions were conducted in flame dried glass apparatus. Reactions were followed by TLC analysis using silica plates visualized with a UV lamp. 1H, 13C, and 19F NMR spectra were recorded in Fourier transform mode. Chemical shifts are given in ppm, the internal standard reference is Trimethylsilane for 1H and 13C, and CFCl3 for 19F spectra. Multiplicities in the 1H NMR spectra are described as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad; coupling constants are reported in Hz. For low (MS) and high (HRMS) resolution mass spectra ion mass/charge (m/z) ratios are reported as values in atomic mass units. Melting points were recorded on a Toshniwal melting point apparatus. CAN was activated at 100°C under vacuum for 1h. All commercially available reagents were purchased and used as supplied. DCM was freshly distilled from P2O5.General procedure for the electrophilic cyclization of N-Aryl perfluoroalkyl propargyl imines by I2–CAN.
N-aryl perfluoroalkyl acetylenic imines 3 were prepared by Sonogashira coupling of imidoyl iodide and alkyne according to the literature procedure.15 To a solution of N-aryl perfluoroalkyl propargyl imine, 3 (0.5 mmol) in CH3CN (5 mL), was added finely grounded I2 (2 equiv) and CAN (2 equiv) successively and the mixture was stirred at room temperature. After completion of the reaction as monitored by TLC, the mixture was diluted with 30 mL of EtOAc, and washed with saturated aqueous solution of Na2S2O3 (25 mL). The organic layer was separated and the aqueous layer was extracted with fresh EtOAc (30 mL). The combined organic layers were dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography on silica gel using 10:1 hexane/EtOAc as eluent unless otherwise stated to get pure 5.3-Iodo-6-methyl-4-phenyl-2-(trifluoromethyl)quinoline (5a)
Pale yellow solid: mp 112–113 °C; 1H NMR (300 MHz, CDCl3): δ 8.09 (d, J = 8.7 Hz, 1H), 7.62-7.52 (m, 4H), 7.22-7.17 (m, 2H), 7.11-7.09 (m, 1H), 2.44 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 156.2, 146.5 (q, J = 32.9 Hz), 143.7, 141.4, 140.0, 133.1, 129.6, 128.93, 128.88, 128.70, 125.6, 121.4 (q, J = 276.6 Hz), 89.7, 21.9, (one sp2carbon missing due to overlap). 19F NMR (376.3 MHz, CDCl3) δ−65.20 (s, 3F). MS (ESI): m/z = 414 [M + H]+. HRMS: m/z calcd for C17H12NF3I [M + H]+ 413.9966, found 413.9952.3-Iodo-4-phenyl-2-trifluoromethyl-1-azaspiro[4.5]deca-1,3,6,9-tetraen-8-one (6a)
White solid: mp 162–163 °C; 1H NMR (200 MHz, CDCl3): δ 7.44-7.24 (m, 5H), 6.48 (d, 2H, J = 10.3 Hz), 6.17 (d, 2H, J = 10.3 Hz). 13C NMR (75.5 MHz, CDCl3): δ 184.2, 173.0, 167.7 (q, J = 37.3 Hz), 139.6, 132.6, 131.5, 130.2, 128.8, 127.3, 118.5 (q, J = 276.6 Hz), 83.7, 83.2. 19F NMR (376.3 MHz, CDCl3) δ−67.80 (s, 3F). MS (ESI): m/z = 438 [M + Na]+. HRMS: m/z calcd for C16H9F3INO [M + Na]+ 437.9579, found 437.9581.3-Iodo-4-(4-methoxyphenyl)-2-trifluoromethyl-1-azaspiro[4.5] deca-1,3,6,9-tetraen-8-one (6b)
White solid: mp 161–162°C; 1H NMR (300 MHz, CDCl3): δ 7.35 (d, 2H, J = 9.1 Hz), 6.87 (d, 2H, J = 9.1 Hz), 6.49 (d, 2H, J = 10.0 Hz), 6.17 (d, 2H, J = 10.0 Hz), 3.82 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 184.4, 172.0, 167.8 (q, J = 37.3 Hz), 161.0, 140.3, 132.4, 129.0, 123.6, 118.6(q, J = 276.1 Hz), 114.2, 82.7, 81.9, 55.3. 19F NMR (376.3 MHz, CDCl3) δ−67.76 (s, 3F). MS (ESI): m/z = 445 [M + Na]+. HRMS: m/z calcd for C17H11F3INO2 [M + Na]+ 467.9684, found 467.9688.General procedure for the synthesis of N-Aryl perfluoroalkyl propargyl amines (4)
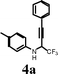
To a solution of N–aryl perfluoroalkyl acetylenic imine 3 (1 mmol) in glacial acetic acid (4 mL) under nitrogen atmosphere at 0 °C was added NaBH3CN (3 equiv.) in portions. The mixture was stirred at room temperature for 30 min. The reaction mixture was diluted with EtOAc (50 mL) and washed with saturated aqueous solution of NaHCO3 until effervescence ceased, followed by saturated aqueous solution of NaCl. The organic layer was dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was purified by column chromatography on silica gel using 10:1 hexane/EtOAc as eluent.(3-Phenyl-1-trifluoromethyl-prop-2-ynyl)-p-tolylamine (4a)
The indicated compound was obtained in 98% yield as pale yellow oil. 1H NMR (300 MHz, CDCl3): δ 7.44-7.37 (m, 2H), 7.34-7.23 (m, 3H), 7.0 (d, 2H, J = 8.2 Hz), 6.65 (d, 2H, J = 8.2 Hz), 4.85-4.74 (m, 1H), 3.80 (d, 1H, J = 9.6 Hz), 2.26 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 142.7, 132.0, 129.9, 129.1, 128.3, 123.8 (q, J = 281.0 Hz), 121.4, 114.9, 86.1, 80.7, 51.1 (q, J = 35.1 Hz), 20.4, (one sp2carbon missing due to overlap). 19F NMR (376.3 MHz, CDCl3) δ−76.05 (d, 3F, J = 6.1 Hz).MS (ESI): m/z = 290 [M + H]+. HRMS: m/z calcd for C17H15F3N [M + H]+ 290.1156, found 290.1163.Synthesis of 5,5,5-Trifluoro-4-(4-methoxyphenylamino)-pent-2-yn-1-ol (4s)
To 3s (1 mmol) (prepared by Sonagashira coupling of N-(4-methoxyphenyl)-2,2,2-triflyroacetimidoyl iodide and THP protected propargyl alcohol) in glacial acetic acid (4 mL) under nitrogen atmosphere at 0 °C was added NaBH3CN (3 equiv.) in portions. The mixture was stirred at room temperature for 30 min. The reaction mixture was again cooled to 0 °C and was added p-TsOH (0.01mmol). The reaction mixture was then stirred at room temperature for the required time. After completion of the reaction as monitored by TLC, the reaction mixture was then diluted with EtOAc (50 mL), and washed with saturated aqueous solution of NaHCO3 until effervescence ceased, followed by saturated aqueous solution of NaCl. The organic layer was dried over Na2SO4 and filtered. The solvent was evaporated under reduced pressure and the product was purified by column chromatography on silica gel using 10:2 hexane/EtOAc as eluent to afford the analytically pure product (4s) in 236 mg (91%).Pale brown oil. 1H NMR (200 MHz, CDCl3): δ 6.82-6.76 (m, 2H), 6.73-6.67 (m, 2H), 4.60-4.50 (m, 1H), 4.27 (d, 2H, J = 2.3 Hz), 3.77 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 154.0 138.6, 123.6 (q, J = 281.5 Hz), 116.7, 114.8, 84.6, 77.4, 55.6, 51.3 (q, J = 33.9 Hz), 50.5.19F NMR (376.3 MHz, CDCl3) δ−76.05 (d, 3F, J = 7.4 Hz). MS (ESI): m/z = 260 [M + H]+. HRMS: m/z calcd for C12H13F3NO2 [M + H]+ 260.0898, found 260.0888.
General procedure for the electrophilic cyclization of N-Aryl perfluoroalkylated propargyl amines by molecular iodine.
To a mixture of N-aryl perfluoroalkylated propargylamine 4 (0.5 mmol), NaHCO3 (2 equiv.) and CH3CN (5 mL) in a 25 mL round-bottomed flask, 3 equiv. of finely grounded iodine was added and the mixture was stirred at room temperature for the required time. After completion of the reaction as monitored by TLC, the mixture was diluted with 30 mL of EtOAc, and washed with saturated aqueous solution of Na2S2O3 (25 mL). The organic layer was separated and the aqueous layer was extracted with fresh EtOAc (30 mL). The combined organic layers were dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography on silica gel using 10:1 hexane/EtOAc as eluent unless otherwise stated to get pure 5.General procedure for the electrophilic cyclization of N-Aryl perfluoroalkylated propargyl amines by ICl.
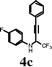
0.5 mmol of the N-aryl perfluoroalkyl propargyl amine 4, 2 equiv of NaHCO3 and 4 mL of dry CH2Cl2 were placed in a 25 mL round bottom flask at 0 °C under the nitrogen atmosphere. 2 Equiv of ICl in 1 mL of dry CH2Cl2 were added dropwise to the flask. The mixture was stirred at room temperature for the required time and was then diluted with 30 mL of EtOAc, and washed with 25 mL of saturated aqueous solution of Na2S2O3. The organic layer was separated and the aqueous layer was extracted with fresh EtOAc (30 mL). The combined organic layers were dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was purified by column chromatography on silica gel using 10:1 hexane/EtOAc as eluent unless otherwise stated to get pure 5.3-Iodo-6-fluoro-4-phenyl-2-(trifluoromethyl)quinoline (5c)
White solid: mp 115–116 °C; 1H NMR (200 MHz, CDCl3): δ 8.25 (dd, 1H, J = 9.5, 5.8 Hz), 7.67-7.51 (m, 4H), 7.26-7.19 (m, 2H), 7.01 (dd, 1H, J = 9.5, 2.9 Hz). 13C NMR (75.5 MHz, CDCl3): δ 162.1 (d, J = 252.5 Hz), 156.5 (d, J = 6.6 Hz), 147.0 (q, J = 32.9 Hz), 142.3, 140.9, 132.8 (d, J = 11 Hz), 129.85 (d, J = 11 Hz), 129.11, 129.0, 128.8, 121.2 (q, J = 276.6 Hz), 121.3 (d, J = 26.3 Hz), 110.5 (d, J = 24.1 Hz), 90.8. 19F NMR (376.3 MHz, CDCl3) δ−65.43 (s, 3F), −107.60–107.68 (m, 1F). MS (ESI): m/z = 418 [M + H]+. HRMS: m/z calcd for C16H9F4IN (M + H)+ 417.9715, found 417.9732.Synthesis of 6-methoxy-4-phenyl-3-p-tolyl-2-(trifluoromethyl) quinoline (8)
A 10 mL round bottom flask was charged with iodoquinoline 5k (107 mg, 0.25 mmol), p-tolylboronic acid (54 mg, 0.4 mmol), Pd(PPh3)2Cl2 (7 mg, 0.01 mmol) and K2CO3 (69 mg, 0.5 mmol) in 5 mL of DMF/H2O (v/v = 4/1). The reaction mixture was stirred under N2 at 100 °C for 6h. The mixture was cooled to room temperature and diluted with 25 mL of EtOAc, washed with 25 mL of H2O and 25 mL of saturated aqueous solution of NaCl. The organic layer was dried over Na2SO4 and filtered. The solvent was evaporated under reduced pressure to get the crude product. The crude product was chromatographed on a silica gel column using 10:1 hexane/EtOAc as eluent to afford the analytically pure product (8) in 74 mg (75%).White solid: mp 239–240 °C; 1H NMR (300 MHz, CDCl3): δ 8.16 (d, 1H, J = 9.1 Hz), 7.39 (dd, 1H, J = 9.1, 3.0 Hz), 7.28-7.21 (m, 3H), 7.06-7.00 (m, 2H), 6.97-6.89 (m, 4H), 6.66 (d, 1H, J = 3.0 Hz), 3.69 (s, 3H), 2.28 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 159.4, 148.9, 143.7 (q, J = 32.9 Hz), 141.9, 136.8, 136.1, 132.4, 132.1, 131.7, 130.3, 129.8, 129.6, 127.9, 127.5, 122.1 (q, J = 276.3 Hz), 122.9, 104.2, 55.4, 21.2, (one sp2carbon missing due to overlap).19F NMR (376.3 MHz, CDCl3) δ−61.63 (s, 3F). MS (ESI): m/z = 394 [M + H]+. HRMS: m/z calcd for C24H19F3NO [M + H]+ 394.1418, found 394.1432.
Synthesis of 2-methoxy-7,8-diphenyl-6-(trifluoromethyl)benzo[k]phenanthridine (10)
A 10 mL round-bottomed flask was charged with iodoquinoline 5k (107 mg, 0.25 mmol), diphenylacetylene (89 mg, 0.5 mmol), Pd(OAc)2 (2.8 mg, 0.012 mmol), NaOAc (41 mg, 0.5 mmol) and LiCl (11 mg, 0.25 mmol) in 5 mL of DMF The reaction mixture was stirred at 100 °C under the atmosphere of nitrogen for 20h. The mixture was cooled to room temperature and diluted with 25 ml of EtOAc, washed with H2O (25 mL) and saturated aqueous solution of NaCl (25 mL). The organic layer was dried over Na2SO4. The solvent was evaporated under reduced pressure. The reaction mixture was chromatographed on a silica gel column using 10:1 hexane/EtOAc as eluent to afford the analytically pure product (10) in 84 mg (70%).White solid: mp 214–215 °C; 1H NMR (300 MHz, CDCl3): δ 9.03 (d, 1H, J = 8.3 Hz), 8.30 (d, 1H, J = 2.3 Hz), 8.23 (d, 1H, J = 9.1 Hz), 7.75-7.56 (m, 3H), 7.49 (dd, 1H, J = 9.1, 3.0 Hz), 7.29-7.21 (m, 3H), 7.12-7.00 (m, 5H), 6.98-6.92 (m, 2H), 4.03 (s, 3H). 13C NMR (75.5 MHz, CDCl3): δ 159.8, 144.1 (q, J = 32.9 Hz), 139.6, 139.4, 138.6, 138.3, 135.1, 134.8, 133.7, 132.6, 131.2, 130.8, 128.5, 127.83, 127.75, 127.70, 126.91, 126.85, 126.6, 125.3, 121.4, 121.2 (q, J = 276.6 Hz), 119.5, 108.0, 55.8, (two sp2carbon missing due to overlap). 19F NMR (376.3 MHz, CDCl3) δ−59.34 (s, 3F). MS (ESI): m/z 480 [M + H]+. HRMS: m/z calcd for C31H21F3NO [M + H]+ 480.1575, found 480.1597.
Synthesis of 6-methoxy-4-phenyl-2-(trifluoromethyl)quinoline (11)
To a two-neck round-bottomed flask, under nitrogen, containing a solution of 5k (107 mg; 0.25 mmol) in dry THF (2 mL) at −78 °C was added n-BuLi (0.16 mL of a 1.6 M solution in hexane, 0.25 mmol) dropwise. The reaction mixture was stirred for 10 min and allowed to stir at room temperature. Then water (2 mL) was added and the reaction mixture was diluted with 25 mL of EtOAc and washed with 25 mL saturated aqueous solution of NaCl. The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel using 10:1 hexane/EtOAc as the eluent to afford the pure product (11) in 72 mg (95%).Pale brown solid: mp 75–76 °C; 1H NMR (300 MHz, CDCl3): δ 8.15 (d, 1H, J = 9.8 Hz), 7.58 (s, 1H), 7.57-7.48 (m, 5H), 7.42 (dd, 1H, J = 9.8, 2.6 Hz), 7.15 (d, 1H, J = 2.6 Hz), 3.80 (s, 3H) 13C NMR (75.5 MHz, CDCl3): δ 159.4, 149.0, 145.1 (q, J = 32.9 Hz), 143.9, 137.5, 131.9, 129.2, 128.9, 128.7, 123.3, 121.8 (q, J = 274.4 Hz), 117.4, 103.4, 55.5 (one sp2carbon missing due to overlap). 19F NMR (376.3 MHz, CDCl3) δ−67.44 (s, 3F). MS (ESI): m/z = 304 [M + H]+. HRMS: m/z calcd for C17H13F3NO (M + H)+ 304.0949, found 304.0950.
Data for 6-methoxy-4-phenyl-2-(trifluoromethyl)-3-uinolinecarboxylic acid (12)
To a two-neck round-bottomed flask, under nitrogen, containing a solution of n-BuLi (0.16 mL of a 1.6 M solution in hexane, 0.25 mmol) was added a solution of 5k (107 mg; 0.25 mmol) in dry THF (2 mL) at −78 °C. The reaction was stirred for 45 min. and then poured on to an excess of freshly crushed dry ice. The reaction mixture was diluted with water (10 mL) and the aqueous phase was washed with diethyl ether (3 × 25 mL). The aqueous layer was acidified with hydrochloric acid to pH 2 and extracted with ethyl acetate (3 × 25 mL). The combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure to get the crude product, which was crystallized from mixture of chloroform and hexane 1:2 (v/v) to afford the analytically pure product (12) in 54 mg (62%). White solid; mp 196–197 °C; 1H NMR (200 MHz, CDCl3): δ 8.13 (d, 1H, J = 9.4 Hz), 7.57-7.43 (m, 4H), 7.42-7.33 (m, 2H), 6.82 (d, 1H, J = 3.1 Hz), 3.72 (s, 3H). 13C NMR (75.5 MHz, CD3OD + CH3OH): δ 169.4, 161.6, 147.8, 143.8, 141.5 (q, J = 35.1 Hz), 136.0, 132.4, 130.6, 130.4, 130.1, 129.6, 127.2, 125.5, 123.0 (q, J = 274.4 Hz), 105.0, 56.0. 19F NMR (376.3 MHz, CDCl3) δ−58.62 (s, 3F). MS (ESI): m/z 348 [M + H]+. HRMS: m/z calcd for C18H13F3NO3 [M + H]+ 348.0847. The structure of 12 was further confirmed by preparing the methyl ester derivative (13) of corresponding acid using CH2N2. White solid; mp 75–76 °C; 1H NMR (200 MHz, CDCl3): δ 8.15 (d, 1H, J = 8.5 Hz), 7.62-7.28 (m, 6H), 6.78 (d, 1H, J = 3.1 Hz), 3.73 (s, 3H), 3.58 (s, 3H).Acknowledgements
M. S. S. and S. R. thank CSIR for providing a research fellowship. We are thankful to NMR, MS, and Analytical Division, IICT for providing spectral data. We also thank R. L. Wilson of RMIT University, Australia, for proof-reading the final manuscript.References
- (a) M. Schlosser, Angew. Chem. Int. Ed., 2006, 45, 5432 CrossRef; (b) M. Hudlicky, Chemistry of Organic Fluorine Compounds, Ellis Horwood Ltd, Chichester, 1992 Search PubMed; (c) I. Ojima, J. R. McCarthy, J. T. Welch, Biomedical Frontiers of Fluorine Chemistry, ACS Symposium Series, no. 639, Washington, 1996 Search PubMed.
- C. J. Ohnmatch, A. R. Patel and R. E. Lutz, J. Med. Chem., 1971, 14, 926–928 CrossRef.
- (a) H. J. Dyke, J. G. Montana, WO 2000026208, 2000; (b) R. Kuang, D. Blythin, N. Y. Shih, H. J. Shue, X. Chen, J. Cao, D. Gu, Y. Huang, J. H. Schwerdt, P. C. Ting, S. C. Wong, L. Xiao, WO 2005116009, 2005.
- H. Sakashita, T. Yoshida, H. Kitajima, M. Takeuchi, Y. Tanaka, T. Yoshimura, F. Akahoshi, Y. Hayashi, WO 2003024942, 2003.
- J. Dade, O. Provot, H. Moskowitz, J. Mayrargue and E. Prina, Chem. Pharm. Bull., 2001, 49, 480–483 CrossRef CAS.
- J. B. Dickey, J. G. McNally, US 2432393, 1947.
- M. S. Raasch, J. Org. Chem., 1962, 27, 1406–1409 CrossRef CAS.
- Y. Kobayashi and I. Kumadaki, Tetrahedron Lett., 1969, 10, 4095–4097 CrossRef.
- (a) R. J. Linderman and K. S. Kirollos, Tetrahedron Lett., 1990, 31, 2689–2692 CrossRef CAS; (b) I. I. Gerus, L. V. Kropachev, M. G. Gorbunova, A. Y. Ilchenko and V. P. Kukhar, Ukr. Khim. Zh. (Russ. Ed.), 1993, 59, 408–411 Search PubMed; (c) H. Keller and M. Schlosser, Tetrahedron, 1996, 52, 4637–4644 CrossRef CAS; (d) M. Schlosser, H. Keller, S. Sumida and J. Yang, Tetrahedron Lett., 1997, 38, 8523–8526 CrossRef CAS; (e) M. Hojo, R. Masuda, Y. Kokuryo, H. Shioda and S. Matsuo, Chem. Lett., 1976, 499–502 CAS; (f) T. Moriguchi, T. Endo and T. Takata, J. Org. Chem., 1995, 60, 3523–3528 CrossRef CAS; (g) A. Colla, M. A. P. Martins, G. Clar, S. Krimmer and P. Fischer, Synthesis, 1991, 483–486 CrossRef CAS; (h) S. I. Vdovenko, I. I. Gerus and M. G. Gorbunova, J. Chem. Soc. Perkin Trans. 2, 1993, 559–562 RSC.
- I. L. Baraznenok, V. G. Nenajdenko and E. S. Balenkova, Eur. J. Org. Chem., 1999, 937–941 CrossRef CAS.
- H. Amii, Y. Kishikawa and K. Uneyama, Org. Lett., 2001, 3, 1109–1112 CrossRef CAS.
- (a) A. Sniady, K. A. Wheeler and R. Dembinski, Org. Lett., 2005, 7, 1769–1772 CrossRef CAS; (b) X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763–766 CrossRef CAS; (c) A. Arcadi, S. Cacchi, G. Fabrizi, F. Marinelli and L. Moro, Synlett., 1999, 9, 1432–1434 CrossRef; (d) D. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905–1909 CrossRef CAS; (e) D. Yue and R. C. Larock, Org. Lett., 2004, 6, 1037–1040 CrossRef CAS; (f) Q. Huang, J. A. Hunter and R. C. Larock, J. Org. Chem., 2002, 67, 3437–3444 CrossRef CAS; (g) A. Arcadi, S. Cacchi, S. Di, Giuseppe, G. Fabrizi and F. Marinelli, Org. Lett., 2002, 4, 2409–2412 CrossRef CAS.
- (a) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS; (b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2310 CrossRef CAS.
- (a) Y. M. Wu, M. Zhang and Y. Q. Li, J. Fluorine Chem., 2006, 127, 218–222 CrossRef CAS; (b) H. B. Yu and W. Y. Huang, J. Fluorine Chem., 1998, 87, 69–73 CrossRef CAS; (c) K. Uneyama and H. Watanabe, Tetrahedron Lett., 1991, 32, 1459–1462 CrossRef CAS.
- (a) H. Amii, M. Kohda, M. Seo and K. Uneyama, Chem. Commun., 2003, 1752–1753 RSC; (b) K. Tamura, H. Mizukami, K. Maeda, H. Watanabe and K. Uneyama, J. Org. Chem., 1993, 58, 32–35 CrossRef CAS; (c) Y. M. Wu, Y. Li and J. Deng, J. Fluorine Chem., 2005, 126, 791–795 CrossRef CAS; (d) K. Uneyama, H. Amii, T. Katagiri, T. Kobayashi and T. Hosokawa, J. Fluorine Chem., 2005, 126, 165–171 CrossRef CAS; (e) K. Uneyama, J. Fluorine. Chem., 1999, 97, 11–25 CrossRef CAS; (f) H. B. Yu and W. Y. Huang, Tetrahedron Lett., 1996, 37, 7999–8000 CrossRef CAS.
- P. R. Likhar, M. S. Subhas, M. Roy, S. Roy and M. L. Kantam, Hel. Chim. Acta, 2008, 91, 259–264 Search PubMed.
- (a) For ipso iodocyclizations see: Q. F. Yu, Y. H. Zhang, Q. Yin, B. X. Tang, R. Y. Tang, P. Zhong and J. H. Li, J. Org. Chem, 2008, 73, 3658–3661 Search PubMed; (b) X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2005, 127, 12230–12231 CrossRef CAS; (c) C. W. Li, C. I. Wang, H. Y. Liao, R. Chaudhuri and R. S. Liu, J. Org. Chem., 2007, 72, 9203–9207 CrossRef CAS.
- CCDC 696933 (6a) and 696932 (5l) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- (a) For molecular iodine as a oxidising agent see H. Togo and S. Iida, Synlett., 2006, 14, 2159–2175 Search PubMed and references cited therein; (b) C. Lin, P.-T. Lai, S. K.-S. Liao, W.-T. Hung, W.-B. Yang and J.-M. Fang, J. Org. Chem., 2008, 73, 3848–3853 CrossRef CAS.
- N. Lebouvier, C. Laroche, F. Huguenot and T. Brigaud, Tetrahedron Lett., 2002, 43, 2827–2830 CrossRef CAS.
- Y. Xu and W. R. Dolbier, Jr., J. Org. Chem., 2000, 65, 2134–2137 CrossRef CAS.
- G. Magueur, B. Crousse and D. B. Delpon, Tetrahedron Lett., 2005, 46, 2219–2221 CrossRef CAS.
- (a) M. I. Rodrı'guez-Franco, I. Dorronsoro, A. I. Herna'nde-Higueras and G. Antequera, Tetrahedron Lett., 2001, 42, 863–865 CrossRef CAS; (b) V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862–1891 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- R. C. Larock, M. J. Doty, Q. Tian and J. M. Zenner, J. Org. Chem., 1997, 62, 7536 CrossRef CAS.
- (a) M. Aso, T. Kaneko, M. Nakamura, N. Koga and H. Suemune, Chem. Commun., 2003, 1094–1095 RSC; (b) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem. Int. Ed., 2003, 42, 4302–4320 CrossRef CAS.
- F. Cottet, M. Marull, O. Lefebvre and M. Schlosser, Eur. J. Org. Chem., 2003, 1559–1568 CrossRef CAS.
- (a) W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS; (b) S. Biswas, J. Postgrad. Med., 2001, 47, 240–243 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the compounds, spectral data and antimalarial study data. CCDC reference numbers 696932 and 696933. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b815398a |
| This journal is © The Royal Society of Chemistry 2009 |