A straightforward approach towards glycoamino acids and glycopeptidesviaPd-catalysed allylic alkylation†
Katja
Krämer
,
Jan
Deska
,
Christina
Hebach
and
Uli
Kazmaier
*
Institut für Organische Chemie, Universität des Saarlandes, D-66123, Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de; Fax: +49 681 302 2409; Tel: +49 681 302 3409
First published on 31st October 2008
Abstract
Chelated enolates are versatile nucleophiles for palladium-catalysed allylic alkylations. Even with complex allylic substrates the reaction proceed without significant isomerisation. This allows the stereoselective introduction of polyhydroxylated allylic sidechains into amino acids and peptides with retention of the olefin geometry.
Introduction
Glycopeptides play a central role in intercellular processes such as cell-cell recognition or ligand-receptor interaction.1 Biosynthetically these glycopeptides and -proteins are the result of post-translational peptide modifications, managed by numerous glycosyltransferases.2 In general, the sugar moieties are connected to the peptide chain via N- or O-acetals, preferentially towards serines or threonines. The acetal linkage is easily to realize for the enzymes involved, but on the other hand is also sensitive towards hydrolytic cleavage. With respect, that the building blocks of glycopeptides, the glyco amino acids are also interesting drug candidates, this lability is a significant limitation.3 Replacing the connecting oxygen by a methylene unit offers a great deal of stability without changing the steric properties of this so-called C-glycosides. Not surprisingly, a lot of different synthetic approaches have been developed in recent years,4 using electrophilic,5 nucleophilic6 or radical glycal precursors.7 Recently, several approaches also used ruthenium-catalysed olefin cross-metathesis to generate unsaturated glycosyl amino acids or derivatives thereof.8 For example, Nolan et al. reported on the coupling of sugar-derived allyl alcohols with protected allyl glycine giving rise to the γ,δ-unsaturated polyhydroxylated amino acid,9 which could by cyclized to the C-linked glycosyl amino acid in the presence of Hg-salts (Scheme 1).10 Best results were obtained with the second generation Grubbs-catalyst (G2)11 which gave yields around 50%. The major problem was the formation of homocoupling products in the metathesis step.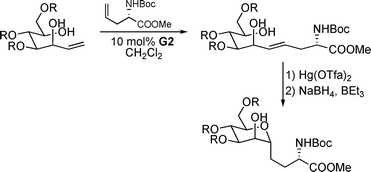 | ||
| Scheme 1 Synthesis of glycosylamino acids via cross-metathesis. | ||
In principle, the double bond obtained can be subjected to other addition reactions to generate modified glycosyl amino acids. Therefore, it would be important to generate the double bond in a highly stereoselective fashion (E or Z), which is not a trivial issue under the metathesis conditions.
Based on our interest in the synthesis of azasugars12 and polyhydroxylated amino acids,13 we developed an alternative protocol towards these unsaturated amino acids, taking advantage of a chelate-enolate Claisen rearrangement.14 If ester of chiral allyl alcohols15 are subjected to this version of the Claisen rearrangement, an excellent chirality transfer is observed towards the α-position of the amino acid (Scheme 2). Based on the chair-like transition state, the (E)-configured double bond is formed exclusively in high yield.14 The free carboxylic acid obtained can be coupled directly with other amino acids and peptides, giving rise to glycopeptide-type structures.
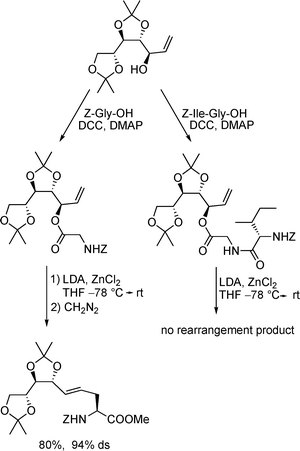 | ||
| Scheme 2 Synthesis of glycosyl amino acids via chelate-Claisen rearrangements. | ||
Interestingly, incorporation of the allylic alcohol into a dipeptide and attempts to rearrange these peptide esters were unsuccessful, although unfunctionalized allylic esters gave rise to modified peptides in high yield.16 In principle, also chirality transfer from the peptide chain towards the newly formed amino acid is possible,17 but with these rather complex substrates only isomerisation of the double bond was observed.
Results and discussion
To figure out if this Claisen protocol is also suitable to introduce not only a polyhydroxylated side chain, but also a glycoside unit directly, we synthesized an allylic alcohol derived from the protected furanoside aldehyde 1 (Scheme 3).18Vinyl magnesium bromide addition gave rise to the required allyl alcohols (S)-2 and (R)-2, which could be separated by flash chromatography.6 Coupling of (S)-2 with Z-glycine gave rise to ester (S)-3 as monoclinic crystals, which allowed the determination of the newly formed stereogenic center via X-ray structure analysis. Subsequent chelate-enolate Claisen rearrangement under standard conditions gave disappointing results. Only 10% of the rearrangement product (R)-4 could be obtained, together with unreacted allylic ester (S)-3 (40%) and (S)-2, resulting from decomposition of the enolate. Obviously, with this sterically demanding allylic ester, the rearrangement is getting very slow and side reactions (such as elimination) have to be considered to an increased extent. By prolonging the reaction time the yield could be increased to 32%, but still, with these highly functionalized substrates the chelate-Claisen rearrangement gets to its bounds.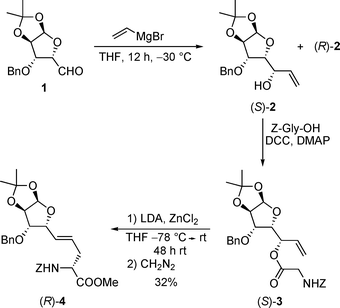 | ||
| Scheme 3 Chelate-Claisen rearrangements of glycosylallylic esters. | ||
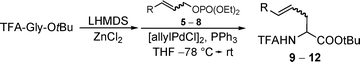
During recent years we have developed an independent approach towards these γ,δ-unsaturated amino acids based on Pd-catalysed allylic alkylations.19 We stumbled into this field during our peptide Claisen rearrangements,20 and nowadays this synthetic pathway has developed to be a good alternative to the Claisen rearrangement. Both protocols complement each other, giving rise to the opposite diastereomers if substituted allylic substrates are used.21 If chelated enolates of TFA-protected glycinates (generated in situ via deprotonation/transmetallation) are subjected to Pd-catalysed allylic alkylations,22 the reaction proceeds under very mild conditions, even at −78 °C. A wide range of allylic substrates can be used, also highly functionalized ones such as 5.23 Therefore, this approach is also suitable for the synthesis of polyhydroxylated amino acids. Also here, the (E)-double bond is formed nearly exclusively, what can be explained by a reaction of the thermodynamically more stable syn-π-allyl complex. In general, π-allyl complexes can undergo fast π–σ–π-isomerisation, what makes it difficult to transfer the olefin geometry from the allylic substrate into the product.24 This is a major issue especially with (Z)-allyl compounds, which also give rise to the (E)-product under standard conditions for allylic alkylations.
Based on the high reactivity of the chelated enolates this isomerisation can be suppressed almost completely for 1,3-disubstituted allylic substrates,25 and to some extend also for terminal π-allyl complexes.26 In many cases, (Z)-allylic substrates reacted more selectively than the (E)-analogues. Therefore, we were interested to see, if we can use the higher selectivity of (Z)-substrates also for the synthesis of polyhydroxylated and glycosyl amino acids, preferentially those containing a (Z)-double bond. The synthesis of several (Z)-allylic substrates, especially phosphates is summarized in Scheme 4. Phosphates showed a higher reactivity compared to the commonly used acetates and carbonates, not only in Pd- but also Rh-catalysed reactions.22
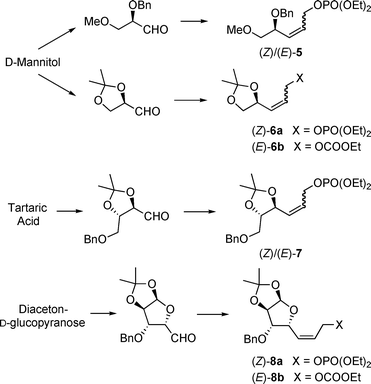 | ||
| Scheme 4 Synthesis of carbohydrate-derived allylic substrates. | ||
Starting from readily available chiral pool materials the (Z)-configured allylic substrates were prepared from the corresponding aldehydes via an Ando version of the Horner–Wadsworth–Emmons reaction.27 Dibal-reduction of the α,β-unsaturated ester formed and subsequent esterification of the resulting allyl alcohol with diethyl chlorophosphate gave rise to the (Z)-allyl phosphates 5–8. For analytical purposes and to investigate the difference between (E)- and (Z)-substrates, the corresponding (E)-substrates were obtained in an analogous way using a standard phosphonate condensation in the first step. The (E)-configured allyl carbonates (E)-6b and (E)-8b were prepared previously.
Interesting results were obtained in the comparison of the (E)- and (Z)-phosphates 5 (Table 1, entries 1 and 2). With (E)-5 the linear product having a (E)-double bond was formed exclusively with high yield and excellent chiral induction. The chiral center at the α-position of the amino acid is exclusively controlled by the stereogenic center in the allyl moiety. With respect, that these two stereogenic centers are separated by three atoms, an induction of 90% is remarkable. Even better results were obtained with the (Z)-substrate. The yield was excellent, and the (Z)-double bond could be conserved to more than 90%, indicating that π–σ–π-isomerisation could be suppressed efficiently. Surprisingly, the induction for the (Z)-product was significantly lower compared to the previous experiment, but the (E)-product (resulting from the isomerisation) was obtained as a single stereoisomer.
| Entry | Subs. | Prod. | Yield (%) | (Z:E) | (Z) (% ds) | (E) (% ds) |
|---|---|---|---|---|---|---|
| 1 | (Z)-5 | 9 | 99 | 91:9 | 61 (S,S) | 99 (R,S) |
| 2 | (E)-5 | 9 | 79 | 1:99 | — | 90 (R,S) |
| 3 | (Z)-6a | 10 | 92 | 85:15 | 76 (R,S) | 99 (R,S) |
| 4 | (E)-6b | 10 | 77 | 1:99 | — | 81 (R,S) |
| 5 | (Z)-7 | 11 | 93 | 98:2 | 95 (R,S,S) | — |
| 6 | (E)-7 | 11 | 69 | 1:99 | — | 87 (R,S,S) |
| 7 | (Z)-8a | 12 | 99 | 90:10 | 99 (R,R,R,S,R) | 65 (R,R,R,S,R) |
| 8 | (E)-8b | 12 | 98 | 1:99 | — | 68 (R,R,R,S,R) |
To prove the generality of this observation, we also subjected the other substrate couples to the same reaction conditions. Comparable results were obtained with substrates 6 (entries 3, 4). Again the yield and the ds for the (E)-product was significantly better if (Z)-6 was used, although the (Z)-selectivity was slightly worse than with the first substrate. Interestingly the selectivity the (Z)-product was formed with increased, probably because of the increased sterical hindrance of the allylic substituent. This tendency was confirmed by using the even more hindered substrates 7 and 8. With (Z)-7 the olefin geometry could be conserved nearly completely (no isomerisation!) and both, yield and stereoselectivity were excellent. By far the best results in this respect were obtained with the glycosidic substrate 8, giving quantitative yield and stereoselectivity. The stereoisomerically pure product was obtained by crystallization.
This clearly indicates that, for the synthesis of complex highly functionalized amino acids, the Pd-catalysed allylic alkylation is definitely more suitable than the chelate-Claisen rearrangement. To prove if this methodology can also be used for the synthesis of glycopeptides by direct introduction of functionalized side chains into peptides,28 we subjected dipeptide enolates towards allylic alkylation using 6 and 7 as substrates (Scheme 5). Peptides are slightly less reactive than the corresponding amino acid enolates, requiring higher reaction temperatures. Under these conditions the olefin geometry of the allylic substrates does not play any role, because the π-allyl complexes formed are in an equilibrium. The same results are obtained if the branched allylic substrates with a terminal double bond are used.23 In both cases the allylated products were obtained in good yield and excellent diastereoselectivity, while the stereochemical outcome of the reaction is controlled exclusively by the stereogenic center in the peptide chain. An (S)-amino acid in the peptide chain induces a (R)-amino acid at the C-terminus of the chain. The stereochemical outcome can be explained by the formation of peptide-metal complexes and the shielding of one face of the enolate formed by the side chain of the adjacent amino acid.28
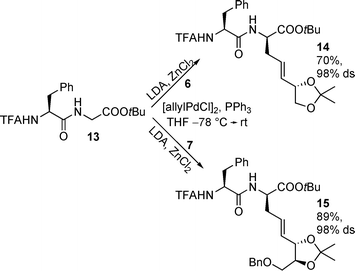 | ||
| Scheme 5 Allylic alkylations of dipeptide enolates. | ||
Conclusion
In conclusion, we have shown that the Pd-catalysed allylic alkylation is a powerful tool for the synthesis of complex amino acids. Even with sterically highly hindered allylic substrates, excellent yields can be obtained not only with amino acid but also peptide enolates.Experimental
General remarks
All the reactions were carried out in oven-dried glassware (100 °C) under nitrogen. All the solvents were dried before use: THF was distilled from LiAlH4 and CH2Cl2 from CaH2. The products were purified by flash chromatography on silica gel (0.063–0.2 mm). Mixtures of ethyl acetate and hexanes were generally used as the eluents. Analysis by TLC was carried out on commercially precoated Polygram SIL-G/UV 254 plates (Machery-Nagel, Dueren). Visualization was accomplished with UV light, KMnO4 solution, or iodine. Melting points were determined on a Büchi melting-point apparatus and are uncorrected. 1H- and 13C-NMR spectroscopic analysis was performed on Bruker AC-500 or Bruker DRX-500 spectrometers. Selected signals for the minor diastereomers are extracted from the spectra of the diastereoisomeric mixture. The values for diastereoisomeric excess were determined by analytical HPLC using a Trentec Reprosil-100 Chiral-NR 8 mm-column and a Shimadzu UV detector. Optical rotations were measured on a Perkin-Elmer polarimeter PE 341. Chemical ionisation (CI) mass spectra were performed on a Finnigan MAT 95, FAB spectra on a Joel JMS-700 mass spectrometer. Elemental analyses were carried out at the Department of Chemistry at Saarland University.General procedure of the preparation of the allyl phosphates 5–8
Pyridine (2.5 eq.), DMAP (5 mol%) and diethyl chlorophosphate (1.2 eq.) were added to a solution of the corresponding allyl alcohol in dry dichloromethane (2 mL/mmol) at 0 °C and the mixture was stirred over night. The solution was diluted with dichloromethane before 1 N KHSO4 was added. The layers were separated, the aqueous phase was extracted three times with diethyl ether and the combined organic layers were dried over Na2SO4. After evaporation of the solventin vacuo the crude product was purified by flash chromatography (hexanes/ethyl acetate = 1:1).General procedure for the allylic alkylation using polyhydroxylated allylic substrates
In a Schlenk flask hexamethyldisilazane (222 mg, 1.38 mmol) was dissolved in dry THF (2 mL) under argon. After cooling the solution to −78 °C, n-BuLi (1.6 M in hexanes, 0.78 mL, 1.25 mmol) was added slowly. The cooling bath was removed, and the solution was stirred 10 min. In a second Schlenk flask ZnCl2 (76 mg, 0.55 mmol) was carefully dried in vacuo. After cooling to room temperature, TFA-Gly-OtBu (114 mg, 0.50 mmol) was added dissolved in dry THF (1 mL). The solution was cooled to −78 °C, before the freshly prepared LHMDS solution was added and stirring was continued for 20 min. In a third flask, [allylPdCl]2 (1 mg, 0.0025 mmol) and PPh3 (3 mg, 0.0113 mmol) were dissolved in dry THF (0.5 mL). After stirring for 15 min at room temperature, this solution was added to the chelated enolate at −78 °C. At the same temperature the allylic substrate (0.25 mmol) was added drop wise in THF (1 mL). Excess of dry ice was added to the cooling bath, before the mixture was allowed to warm to room temperature overnight. The solution was diluted with diethyl ether and then quenched by the addition of 1 N KHSO4. The layers were separated, the aqueous layer was extracted three times with diethyl ether and the combined organic layers were dried over MgSO4. The solvent was evaporated under vacuum and the residue was purified by flash-chromatography.General procedure for allylic alkylations of peptides
n-BuLi (1.6M in hexanes, 0.82 mL, 1.31 mmol) was added drop wise to a solution of hexamethyldisilazane (233 mg, 1.44 mmol) in dry THF (2 mL) at −78 °C. The cooling bath was removed, and the solution was allowed to warm up to room temperature. In a second flask, ZnCl2 (57 mg, 0.42 mmol) was carefully dried in vacuo with a heat gun. After the mixture was cooled to room temperature, the dipeptide 13 (140 mg, 0.375 mmol) was added dissolved in dry THF (2 mL). The freshly prepared LHMDS solution was cooled to −78 °C and the ZnCl2/peptide solution was slowly added by syringe. In a third flask, [allylPdCl]2 (1.8 mg, 5.0 mmol) and PPh3 (5.9 mg, 22.5 mmol) were dissolved in dry THF (0.5 mL), allyl carbonate (0.25 mmol) was added, and the catalyst/carbonate solution was transferred to the cold solution of the zinc enolate by syringe. The excess dry ice was removed from the cooling bath, and the reaction mixture was allowed to warm up. After diluting with diethyl ether, the mixture was quenched by the addition of 1 N HCl. The aqueous layer was extracted twice with diethyl ether and the combined organic layers were dried over Na2SO4. The solvent was evaporated and the residue was purified by column chromatography.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft as well as the Fonds der Chemischen Industie is gratefully acknowledged.References
- (a) R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS; (b) T. K. Lindhorst, in Essentials of Carbohydrate Chemistry and Biochemistry, ed. T. K. Lindhorst, Wiley-VCH, Weinheim, 2000, chapter 7 Search PubMed; (c) Examples for participation of glycopeptides in viral infections: R. L. Willey, D. H. Smith, L. A. Lasky, T. S. Theodore, P. L. Earl, B. Moss, D. J. Capon and M. A. Martin, J. Virol., 1988, 62, 139–147 Search PubMed; (d) W. R. Lee, X.-F. Yu, W.-J. Syu, M. Essex and T. -H. Lee, J. Virol., 1992, 66, 1799–1803 CAS.
- (a) Marth, J. B. Lowe, A. Varki, in Essentials of Glycobiology, ed. A. Varki, R. Lummins, J. Esko, H. Freeze, G. Hart, J. Marth, Cold Spring Harbor Press, New York, 1999, chapters 8 and 17 Search PubMed; (b) P. Sears and C. H. Wong, Cell. Mol. Life Sci., 1998, 54, 223–252 CrossRef CAS.
- F. Schweizer, Angew. Chem., 2002, 114, 240–264 CrossRef; F. Schweizer, Angew. Chem. Int. Ed., 2002, 41, 230–253 CrossRef CAS.
- M. H. D. Postema, in C-Glycoside Synthesis, ed. M. H. D. Postema, CRC Press, Boca Raton, 1995, and references cited therein Search PubMed.
- (a) A. Dondoni, A. Marra and A. Massi, J. Chem. Soc. Chem. Commun., 1998, 1741–1742 RSC; (b) A. Dondoni, A. Marra and A. Massi, J. Org. Chem., 1999, 64, 933–944 CrossRef CAS; (c) T. Fuchs and R. R. Schmidt, Synthesis, 1998, 753–775 CrossRef; (d) C. H. Rohrig, M. Takki and R. R. Schmidt, Synlett, 2001, 1170–1172 CrossRef CAS; (e) A. Dondoni, N. Catozzi and A. Marra, J. Org. Chem, 2004, 69, 5023–5036 CrossRef CAS; (f) T. G. Marron, T. J. Woltering, G. Weitz-Schmidt and C. H. Wong, Tetrahedron Lett., 1996, 37, 9037–9039 CrossRef CAS.
- D. Urban, T. Shrydstrup and J. Bean, J. Chem. Soc. Chem. Commun., 1998, 955–956 RSC.
- G. E. Keck, E. J. Engholm, J. B. Yates and M. R. Wiley, Tetrahedron, 1985, 41, 4079–4094 CrossRef CAS.
- (a) A. Dondoni and P. P. Marra, J. Chem. Soc. Perkin Trans. 1, 2001, 2380–2388 RSC; (b) G. J. McGavreg, T. E. Benedum and F. W. Schmidtmann, Org. Lett., 2002, 4, 3591–3594 CrossRef CAS; (c) E. G. Nolen, A. J. Kurish, K. A. Wong and M. D. Orlando, Tetrahedron Lett., 2003, 44, 2449–2453 CrossRef CAS.
- E. G. Nolan, A. J. Kurish, J. M. Potter, L. A. Donahue and M. D. Orlando, Org. Lett., 2005, 7, 3383–3386 CrossRef.
- A. Boschetti, F. Nicotra, L. Panza and G. Russo, J. Org. Chem., 1988, 53, 4181–4185 CrossRef CAS.
- (a) H. E. Blackwell, D. J. O'Leary, A. K. Chatterjee, R. A. Washenfelder, D. A. Bussmann and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 58–71 CrossRef CAS; (b) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- (a) R. Grandel and U. Kazmaier, Tetrahedron Lett., 1997, 38, 8009–8012 CrossRef CAS; (b) C. Schneider and U. Kazmaier, Eur. J. Org. Chem., 1998, 1155–1159 CrossRef CAS; (c) U. Kazmaier and R. Grandel, Eur. J. Org. Chem., 1998, 1833–1840 CrossRef CAS.
- (a) R. Grandel, U. Kazmaier and F. Rominger, J. Org. Chem., 1998, 63, 4524–4528 CrossRef CAS; (b) U. Kazmaier and C. Schneider, Synthesis, 1998, 1321–1326 CrossRef CAS.
- (a) U. Kazmaier, Amino Acids, 1996, 11, 283–299 CrossRef CAS; (b) U. Kazmaier and C. Schneider, Synlett, 1996, 975–977 CrossRef; (c) U. Kazmaier, Lieb. Ann./Rec., 1997, 285–295 Search PubMed.
- C. Schneider and U. Kazmaier, Synthesis, 1998, 1314–1320 CrossRef CAS.
- (a) U. Kazmaier and S. Maier, J. Chem. Soc. Chem. Commun., 1998, 22535–2536 Search PubMed; (b) U. Kazmaier and S. Maier, Org. Lett., 1999, 1, 1763–1766 CrossRef CAS; (c) S. Maier and U. Kazmaier, Eur. J. Org. Chem., 2000, 1241–1251 CrossRef CAS.
- (a) U. Kazmaier and S. Maier, J. Org. Chem., 1999, 64, 4573–4575; (b) U. Kazmaier, S. Maier and F. Zumpe, Synlett, 2000, 1523–1535 CAS.
- R. C. Anderson and B. Fraser-Reid, J. Org. Chem., 1985, 50, 4781–4786 CrossRef CAS.
- (a) U. Kazmaier, Current Org. Chem., 2003, 317–328 Search PubMed; (b) M. Bauer and U. Kazmaier, Recent Res. Dev. Org. Chem., 2005, 9, 49–69 Search PubMed.
- U. Kazmaier, J. Org. Chem., 1994, 59, 6667–6670 CrossRef CAS.
- U. Kazmaier and F. Zumpe, Angew. Chem., 1999, 111, 1572–1574 CrossRef; U. Kazmaier and F. Zumpe, Angew. Chem. Int. Ed., 1999, 38, 1468–1470 CrossRef CAS.
- (a) For comparable Rh-catalysed allylations see: U. Kazmaier and D. Stolz, Angew. Chem., 2006, 118, 3143–3146 Search PubMed; U. Kazmaier and D. Stolz, Angew. Chem. Int. Ed., 2006, 45, 3072–3075 CrossRef; (b) D. Stolz and U. Kazmaier, Synthesis, 2008, 2288–2292 CAS.
- (a) U. Kazmaier and T. Lindner, Angew. Chem., 2005, 117, 3368–3371 CrossRef; U. Kazmaier and T. Lindner, Angew. Chem. Int. Ed., 2005, 44, 3303–3306 CrossRef CAS; (b) T. Lindner and U. Kazmaier, Adv. Synth. Catal., 2005, 1687–1695 CrossRef CAS.
- (a) P. R. Auburn, P. B. Mackenzie and B. Bosnich, J. Am. Chem. Soc., 1985, 107, 2033–2046 CrossRef CAS; (b) P. B. Mackenzie, J. Whelan and B. Bosnich, J. Am. Chem. Soc., 1985, 107, 2046–2054 CrossRef CAS; (c) P. Corradini, G. Maglio, A. Musco and G. Paiaro, J. Chem. Soc. Chem. Commun., 1996, 618–619 Search PubMed; (d) N. Solin and K. J. Szabo, Organometallics, 2001, 20, 5464–5471 CrossRef CAS; (e) J. Yu, M. J. Gaunt and J. B. Spencer, J. Org. Chem., 2002, 67, 4627–4629 CrossRef CAS; (f) M. Ogasawara, K. -i. Takizawa and T. Hayashi, Organometallics, 2002, 21, 4853–4861 CrossRef CAS.
- (a) U. Kazmaier and F. Zumpe, Angew. Chem., 2000, 112, 805–807 CrossRef; U. Kazmaier and F. Zumpe, Angew. Chem. Int. Ed., 2000, 39, 802–804 CrossRef CAS; (b) U. Kazmaier and F. Zumpe, Eur. J. Org. Chem., 2001, 4067–4076 CrossRef CAS; (c) U. Kazmaier and M. Pohlman, Synlett, 2004, 623–626 CrossRef CAS.
- (a) U. Kazmaier and K. Krämer, J. Org. Chem., 2006, 71, 8950–8953 CrossRef; (b) U. Kazmaier, D. Stolz, K. Krämer and F. Zumpe, Chem. Eur. J., 2008, 14, 1322–1329 CrossRef CAS.
- (a) K. Ando, J. Org. Chem., 1999, 64, 8406–8408 CrossRef CAS; (b) P. Pihko and T. M. Salo, Tetrahedron Lett., 2003, 44, 4361–4364 CrossRef CAS.
- (a) U. Kazmaier, J. Deska and A. Watzke, Angew. Chem., 2006, 118, 4973–4976 CrossRef; U. Kazmaier, J. Deska and A. Watzke, Angew. Chem. Int. Ed., 2006, 45, 4855–4858 CrossRef CAS; (b) Deska and U. Kazmaier, Angew. Chem., 2007, 119, 4654–4657 CrossRef; Deska and U. Kazmaier, Angew. Chem. Int. Ed., 2007, 46, 4570–4573 CrossRef; (c) J. Deska and U. Kazmaier, Chem. Eur. J., 2007, 13, 6204–6211 CrossRef CAS.
- S. J. Danishefsky, M. P. DeNinno, G. B. Phillips and R. E. Zelle, Tetrahedron, 1986, 42, 2809–2819 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and 13C-NMR spectra of compounds 5–12. See DOI: 10.1039/b813978d |
| This journal is © The Royal Society of Chemistry 2009 |