A divergent synthesis of minor groove binders with tail group variation†
David
Breen
,
Alan R.
Kennedy
and
Colin J.
Suckling
*
WestCHEM, Department of Pure & Applied Chemistry, University of Strathclyde, 296 Cathedral Street, Glasgow, G1 1XL, Scotland. E-mail: c.j.suckling@strath.ac.uk; Fax: +44 141 548 5743; Tel: +44 141 548 2271
First published on 7th November 2008
Abstract
A new synthesis of polyamide minor groove binders in which diversity is introduced by the nucleophilic substitution of a 2-sulfido-1,3,2-diazaphospholidinyloxy substituent by volatile secondary amine nucleophiles is described. Such a method has potential value for economically investigating structure–activity relationships in this important class of compounds through library synthesis. As an example using this method are prepared two new minor groove binders with pyrrolidinyl or piperidinyl tail groups that are close relatives of highly active antibacterial minor groove binders with morpholinyl tail groups. The antibacterial activity found against Staphylococcus aureus and Mycobacterium spp. indicates that the pKa of this set of compounds is not the dominant factor in determining the antibacterial activity.
Introduction
Polyamide minor groove binders (MGBs) have been found to have significant anti-infective1,2 and anticancer activity.3,4 These compounds are composed of aromatic and heteroaromatic amino acid amides usually with an aromatic ring at the N-terminus and a tertiary alkylamino, amidino, or guanidine group at the C-terminus, the so-called tail group. The importance of the pKa of the tail group has been suggested to be of major importance in the selectivity and potency of the biological activity of such MGBs.5 In most cases, the synthesis of such compounds has required the preparation of many intermediates with the specific tail group of interest.6 In order to investigate more rapidly the variation of tail groups, it would be advantageous to have a method of synthesis that inserted the amino tail group at the end of the synthesis of the MGB. Such a method must be compatible with critical functional groups in the MGB such as the alkene, which has a major role in promoting antibacterial activity.2 We describe here a method suitable for multiple parallel synthesis based upon phosphorus chemistry that meets these requirements, together with the biological activity of some of the products.Synthesis
The starting point was the possibility that a precursor with a P(III) tail group might be carried through to an MGB oligomer and the various amino tail groups introduced by a variation of the Micahelis–Arbuzov reaction (Scheme 1). Many variations of this strategy were tried including alkyl and aryl phosphites and phosphoramidates. None was successful either because of poor reactivity or more commonly because of ready oxidation from P(III) to P(V) during several of the synthetic steps. Eventually, the solution to this problem was found through the use of diazaphosphacyclopentanes, which were found to have high stability to oxidation and to provide derivatives capable of substitution. The preparation and substitution of the diazaphosphocyclopentanes was developed first on monomeric precursors of MGBs and simple benzenoid analogues. | ||
| Scheme 1 Concept for divergent amino tail synthesis. | ||
Chloro-1,3-dimethyl-1,3,2-diazaphosphacyclopentane, 1, was prepared as described by Anson and McGuigan7 in 62% yield free from P(V) impurities as shown by the single 31P NMR resonance at 171.76 ppm. The intermediate phosphite esters, for example 2, were obtained in 92–95% yield from the corresponding alcohol in the presence of triethylamine; again, significant oxidation did not occur as shown by the single 31P resonance at close to 128 ppm (Scheme 2). The choice of substituted ethanols as substrates is not essential for the chemistry of the reactions but derives from the fact that aminoethyl substituted tail groups afford the strongest antibacterially active compounds;2 this was regarded as an essential feature for the MGB synthesis.
 | ||
| Scheme 2 | ||
In accordance with the original concept (Scheme 1), the two-step cleavage of the phosphite was attempted. Alkylation with methyl triflate at −75 °C afforded the phosphonium salt 3; clear evidence for alkylation at phosphorus and not on the ring nitrogen atoms came from the new methyl resonance which appears as a doublet due to 31P–1H coupling (δH = 1.34, J = 22.5 Hz). Subsequent nucleophilic displacement of the phosphonium salt unfortunately was not successful with morpholine at −78 °C, −40 °C, 0 °C, and room temperature nor with pyrrolidine, in which case the 2-phenyltetrahydro-oxazole 4 was obtained at −78 °C and at room temperature together with the phosphonamidate 5 (δP = 40.93); intramolecular nucleophilic displacement by the amide carbonyl group accounts well for this observation. This reaction clearly demonstrates the thermodynamic driving force for the C–O cleavage and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O formation planned but the substrates require modification in order to develop the MGB synthesis. The phenylethanol derivative 6 (Scheme 3) was used as a test substrate to avoid the intramolecular cleavage but unfortunately, clean products were not isolable under a wide range of reaction conditions using methyl triflate followed by pyrrolidine. The required transformation was, however, effected with the phenylethanol derivative 6 using methyl iodide as an alkylating agent followed by pyrrolidine. Substitution to afford the intermediate phosphonium salt occurred in close to quantitative yield and substitution with an excess of pyrrolidine afforded N-(2-phenyl)ethylpyrrolidine in 28% isolated yield, over the two steps of P–O cleavage and substitution of the intermediate alkyl iodide by pyrrolidine. The alkyl iodide is a potential intermediate for the synthesis of MGBs; however the risk of oxidation to iodine and subsequent addition to alkene links in the MGBs argued for the investigation of further possibilities.
O formation planned but the substrates require modification in order to develop the MGB synthesis. The phenylethanol derivative 6 (Scheme 3) was used as a test substrate to avoid the intramolecular cleavage but unfortunately, clean products were not isolable under a wide range of reaction conditions using methyl triflate followed by pyrrolidine. The required transformation was, however, effected with the phenylethanol derivative 6 using methyl iodide as an alkylating agent followed by pyrrolidine. Substitution to afford the intermediate phosphonium salt occurred in close to quantitative yield and substitution with an excess of pyrrolidine afforded N-(2-phenyl)ethylpyrrolidine in 28% isolated yield, over the two steps of P–O cleavage and substitution of the intermediate alkyl iodide by pyrrolidine. The alkyl iodide is a potential intermediate for the synthesis of MGBs; however the risk of oxidation to iodine and subsequent addition to alkene links in the MGBs argued for the investigation of further possibilities.
 | ||
| Scheme 3 | ||
The alternative strategy was the activation of the phosphorus leaving group by oxidation with sulphur.8 In order to replicate the requirements for MGB synthesis as closely as possible in the development, the hydroxyethylamidopyrrole 8 was used as a substrate (Scheme 4). This compound was converted into the diazaphosphocyclopentyl ester 9 (98%) which was oxidised by sulfur flowers to afford the P(V) intermediate 10, the structure of which was confirmed by X-ray crystallography (Fig. 1). In the crystal, the unit cell contains three molecules, one of which is shown in Fig. 1.
 | ||
| Scheme 4 | ||
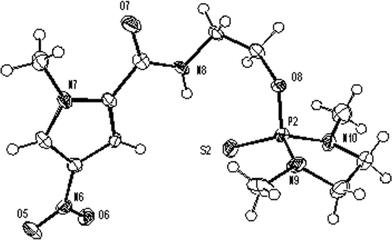 | ||
| Fig. 1 View of one of the three crystallographically independent molecules present in the asymmetric unit of 10. Ellipsoids are drawn at the 50% probability level. | ||
This method potentially offers several advantages over the previous reactions described. Firstly there is no need for a strong methylating reagent which could be problematic with certain MGBs such as those containing thiazoles or imidazoles. Secondly oxidation to P(V) with sulfur leads to a much more stable intermediate, a white crystalline solid as opposed to an oil for the previous method. A balancing disadvantage is that the cleavage step now requires forcing conditions, the phosphorus already being oxidised to P(V). The conversion of 10 into 12, for example, required reaction in a sealed tube under reflux for 24–48 h with pyrrolidine as solvent (43%). This may limit the number of nucleophiles that can be used.
Having demonstrated that suitable cleavage chemistry for introducing the tail group was available, it was now necessary to incorporate it into a full MGB synthesis sequence (Scheme 5). Using the hydroxyethyl nitropyrrole derivative 9 the nitro group was reduced in high yield to afford the corresponding aminopyrrole 13, but it proved impossible to acylate this amine with pyrrole acyl chlorides without complete oxidation of phosphorus to P(V) (14) as shown by NMR. Therefore reduction and oxidation were attempted with the P(V) thiophosphonamidate 10 previously prepared. Reduction with palladium on charcoal and hydrogen gas was slow and incomplete, no doubt due to the presence of sulfur, but reduction with sodium borohydride as the hydrogen source in the presence of palladium on carbon was successful, with reduction complete in 25 min. A demonstration coupling reaction with 3-nitropyrrole-2-carbonyl chloride to give 16 was achieved, albeit in low yield (11%). With the limitations of these reactions in mind, it was decided to prepare a complete MGB delaying the phosphorus chemistry until later in the sequence with a protected hydroxyl group (acetate, 17) as a precursor. The target molecule thus became the MGB acetate 17. This compound was selected because it embodies features of some of our most active antibacterial MGBs.2
 | ||
| Scheme 5 | ||
The alkenyl head group 19 was prepared from 4-methoxyacetophenone and methyl 6-methylnicotinate using acetic anhydride as the solvent and a catalytic amount of zinc chloride (Scheme 6);2,9 the methyl ester was then hydrolysed under standard basic conditions to give 19 and the required acid chloride, 20, prepared using thionyl chloride. The pyrrolyl acetate dimer 21 was prepared from the nitrotrichloroacetylpyrrole 22 and ethanolamine in good yield. The alcohol formed was then protected with acetyl chloride to give 23. The nitropyrrole dimer 21 was reduced under standard hydrogenation conditions and coupled with the acid chloride 20 to give the MGB acetate 17, again using a standard procedure. The acetate protecting group was hydrolysed in base to give the alcohol 24, the phosphorus tail group was then attached followed by oxidation with sulphur to give the key intermediate 18. Attempts to isolate and fully characterise 18 were carried out on HPLC using our standard conditions; this however led to the hydrolysis of the phosphorus containing ring to give 25. However isolation of 18 was not necessary and substitution with pyrrolidine and piperidine gave two target MGBs, 26 and 27, respectively in sufficient yield for initial investigation of biological activity in comparison with compounds previously prepared by non-diversifying routes.221 forms a key intermediate in this new procedure, as it is a readily prepared stable crystalline solid which is stable on the bench for months rather than weeks, and provides a rapid route for the preparation of a range of final products as demonstrated.
 | ||
| Scheme 6 | ||
Biological evaluation
The value of this approach is evidenced from the biological activity of the two test compounds 26 and 27 against Gram positive organisms, the class of bacteria for which we observe the highest antibacterial effects.1,2 The acetate and alcohol intermediates 17 and 24 and the by-product 25 importantly showed no antibacterial activity, emphasising the importance of a cationic group in the presumed interaction of MGBs with the biological target DNA. On the other hand, the pyrrolidinoethyl tail group in 26 was associated with a minimum inhibitory concentration (MIC) of 15.6 µM against Staphylococcus aureus, 7.8 µM against Staphylococcus epidermis, and 15.6 µM against Mycobacterium aureum. The corresponding values for 27 were 3.9, 1.9, and 15.6 µM. The activity of the more potent compound 27 is comparable with that of some of our similar antibacterial MGBs such as 28, 29, and 30. Comparable data for 28 are 6.3 µM for S. aureus but no activity against Mycobacterium fortuitum; similarly for 29, activity against S. aureus was also high (3 µM) and activity against M. fortuitum was poorer (50 µM). 30 had a similar profile to 28 and 29 (6.3 µM against S. aureus and 50 µM against M. fortuitum). In the context of the development of antibacterial MGBs, both of the new compounds 26 and 27 are significantly active especially with respect to Mycobacterium aurum, which is of interest in view of the recurrence of tuberculosis. In making these assessments, differences in MIC greater than a factor of two can be taken as significant and differences of a factor of ten or more highly significant.
Conclusions
Although developed for specific application in minor groove binder chemistry, the new route to aminoalkyl groups may have applications in other fields. In relation to the biological activity, we have discussed elsewhere the importance of the balance of physicochemical properties in terms of hydrophobicity and pKa that promotes antibacterial activity2,4 and others have been concerned with the influence of tail group structure on DNA binding.6 Lee and colleagues have found that tighter binding is found with diaminoalkyl tail groups on the polyamide MGB.6 It is also notable that many anticancer MGBs of this class bear amidine or guanidine tail groups.3,4 From the point of view of selective and strong biological activity, the basicity of the tail group is one factor that plays an important role. Several studies suggest that optimum antibacterial activity is found in compounds with low pKa tail groups such as morpholinoalkyl.1,2 The most active compounds not only possess such a tail group but also a number of hydrophobic components, in contrast to the compounds studied by Lee and colleagues.6 The inference can be drawn that not only is strong binding to DNA important but also that the MGB must have suitable properties to penetrate the bacterial cell. The influence of a specific tail group, however, depends upon the structure of the rest of the MGB. For example we have found that the dimethylaminopropyl analogue (31) of 29 is approximately ten times less active than 29 against Staphylococcus aureus and inactive against Mycobacterium fortuitum. For the new compounds 26 and 27, it turns out that tail group variations within this group of monocyclic alkyl tertiary amines do not have a major influence on antibacterial activity; in this case, the hydrophobic character of the N-terminal alkenyl region of the MGB plays the dominant role in determining antibacterial activity.Experimental
General
1H, 13C, and 31P-NMR were carried out on a Bruker DPX-400 MHz spectrometer with chemical shifts given in ppm (δ values), relative to proton and carbon traces in solvent or in the case of 31P, phosphoric acid was used as an external standard (δ = 0). Coupling constants are reported in Hz. IR spectra were recorded on a Perkin Elmer, 1 FT-IR spectrometer. Elemental analysis was carried out on a Perkin Elmer 2400, analyser series 2. Mass spectra were obtained on a Jeol JMS AX505 mass spectrometer. Anhydrous solvents were obtained from a Puresolv purification system, from Innovative Technologies, or purchased as such from Aldrich. Melting points were recorded on a Reichert hot stage microscope, and are uncorrected. Chromatography was carried out using 200–400 mesh silica gels, or using reverse phase HPLC chromatography on a Waters system using a C18 Luna column with the gradient given in Table 1.| Time/min | A | B | Flow rate (ml/min) |
|---|---|---|---|
| A = Water + 0.1% TFA; B = Acetonitrile + 0.1% TFA | |||
| 0 | 90 | 10 | 4 |
| 28 | 30 | 70 | 4 |
| 33 | 10 | 90 | 4 |
| 38 | 90 | 10 | 4 |
| 40 | 90 | 10 | 0 |
2-Chloro-1,3-dimethyl-1,3,2-diazaphospholidine 17
A solution of N,N′-dimethylethylenediamine (12.10 ml, 113.4 mmol) and triethylamine (11.14 ml, 78 mmol) in dichloromethane (20 ml), and a solution of phosphorus trichloride (11.88 ml, 136 mmol) in dichloromethane (20 ml) were added simultaneously to dichloromethane (60 ml) at −40 °C. After addition, the solution was allowed to warm to −30 °C, and a further solution of triethylamine (11.14 ml, 78 mmol) in dichloromethane (20 ml) added. The solution was then allowed to return to room temperature over 2 h. The solvent was removed, and the residue extracted with diethyl ether (3 × 100 ml). The ether fractions were combined and the solvent removed under reduced pressure to yield the crude product, from which the desired product was distilled, 62%, b.p. 60–62 °C @ 1–2 mmHg. νmax (NaCl): 2933, 2451, 1030 cm−1. δH (CDCl3), 2.70 (6H, d, 2 × CH3, J = 15.9), 3.29 (4H, d, CH2, J = 7.3). δP (CDCl3), 171.76.General coupling procedure of 2-chloro-1,3-dimethyl-1,3,2-diazaphospholidine with an alcohol7
A solution containing the alcohol (6.5 mmol) and triethylamine (6.5 mmol) in dichloromethane (10 ml) was added dropwise to a solution of 1 (65 mmol) in dichloromethane (10 ml) at −60 °C under nitrogen. After addition the solution was allowed to return to room temperature over 40 min. The solution was then extracted with saturated aqueous sodium hydrogen carbonate (40 ml), the dichloromethane fraction was dried (Mg2SO4), the residue was then dissolved in anhydrous diethyl ether (40 ml) filtered and the solvent removed under reduced pressure to yield the desired product.N-{2-[(1,3-Dimethyl-1,3,2-diazaphospholidin-2-yl)oxy]ethyl}benzamide 2
Obtained in 93% yield. νmax (NaCl): 3311, 3062, 2873, 1643, 1531, 1487, 1222, 1023 cm−1. δH (CDCl3), 2.65 (3H, s, CH3), 2.69 (3H, s, CH3), 3.03 (2H, m, CH2), 3.24 (2H, m, CH2), 3.57 (2H, q, CH2, J = 5.4), 3.72 (2H, q, CH2, J = 5.2), 6.74 (1H, s, NH), 7.36 (2H, t, Ar–H, J = 6.9), 7.44 (1H, t, Ar–H, J = 7.0), 7.75 (2H, m, Ar–H). δP (CDCl3), 128.44.1,3-Dimethyl-2-(2-phenylethoxy)-1,3,2-diazaphospholidine 6
Obtained in 88% yield as an oil. νmax (NaCl): 3062, 1603, 1583, 1496, 1453, 1231, 1015 cm−1. δH (CDCl3), 2.68 (3H, s, CH3), 2.70 (3H, s, CH3), 2.89 (2H, t, CH2, J = 7.2), 3.01 (2H, m, CH2), 3.29 (2H, m, CH2), 3.88 (2H, q, CH2, J = 7.2), 7.42–7.17 (5H, m, Ar–H). δP (CDCl3), 129.20.Procedure for the cleavage step with methyl iodide
The above phenylethanol derivative 6 (1.05 g, 4.42 mmol), and methyl iodide (277 µl, 4.42 mmol) were placed in toluene (15 ml). The solution was heated to reflux for 4 h under nitrogen, the solution was then allowed to cool and the solvent removed under reduced pressure to yield the crude product. This was purified on silica gel using 1:1 ethyl acetate:hexane, to yield the products 5 and (2-iodoethyl)benzene:1,2,3-Trimethyl-1,3,2-diazaphospholidine 2-oxide 5
Obtained as a white, low-melting solid, m.p. 36–37 °C in 83% yield. δH (CDCl3), 1.34 (3H, d, CH3, J = 22.5), 2.52 (3H, s, CH3), 2.56 (3H, s, CH3), 2.91 (2H, m, CH2), 3.09 (2H, m, CH2). δP (CDCl3), 40.36. LRMS: Found 149.1 (M + H) calculated for C5H13N2OP 148.0.2-(Iodoethyl)benzene
Obtained in 74% yield as an oil. νmax (NaCl): 3061, 2835, 1601, 1584, 1495, 1453, 1236, 618 cm−1. δH (CDCl3), 3.19 (2H, t, CH2, J = 11.1), 3.38 (2H, t, CH2, J = 10.5), 7.41–7.15 (5H, m, Ar–H). δC (CDCl3), 5.80 (CH2), 40.52 (CH2), 127.02 (2C), 128.48 (2C), 128.80, 140.77, (Ar). LRMS: Found 233.0 (M + H), 105.1 (M − I) calculated for C13H19N6+ 231.9, 105.0.Preparation of 1-(2-phenylethyl)pyrrolidine 7
The above cleavage step with methyl iodide was carried out with the diazaphospholidine 6 (1.00 g, 4.42 mmol) and iodomethane (275 µl, 4.42 mmol), then before workup a 10 fold excess of pyrrolidine (3.67 ml, 44.54 mmol) was added and the solution allowed to stir overnight at room temperature. The solvent was then removed under reduced pressure, and the residue dissolved in dichloromethane (20 ml). The solution was then acidified with dilute hydrochloric acid, and extracted with distilled water (2 × 20 ml), the dichloromethane layer was dried and the solvent removed under reduced pressure to yield 5. The aqueous solution was then made alkaline with saturated sodium bicarbonate solution, and extracted with dichloromethane (2 × 20 ml). The dichloromethane fraction was then dried (Mg2SO4), and the solvent removed under reduced pressure to yield the desired product 7 (0.211 g, 27%). νmax (NaCl): 3063, 2788, 1603, 1584, 1496, 1454, 1235 cm−1. δH (CDCl3), 1.82 (4H, m,CH2), 2.61 (4H, m, CH2), 3.75 (2H, m, CH2), 3.89 (2H, m, CH2), 7.40–7.12 (5H, m, Ar-H). LRMS: Found 176.1 (M + H) calculated for C12H17N 175.1N-(2-Hydroxyethyl)-1-methyl-4-nitro-1H-pyrrole-2-carboxamide 8
2,2,2-Trichloro-1-(1-methyl-4-nitro-1H-pyrrol-2-yl)ethanone (1.00 g, 3.70 mmol) was dissolved in dichloromethane (15 ml), and a solution of ethanolamine (225 µl, 3.70 mmol) in dichloromethane (5 ml) added. The solution was allowed to stir for 1 h after which the solvent was removed to yield the product 8 as a pale yellow solid (0.772 g, 98%) m.p. = 158–160 °C. νmax (KBr): 3500–2500, 3358, 3268, 3141, 2947, 1644, 1566, 1313 cm−1. δH (DMSO): 3.25 (2H, q, CH2, J = 6.0), 3.2 (2H, m, CH2), 3.90 (3H, s, NCH3), 4.69 (1H, s, O–H), 7.44 (1H, d, Ar–H, J = 1.9), 8.10 (1H, d, Ar–H, J = 1.90), 8.36 (1H, s, NH,). LRMS: Found 214.1 calculated for C8H12N3O4+, 214.1.N-{2-[(1,3-Dimethyl-1,3,2-diazaphospholidin-2-yl)oxy]ethyl}-1-methyl-4-nitro-1H-pyrrole-2-carboxamide 9
The diazaphospholidine 1 (0.152 g, 1.00 mmol) was dissolved in dichloromethane (15 ml) and cooled to −60 °C under nitrogen. The pyrrole 8 (0.230 g, 1.00 mmol) and triethylamine (154 µl, 1.20 mmol) in dichloromethane (15 ml) were then added together dropwise and the solution allowed to return to room temperature over 40 min. Sat. sodium bicarbonate solution (20 ml) and brine (30 ml) were then added and the dichloromethane layer separated and dried (Mg2SO4). The solvent was removed under reduced pressure to yield the product 9 as a pale yellow oil (0.322 g, 98%). νmax (NaCl): 3311, 2873, 1643, 1531, 1222, 1023 cm−1. δH (CDCl3): 2.69 (3H, s, N–Me), 2.73 (3H, s, NCH3), 3.10 (2H, m, CH2), 3.31 (2H, m, CH2), 3.52 (2H, m, CH2), 3.78 (2H, m, CH2), 3.99 (3H, s, N–Me), 7.07 (1H, d, Ar–H, J = 1.8), 7.55 (2H, m, Ar–H and NH). δP (CDCl3): 131.26. HRFABMS: Found 330.1283; C12H21N5O4P+ requires 330.1286.Preparation of N-{2-[(1,3-dimethyl-2-sulfido-1,3,2-diazaphospholidin-2-yl)oxy]ethyl}-1-methyl-4-nitro-1H-pyrrole-2-carboxamide 10
The pyrrolyl diazaphospholidine 9 (0.417 g, 1.30 mmol), was dissolved in toluene (10 ml). Sulfur (flowers 0.043 g, 1.30 mmol) was then added and the solution allowed to stir for 15 min. The solvent was then removed under reduced pressure and the residue purified by flash chromatography eluting with ethyl acetate/hexane (1:1 v/v) to yield the product 10 as a white solid (0.460 g, 98%) m.p. = 118–120 °C. νmax (KBr): 3321, 2870, 1661, 1522, 1309, 1030 cm−1. δH (DMSO): 2.70 (3H, s, N–Me), 2.73 (3H, s, NCH3), 3.21 (2H, m, CH2), 3.33 (2H, m, CH2), 3.67 (2H, m, CH2), 4.05 (3H, s, N–Me), 4.21 (2H, m, CH2), 6,89 (1H, broad s, NH), 7.27 (1H, d, Ar–H, J = 1.8), 7.65 (1H, d, Ar–H, J = 1.8). δP (CDCl3): 84.07. HRFABMS: Found 362.1050; C12H20N5O4PS+ requires 362.1052.1-Methyl-4-nitro-N-[2-(1-pyrrolidinyl)ethyl]-1H-pyrrole-2-carboxamide 128
The pyrrolyl sulfidodiazaphospholidine 10 (0.200 g, 0.51 mmol) was placed in a sealed thick walled Pyrex tube, pyrrolidine (5 ml) was then added and the solution heated to 100 °C and allowed to stir for 48 h, after which time the solvent was removed under reduced pressure, and the residue purified by flash chromatography to yield the product 12 as an off-white solid (0.133 g, 43%) m.p. = 133–135 °C. νmax (KBr): 3321, 2955, 1651, 1306 cm−1. δH (DMSO): 2.78 (4H, m, 2(CH2)), 3.10 (4H, m, 2(N–CH2)), 3.30 (2H, m, CH2), 3.38 (2H, m, CH2), 3.91 (3H, s, N–Me), 6.89 (1H, broad s, NH), 7.44 (1H, d, Ar–H, J = 2.0), 8.13 (1H, d, Ar–H, J = 2.0). LRMS: Found 267.2 calculated for C12H19N4O3+, 267.1. Found: C, 53.7; H, 6.5; N, 21.5; C12H18N4O3 requires C, 54.1; H, 6.8; N, 21.0%.N-{2-[(1,3-Dimethyl-2-sulfido-1,3,2-diazaphospholidin-2-yl)oxy]ethyl}-1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1H-pyrrole-2-carboxamide 16
The pyrrolyl sulfidodiazaphospholidine 10 (0.200 g, 0.56 mmol) was dissolved in dioxane (10 ml), Pd/C (10%, 0.100 g) was then added, followed by a solution of sodium borohydride (0.040 g, 0.70 mmol) in water (5 ml). The solution was then allowed to stir for 1 h. The solution was then filtered directly into a flask containing 1-methyl-4-nitro-1H-pyrrole-2-carbonyl chloride (0.104 g, 0.56 mmol). Sat. sodium carbonate solution (2 ml) was then added and the solution allowed to stir overnight. Water (5 ml) was added and the solution extracted with ethyl acetate (2 × 20 ml); the organic layer was dried and the solvent removed under reduced pressure. The residue was purified by chromatography on neutral alumina to yield the product 16 as a bright yellow solid (0.03 g, 11%) m.p. = sublimes 90 °C, m.p. 149–151 °C. νmax (KBr): 3421, 2928, 1666, 1305, 1028 cm−1. δH (DMSO): 2.50 (3H, s, N–Me), 2.53 (3H, s, NCH3), 3.06 (2H, m, CH2), 3.23 (2H, m, CH2), 3.33 (2H, m, CH2), 3.81 (3H, s, N–Me), 3.87 (2H, m, CH2), 3.95 (3H, s, N–Me), 6.87 (1H, d, Ar–H, J = 1.8 Hz), 7.24 (1H, d, Ar–H, J = 1.8 Hz), 7.58 (1H, d, Ar–H, J = 1.8 Hz), 8.15 (1H, t, NH, J = 5.8 Hz), 8.18 (1H, d, Ar–H, J = 1.8 Hz), 10.26 (1H, s, NH), δP (CDCl3): 81.51, HRFABMS: Found 484.1533 calculated for C18H27N7O5PS+, 484.1532. Found: C, 44.4; H, 5.9; N, 20.1. C18H26N7O5PS requires C, 44.72; H, 5.4; N, 20.3%.Methyl 6-[(E)-2-(4-methoxyphenyl)ethenyl]nicotinate 10
4-Methoxybenzaldehyde (2.120 g, 30.40 mmol) and methyl 6-methylnicotinate (2.305 g, 30.40 mmol) were dissolved in acetic anhydride (10 ml); zinc chloride (0.050 g, 0.38 mmol) was then added and the solution refluxed for 72 h. Water (10 ml) was then added and the solution extracted with ethyl acetate (2 × 15 ml); the aqueous layer was then made alkaline with sat. sodium carbonate solution and extracted with ethyl acetate (2 × 15 ml). The organic layers were combined, dried, and the solvent removed under reduced pressure. The residue was then purified by flash chromatography to yield the product as a brown solid (1.78 g, 23%) m.p. = 173–175 °C. νmax (KBr): 2950, 1717, 1595, 1290, 1254 cm−1. δH (DMSO): 3.80 (3H, s, OCH3), 3.88 (3H, s, OCH3), 7.01 (1H, d, Ar–H, J = 8.8), 7.27 (1H, d, (CC–H), J = 16.0), 7.64 (3H, m, Ar–H), 7.80 (1H, d, (CC–H), J = 16.0), 7.01 (1H, d of d, Ar–H, J = 2.2 and 8.2) 9.04 (1H, s, Ar–H), HRFABMS: Found 270.1129 calculated for C16H16NO3+, 270.1130.
6-[(E)-2-(4-methoxyphenyl)ethenyl]nicotinic acid 1910
Methyl 6-[(E)-2-(4-methoxyphenyl)ethenyl]nicotinate (1.058 g, 4.15 mmol) was dissolved in ethanol (3 ml). A solution of sodium hydroxide (0.332 g, 8.30 mmol) in water (10 ml) was then added and the solution was heated under reflux for 2 h. Water (20 ml) was then added and the solution cooled to 0 °C, followed by acidification using dilute hydrochloric acid, during which the product precipitated as a yellow solid. (0.77 g, 73%), sublimes at 200–202 °C, m.p. >230°C. νmax (KBr): 3600–3000, 3107, 2934, 1719, 1595, 1276 cm−1. δH (DMSO): 3.80 (3H, s, OCH3), 7.03 (2H, d, Ar–H, J = 8.7), 7.36 (1H, d, (CC–H), J = 16.2), 7.67 (2H, d, Ar–H, J = 8.7), 7.93 (1H, d, Ar–H, J = 8.4) 7.98 (1H, d, (CC–H), J = 16.2), 8.44 (1H, d, Ar–H, J = 8.4), 9.01 (1H, d, Ar–H, J = 1.7), HRFABMS: Found 256.0972 calculated for C15H14NO3+, 256.0974; Found: C, 71.0; H, 5.5; N, 5.4; C15H13NO3 requires C, 70.6; H, 5.1; N, 5.5%.
2-{[(1-Methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}ethyl acetate 23
The hydroxyethylpyrrole 8 (0.826 g, 5.42 mmol) was dissolved in dichloromethane (10 ml) and cooled to 0 °C. Acetyl chloride (386 µl, 5.42 mmol) in dichloromethane (2 ml) was then added dropwise followed by triethylamine (550 µl, 6.50 mmol) and the solution allowed to return to room temperature over 2 h. The solvent was then removed under reduced pressure and the residue purified by flash chromatography to give the product 23 as an off-white solid (1.34 g, 97%), m.p. = 127–129 °C. νmax (KBr): 3410, 3110, 2952, 1723, 1661, 1308 cm−1. δH (DMSO): 2.01 (3H, s, CH3), 3.44 (2H, q, CH2, J = 5.7), 3.91 (3H, s, NCH3), 4.10 (2H, t, CH2, J = 5.7), 7.43 (1H, d, Ar–H, J = 2.0), 8.14 (1H, d, Ar–H, J = 1.7), 8.54 (1H, t, NH, J = 6.5), HRFABMS: Found 256.0887 calculated for C10H14N3O5+, 256.0889. Found: C, 47.1; H, 5.3; N, 16.2; C10H13N3O5 requires C, 47.1; H, 5.1; N, 16.5%.2-{[(1-Methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1H-pyrrol-2-yl)carbonyl]amino}ethyl acetate 21
The acetoxyethyl pyrrole 23 (0.900 g, 3.56 mmol) was dissolved in methanol (10 ml); Pd/C (10%, 0.450 g) was then added and the solution was then allowed to stir under hydrogen for 3 h. The solution was then filtered and the solvent removed under reduced pressure. The residue was dissolved in DMF (1.5 ml) and added to a solution of 1-methyl-4-nitro-1H-pyrrole-2-carbonyl chloride (0.600 g, 3.56 mmol) and N-methylmorpholine (431 µl, 4.27 mmol). The solution was allowed to stir overnight, during which time the product 21 precipitated as a bright yellow solid which was collected by filtration (0.86 g, 63%), m.p. = 155–160 °C. νmax (KBr): 3399, 3296, 3129, 2957, 1731, 1657, 1303 cm.−1.δH (DMSO): 2.01 (3H, s, CH3), 3.42 (2H, q, CH2, J = 5.8), 3.81 (3H, s, NCH3), 3.96 (3H, s, NCH3), 4.09 (2H, t, CH2, J = 5.8), 6.87 (1H, d, Ar–H, J = 1.9), 7.23 (1H, d, Ar–H, J = 1.8), 7.58 (1H, d, Ar–H, J = 2.0), 8.14 (2H, m, Ar–H and NH), 10.25 (1H, s, NH), HREIMS: Found 378.1365 calculated for C16H20N5O6+, 378.1369. Found: C, 50.5; H, 5.7; N, 18.2; C16H19N5O6 requires C, 50.9; H, 5.1; N, 18.6%.2-[({4-[({4-[({6-[(E)-2-(4-Methoxyphenyl)ethenyl]-3-pyridinyl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]ethyl acetate 17
The alkene dimer 19 (0.067 g, 0.27 mmol) was dissolved in dichloromethane (2 ml) and thionyl chloride (5 ml) added and the solution refluxed for 1 h, after which the solvent was removed under reduced pressure to give 6-[(E)-2-(4-methoxyphenyl)ethenyl] nicotinoyl chloride 20 as a dark orange solid. The pyrrolyl acetate dimer acetate 21 (0.100 g, 0.27 mmol) was dissolved in methanol (5 ml), Pd/C, (10%, 0.050 g) was then added, the solution was then allowed to stir under hydrogen for 3 h, the solution was then filtered and the solvent removed under vacuum, the residue was dissolved in DMF (1.5 ml) and added to the previously prepared 6-[(E)-2-(4-methoxyphenyl)ethenyl]nicotinic acid chloride; N-methylmorpholine (36 µl, 0.32 mmol) was added and the solution allowed to stir overnight. The product was then purified by HPLC and freeze drying the eluate afforded the pure product as an orange solid (0.04 g, 27%) m.p. >230 °C, purity by HPLC = 97%. νmax (KBr): 3446, 2925, 1637, 1594, 1260 cm.−1δH (DMSO): 2.02 (3H, s, CH3), 3.40 (2H, q, CH2, J = 5.7), 3.81 (6H, m, NCH3 and OCH3), 3.88 (3H, s, NCH3), 4.09 (2H, t, CH2, J = 5.7), 6.89 (1H, d, Ar–H, J = 1.8), 7.00 (2H, d, Ar–H, J = 8.8), 7.10 (1H, d, Ar–H, J = 1.8), 7.23 (1H, d, Ar–H, J = 1.8), 7.26 (1H, d, (CC–H), J = 16.1), 7.37 (1H, d, Ar–H, J = 1.8), 7.66 (3H, m, Ar–H), 7.77 (1H, d, (CC–H), J = 16.1), 8.14 (1H, t, NH, J = 5.6), 8.29 (1H, d of d, Ar–H, J = 2.3 and 8.2 Hz), 9.07 (1H, d, Ar–H, J = 2.2), 9.98 (1H, s, NH), 10.51 (1H, s, NH). HRFABMS: Found 585.2419 calculated for C31H33N6O6+, 585.2417.
N-(5-{[(5-{[(2-Hydroxyethyl)amino]carbonyl}-1-methyl-1H-pyrrol-3-yl)amino]carbonyl}-1-methyl-1H-pyrrol-3-yl)-6-[(E)-2-(4-methoxyphenyl)ethenyl]nicotinamide 24
The tetramer 17 (0.100 g, 0.17 mmol) was dissolved in ethanol (1 ml); a solution of sodium hydroxide (0.020 g, 0.5 mmol) in water (5 ml) was then added and the solution allowed to reflux for 2 h. The whole solution was then freeze dried and the residue purified by HPLC to give the product as an orange solid (0.02 g, 23%), m.p. >230 °C, purity by HPLC = 98%. νmax (KBr): 1342, 2926, 1632, 1593, 1261 cm−1. δH (DMSO): 3.24 (2H, q, CH2, J = 5.5), 3.81 (6H, m, NCH3 and OCH3), 3.87 (3H, s, NCH3), 3.46 (2H, t, CH2, J = 5.5), 6.86 (1H, d, Ar–H, J = 1.9), 7.00 (2H, d, Ar–H, J = 8.8), 7.07 (1H, d, Ar–H, J = 1.9), 7.18 (1H, d, Ar–H, J = 1.8), 7.24 (1H, d, (CC–H), J = 16.1), 7.35 (1H, d, Ar–H, J = 1.8), 7.66 (3H, m, Ar–H), 7.77 (1H, d, (CC–H), J = 16.0), 7.91 (1H, t, NH, J = 5.6), 8.25 (1H, d of d, Ar–H, J = 2.3 and 8.2), 9.06 (1H, d, Ar–H, J = 2.2), 9.93 (1H, s, NH), 10.46 (1H, s, NH), HRFABMS: Found 543.2314 calculated for C31H33N6O6+, 543.2311.
1-{2-[({4-[({4-[({6-[(E)-2-(4-Methoxyphenyl)ethenyl]-3-pyridinyl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]ethyl}pyrrolidinium trifluoroacetate 26 and 1-{2-[({4-[({4-[({6-[(E)-2-(4-methoxyphenyl)ethenyl]-3-pyridinyl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]ethyl}piperidinium trifluoroacetate 27
The tetramer 24 (0.100 g, 0.18 mmol) and triethylamine (40 µl, 0.36 mmol) in dichloromethane (5 ml) was added dropwise to a solution of 1 (0.040 g, 0.22 mmol) in dichloromethane (10 ml) at −60 °C under nitrogen. After addition the solution was allowed to return to room temperature over 40 min. Sulfur (flowers 0.028 g, 0.22 mmol) was then added and the solution allowed to stir for a further 30 min. The dichloromethane was then removed under reduced pressure to yield an orange residue. This material was then divided into two portions. The fractions were then refluxed in either pyrrolidine (2 ml) or piperidine (2 ml) for 24 h, after which the solvent was removed under reduced pressure, and the residues purified by HPLC before being freeze dried to yield 26 and 27 as orange solids.1-{2-[({4-[({4-[({6-[(E)-2-(4-methoxyphenyl)ethenyl]-3-pyridinyl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]ethyl}pyrrolidinium trifluoroacetate 26
Obtained in 26% yield, m.p. >230 °C, purity by HPLC = 98%. νmax (KBr): 3315, 3126, 2984, 1679, 1530, 1310 cm−1. δH (DMSO): 1.88 (4H, m, 2(CH2)), 2.06 (4H, m, 2(CH2)), 3.22 (2H, q, CH2, J = 5.4), 3.38 (2H, t, CH2, J = 5.4), 3.80 (6H, m, NCH3 and OCH3), 3.89 (3H, s, NCH3), 7.02 (3H, m, Ar–H), 7.09 (1H, d, Ar–H, J = 1.9), 7.18 (1H, d, Ar–H, J = 1.8), 7.26 (1H, d, (CC–H), J = 16.0), 7.35 (1H, d, Ar–H, J = 1.8), 7.66 (3H, m, Ar–H), 7.77 (1H, d, (CC–H), J = 16.0), 7.91 (1H, t, NH, J = 5.6), 8.25 (1H, d of d, Ar–H, J = 2.3 and 8.2), 9.06 (1H, d, Ar–H, J = 2.2), 9.93 (1H, s, NH), 10.46 (1H, s, NH). HRFABMS: Found 596.2941 calculated for C33 H38 N7 O4, 596.2941.
1-{2-[({4-[({4-[({6-[(E)-2-(4-methoxyphenyl)ethenyl]-3-pyridinyl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]ethyl}piperidinium trifluoroacetate 27
Obtained in 22% yield, m.p. >230 °C, purity by HPLC = 96%. νmax (KBr): 3296, 3136, 2875, 1701, 1542, 1298 cm−1. δH (DMSO): 1.67 (6H, m, 2(CH2)), 1.81 (4H, m, 2(CH2)), 2.93 (2H, q, CH2, J = 5.4), 3.19 (2H, t, CH2, J = 5.4), 3.79 (6H, m, NCH3 and OCH3), 3.82 (3H, s, NCH3), 6.98 (3H, m, Ar–H), 7.10 (1H, d, Ar–H, J = 1.9), 7.21 (1H, d, Ar–H, J = 1.8), 7.25 (1H, d, (CC–H), J = 16.0), 7.34 (1H, d, Ar–H, J = 1.8), 7.65 (3H, m, Ar–H), 7.75 (1H, d, (CC–H), J = 16.0), 8.26 (2H, m, Ar–H, NH), 9.05 (1H, d, Ar–H, J = 2.2 Hz), 10.00 (1H, s, NH), 10.49 (1H, s, NH). HRFABMS: Found 610.3093 calculated for C34H40N7O4+, 610.3097.
N-[{2-[({4-[({4-[({6-[(E)-2-(4-Methoxyphenyl)ethenyl]-3-pyridinyl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]ethoxy}(sulfanyl)phosphoryl]-N,N′-dimethylmethanediaminium bis(trifluoroacetate) 25
The tetramer 24 (0.100 g, 0.18 mmol) and triethylamine (40 µl, 0.36 mmol) in dichloromethane (5 ml) was added dropwise to a solution of 1 (0.040 g, 0.22 mmol) in dichloromethane (10 ml) at −60 °C under nitrogen. After addition the solution was allowed to return to room temperature over 40 min. Sulfur (flowers 0.028 g, 0.22 mmol) was then added and the solution allowed to stir for a further 30 min. The dichloromethane was then removed under reduced pressure to yield an orange residue which was purified by HPLC to yield the product as a yellow/orange solid (0.02 g, 14%), m.p. >230 °C, purity by HPLC = 94%. νmax (KBr): 3421, 3118, 2487, 1666, 1593, 1261, 1175 cm.−1δH (DMSO): 2.53 (4H, m, CH2), 2.63 (3H, s, N+CH3), 3.17 (3H, s, N+CH3), 3.46 (4H, m, CH2), 3.79 (6H, m, NCH3 and OCH3), 3.87 (3H, s, NCH3), 6.65 (1H, m, Ar–H), 6.88 (2H, d, Ar–H, J = 8.7), 7.09 (1H, d, Ar–H, J = 1.6), 7.18 (2H, m, Ar–H and (CC–H), 7.26 (1H, d, Ar–H, J = 1.5), 7.63 (3H, m, Ar–H), 7.75 (1H, d, (CC–H), J = 16.0), 7.91 (1H, m, NH), 8.25 (1H, d, Ar–H, J = 8.1), 8.58 (2H, broad s, 2(N+H)), 9.06 (1H, s, Ar–H), 9.97 (1H, s, NH), 10.40 (1H, s, NH). HRFABMS: found 709.2688 calculated for C33H43N8O6PS+, 709.2686
Crystal data for 10: C12H20N5O4PS, Mr = 361.36, monoclinic, space group P21/n, a = 18.0006(6), b= 10.8282(3), c= 25.5950(9) Å, β = 98.184(2)°, V = 4938.0(3) Å3, Z = 12, λ = 0.71073 Å, µ = 0.321 mm−1, T = 120 K; data quality was impaired by generic twinning. This model was treated as twinned by a 180° rotation about 1 0 0. Using the SHELXL-97 HKLF 5 formalism11 gave a twin ratio of 0.852:0.148. In addition two of the three crystallographically independent molecules show some disorder. Final refinement to convergence on F2 gave R = 0.0904 (F, 19205 obs. data only) and Rw = 0.2004 (F2, all 28548 data), GOF = 1.085. CCDC reference numbers 701664.†
Biological data
MIC data was obtained as described previously.2 In summary, sample dilutions were typically prepared by dissolving the test sample (2 mg) in sterile water (10 mL) to provide a working concentration of 200 µg mL−1. The test wells on each 96 well microtiter plate were initially inoculated with culture medium (100 µL) using Mueller–Hinton broth. A solution of each test sample (100 µL) was added to one row of each plate, and a series of doubling dilutions was made for successive rows. Incubation was at 37 °C for antibacterial assays and 25 °C for antifungal assays. Plates were inspected visually for growth, and Resazurin was added to each well; a distinct color change from blue to red indicated that growth had occurred in an individual well. From the observed pattern of color, the MIC was determined. All tests included sterility and growth controls.Acknowledgements
Crystallographic data for 10 was kindly collected by the EPSRC National Crystallography Service, University of Southampton.Notes and references
- R. W. Bürli, Y. Ge, S. White, S. M. Touami, M. Taylor, J. A. Kaizerman and H. E. Moser, Bioorg. Med. Chem. Lett., 2002, 12, 2591 CrossRef CAS; R. W. Bürli, D. McMinn, J. A. Kaizerman, W. Hu, Y. Ge, Q. Pack, V. Jiang, M. Gross, M. Garcia, R. Tanaka and H. E. Moser, Bioorg. Med. Chem Lett., 2004, 14, 1253 CrossRef CAS; R. W. Bürli, P. Jones, D. McMinn, Q. Le, J.-X. Duan, J. A. Kaizerman, S. Difuntorum and H. E. Moser, Bioorg. Med. Chem. Lett., 2004, 14, 1259 CrossRef CAS; R. W. Bürli, J. A. Kaizerman, J.-X. Duan, P. Jones, K. W. Johnson, M. Iwamoto, K. Truong, W. Hu, T. Stanton, A. Chen, S. Touami, M. Gross, V. Jiang, Y. Ge and H. E. Moser, Bioorg. Med. Chem. Lett., 2004, 14, 2067 CrossRef CAS.
- C. J. Suckling, D. Breen, A. I. Khalaf, E. Ellis, I. S. Hunter, G. Ford, C. G. Gemmell, N. Anthony, J.-J. Helsebeux, S. P. Mackay and R. D. Waigh, J. Med. Chem., 2007, 50, 6116–6125 CrossRef.
- A. J. Ten Tije, J. Verweij, A. Sparreboom, A. von der Gaast, C. Fowst, F. Fiorentini, J. Tursi, A. Antonellini, M. Mantel, C. M. Hartman, G. Stoter, A. S. T. Planting and M. J. A. de Jonge, Clin. Cancer Res., 2003, 9, 2957 CAS; A. Fedier, C. Fowst, J. Tursi, C. Geroni, U. Haller, S. Marchini and D. Fink, Br. J. Cancer, 2003, 89, 1559 CrossRef CAS; A. Lansiaux, F. Tanious, Z. Mishal, L. Dassoneville, A. Kumar, C. E. Stephens, Q. Hu, W. D. Wilson, D. W. Boykin and C. Bailly, Cancer Research, 2002, 62, 7219 CAS.
- C. J. Suckling, Expert Opinion on Therapeutic Patents, 2004, 14, 1693 Search PubMed.
- C. J. Suckling, J. Phys. Org. Chem., 2008, 21, 575 CrossRef CAS.
- T. Brown, Z. Taherbhai, J. Sexton, A. Sutterfield, M. Turlington, J. Jones, L. Stallings, M. Stewart, K. Buchmueller, H. Mackay, C. O'Hare, J. Kluza, B. Nguyen, D. Wilson, M. Lee and J. A. Hartley, Bioorg. Med. Chem., 2007, 15, 474 CrossRef CAS.
- C. McGuigan and M. S. Anson, J. Chem. Soc., Perkin Trans., 1989, 715 Search PubMed.
- R. J. M. Hermans and H. M. Buck, J. Org. Chem., 1987, 23, 5150 CrossRef.
- C. A. Lipinski, J. L. LaMattina and P. J. Oates, J. Med. Chem., 1986, 29, 2154 CrossRef CAS.
- R. Cluzan and L. Katz, The Boots Company Ltd., 1977, US patent 4,009,174.
- G. M. Sheldrick, SHELXL-97 a program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 2007 Search PubMed.
Footnote |
| † CCDC reference number 701644. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b814452d |
| This journal is © The Royal Society of Chemistry 2009 |