Thio-arylglycosides with various aglyconpara-substituents: a probe for studying chemical glycosylation reactions†
Xiaoning
Li
a,
Lijun
Huang
a,
Xiche
Hu
a and
Xuefei
Huang
*ab
aDepartment of Chemistry, The University of Toledo, 2801 W. Bancroft Street, MS 602, Toledo, OH 43606
bDepartment of Chemistry, Michigan State University, East Lansing, MI 48824. E-mail: xuefei@chemistry.msu.edu
First published on 20th October 2008
Abstract
Three series of thioglycosyl donors differing only in their respective aglycon substituents within each series have been prepared as representatives of typical glycosyl donors. The relative anomeric reactivities of these donors were quantified under competitive glycosylation conditions with various reaction time, promoters, solvents and acceptors. Over three orders of magnitude reactivity difference were generated by simple transformation of the para-substituent on the aglycon with methanol as the acceptor, while chemoselectivities became lower with carbohydrate acceptors. Excellent linear correlations were attained between relative reactivity values of donors and σp values of the substituents in the Hammett plots. This indicates that the glycosylation mechanism remains the same over a wide range of reactivities and glycosylation conditions. The negative slopes of the Hammett plots suggested that electron donating substituents expedite the reactions and the magnitudes of slopes can be rationalized by neighboring group participation as well as electronic properties of the glycon protective groups. Within the same series of donors, less nucleophilic acceptors gave smaller slopes in their Hammett plots. This is consistent with the notion that acceptor nucleophilic attack onto the reactive intermediate is part of the rate limiting step of the glycosylation reaction.
Introduction
With the increasing recognition of the biological importance of carbohydrates, carbohydrate chemistry is gaining more prominence.1–5 While much effort has been spent on synthesis of specific oligosaccharides, relatively little attention has been devoted to systematic exploration of the general aspects of the glycosylation reaction. Such investigations can, however, be very fruitful.It is known that protective groups and aglycon leaving group structures have profound effects on the anomeric reactivities of glycosyl donors.6 Donors bearing electron donating protective groups have much higher anomeric reactivities towards a promoter than those bearing electron withdrawing and conformational restricting protective groups. This phenomenon was first observed with the glycosyl halides,7 which has been subsequently found to be general for a variety of donors including pentenyl glycosides,8 thioglycosides,6,9,10 glycosyl fluorides,11 glycosyl thioimidates12 and glycosyl sulfoxides.13 The understanding of protective group effect on glycosylation rate led to the development of reactivity based chemoselective glycosylation method, where a thioglycoside donor with higher anomeric reactivity (armed) is mixed together with a less reactive (disarmed) thioglycosyl acceptor.6,10 Upon addition of a limiting amount of promoter, the armed donor is preferentially activated, glycosylating the bifunctional thioglycoside acceptor. The newly formed thioglycosyl disaccharide, in turn can be further activated as a donor, leading to a more convergent synthesis of oligosaccharides. Multiple sequential chemoselective glycosylations can be carried out in the sequence of decreasing order of anomeric reactivities in one single reaction flask.6,9,10,14,15
In order to aid in building block selection, Ley and coworkers quantified the relative reactivities of thioglycosyl donors bearing various protective groups towards a carbohydrate acceptor.16 Similarly, a database was established by the Wong group tabulating Relative Reactivity Values (RRVs) of thiotolyl building blocks glycosylating methanol.6,15RRVmethanol values have been employed as a quantitative measure to guide the design of building blocks with requisite anomeric reactivities for reactivity based chemoselective glycosylation, as applied in assemblies of several complex oligosaccharides.6,14,15,17–19 Generally, minimum ten-fold difference in RRVsmethanol between the two competing building blocks is desired so that theoretically greater than 90% glycosylation yield can be obtained. However, the effects of different acceptors on RRVs were not quantitatively evaluated.
Aglycon of thioglycosides can also significantly influence the anomeric reactivity. Roy and colleagues reported that the p-acetamidophenylthioglycoside could be selectively activated and used to glycosylate the electronically deactivated p-nitrophenylthioglycosides.20 The aglycon of the resulting disaccharide was then reduced and acetylated to yield another p-acetamidophenylthioglycosyl donor for convenient chain elongation. Besides electronic effects, the size of the aglycon can play an important role as well. The Boons group took advantage of the slow activation of the bulky dicyclohexylmethylthioglycosides, allowing the selective activation of a sterically less hindered ethylthioglycoside and its subsequent chemoselective glycosylation.21 Oscarson and colleagues quantified the effects of size and electronic properties on the anomeric reactivities of a series of thioglycosides bearing different aglycons and applied the knowledge gained to a trisaccharide synthesis.22 Following this trend of anomeric reactivity tuning, our group has developed a new one pot method utilizing a series of glycosyl donors (1a–1e, 2a–2e) differing only in their aglycon substituents, which were readily acquired through post-synthetic aglycon modification of the common intermediates 1f and 2f (Scheme 1), allowing rapid access to glycosyl donors with multiple levels of anomeric reactivities.23 These building blocks were then used in construction of several tetrasaccharides and pentasaccharides by one pot syntheses.
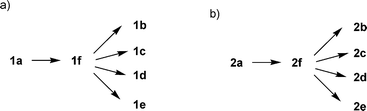 | ||
| Scheme 1 Post-synthetic modification of aglycons rapidly generated a panel of building blocks with multiple levels of anomeric reactivities. | ||
As the donor series 1a–1f and 2a–2f only differed in their aglycon substituents, this provided us unique opportunities to investigate the glycosylation using Hammett analyses. Hammett relationship has been a useful tool for probing reaction mechanisms,24 and in relation to glycoscience, glycosidase mechanisms.25–31 For chemical glycosylations, there are several possible Hammett plot scenarios: 1) a linear Hammett relationship for a wide range of leaving group σp values, indicating that the glycosylation mechanism remains the same for all substituents; 2) a ‘bent’ Hammett relationship consisting of linear correlations with different slopes, suggesting a change in mechanism. Such phenomena have been observed previously with several glycosidases,26,32,33 resulting from a change in rate limiting step with different substituents; 3) Hammett relationship with poor linear correlations and badly scattered points, implying factors other than substituent electronic effects influencing the reaction.
Herein, we report the quantification of anomeric reactivities of three donor series, 1a–1e, 2a–2e and 3a–3e. Hammett analyses were then performed on these donors by plotting reaction rates against σp values under a variety of glycosylation conditions to probe the glycosylation mechanism.
Results
Preparation of three series of donors
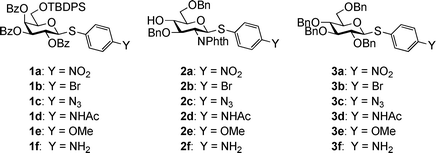
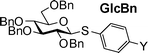
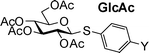
Three donor series 1a–1f, 2a–2f and 3a–3f were selected as representative glycosyl donors for our investigation. Thiogalactosides 1a–1f (Gal series) contain multiple electron withdrawing protective groups on the glycon ring with a participating neighboring group (OBz) on C2. Thioglucosamine series 2a–2f (GlcN series) bear a participating neighboring group phthalimido (NPhth) on C2 and several electron donating groups, while glucosides 3a–3f (GlcBn series) only contain electron donating protective groups with no participating neighboring groups. 1a–1f and 2a–2f are prepared through post-synthetic modifications of aglycons as previously described.23 Instead of spending five synthetic steps on average to synthesize each donor starting from individual aryl thiol, it only took one to two steps to acquire building blocks 1b–1e and 2b–2e divergently from common synthetic intermediates 1f and 2f.With the simpler protective group pattern on glucose series donors 3a–3e, we started the synthesis from per-acetylated glucose 4 and the corresponding thiophenols for 3a,20,343b and 3e (Scheme 2a). Borontrifluoride etherate mediated glycosylation produced thioglycosides 5–735,36 in excellent yields. Replacement of the four acetates on the glycon ring with benzyl groups was accomplished by treatment of 5–7 with NaOMe followed by benzylation. The yield for formation of 3a was lower due to competitive reduction of the nitro moiety by NaH.
 | ||
| Scheme 2 Synthesis of thioglucoside donors 3a–3e. | ||
An initial attempt to synthesize acetamido substituted donor 3d from 4-acetamido thiophenol and glucoside 4 failed due to complexation of tin (IV) chloride by the acetamido moiety. As an alternative, we performed stannous chloride reduction of 3a to produce aminophenyl glucoside 3f, which was found to be very prone to air oxidation. Instead, a one-pot procedure of stannous chloride reduction of 3a followed by immediate acetylation in the same reaction flask led to donor 3d in 84% yield (Scheme 2b). Stannous chloride reduction of 4-nitrophenyl glucoside 5 with subsequent diazonium salt formation and in situ displacement by sodium azide produced the azido bearing glucoside 9 in 59% yield for the two steps. Removal of acetates followed by benzylation gave donor 3c in 78% yield (Scheme 2c).
Determination of the rate of glycosylation by NMR
In order to quantitatively determine donor reactivities, we first investigated the possibility of real time monitoring of glycosylation by NMR. The reaction of 1b and methanol using N-iodosuccinimide (NIS) and catalytic amount of trifluoromethane sulfonic acid (TfOH) as the promoter system37 was performed in CD2Cl2 with a 1H-NMR spectrum of the reaction mixture acquired every eight seconds. As the reaction proceeded, the amount of donor decreased as determined from integrations of the anomeric proton (Fig. 1a). Interestingly, the rate of reaction calculated from the speed of donor consumption rose during the first few minutes of the reaction, before decreasing with time in a manner expected for a reaction exhibiting positive-order kinetics in substrate concentration (Fig. 1b). Similar phenomenon was also observed when dimethyl(methylthio) sulfonium triflate was used as the promoter. The cause of such an induction period38 at the onset of glycosylation is unclear. As the glycosylation progressed passing the induction period, the observed pseudo-linear decrease of the reaction rate indicated that glycosylation is apparent first order to donor under this condition (Fig. 1b,c). However, it was difficult to accurately determine the absolute rate via this method, as subtle variation of the reaction condition could cause substantial rate fluctuation, which led us to focus on measurements of relative rate of glycosylation.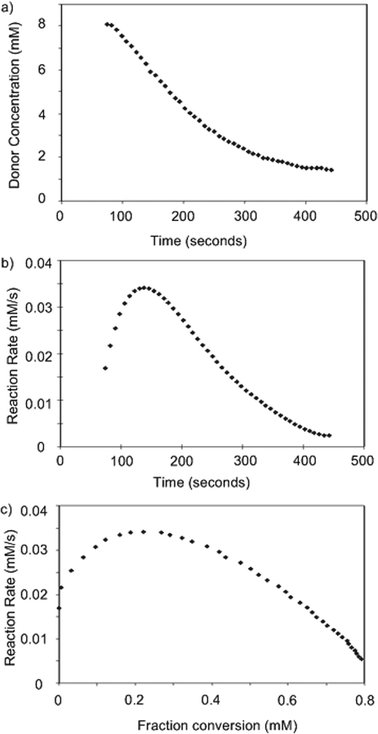 | ||
| Fig. 1 Changes of a) donor concentration, b) reaction rate vs. time; and c) reaction rate vs. donor fraction conversion as measured by 1H-NMR. | ||
Determination of relative reactivity values using methanol as the acceptor
To measure the relative glycosylation rates, we adapted a competitive glycosylation condition where two different glycosyl donors are mixed together with methanol.15,16,22,39 Upon addition of a limiting amount of the promoter, the two donors competed for promoter activation, with the rate of glycosylation determined through the decrease of donor concentration. The measurement of relative reactivities allowed us to avoid the need to measure acceptor concentration as well as rate constant dependence on the identity of the promoter.As glycosylation was apparent first order to donor under the experimental condition, the rate of glycosylation could be described by kobs[D], where kobs is the apparent first order rate constant and [D] is the donor concentration. Thus, the relative reactivity between these two donors in a competition reaction could be derived according to equation (1)
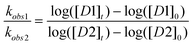 | (1) |
(0 is designated as the starting time and t is designated as the finishing time). Because 1H-NMR of such competition reactions could be fairly complex, we quantified the compounds from chromatogram integration following HPLC separation (Fig. 2).15,22 The concentrations of compounds were calculated from peak integration areas. An internal reference R inert to glycosylation was added to the reaction mixture in order to normalize the amounts of compounds between different experiments and to minimize the errors caused by injected sample volume changes. Thus, equation 1 was revised to equation 2
 | (2) |
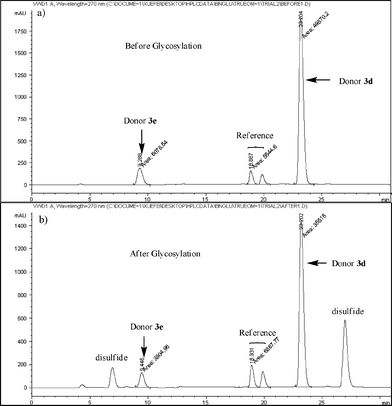 | ||
| Fig. 2 HPLC chromatograms a) before and b) after competitive glycosylation between donors 3d and 3e (1,3,4,6-tetra-O-acetyl-2-deoxy-2-N-phthalimido-α/β-D-glucopyranosides were used as the reference for these reactions. Same amounts of donors 3d and 3e were added before glycosylation. The large disparity in UV absorbance is due to the difference in extinction coefficients. p-Tolyl p-methoxyphenyldisulfide and p-tolyl p-acetamidophenyldisulfide were formed and detected in the chromatogram after glycosylation). | ||
(D and R are the amounts of compounds calculated from the HPLC peak integration area).
In all cases, baseline separation of desired peaks in HPLC chromatogram was achieved. The reactivity differentials between the two competing donors were kept below 20-fold to ensure accuracy of the experiments. When there was a greater than 20-fold donor reactivity differential or chromatogram overlap, a third donor was used to bridge the two donors of interest. For each pair of donors, the reaction was repeated at least three times. Usually, less than 10% differential was obtained among all measurements, with the average value taken as the relative reactivity value between the two donors, an example of which is shown in Table 1.
| Entry | Donor pair (more reactive/less reactive) | Experiment #1 | Experiment #2 | Experiment #3 | Average |
|---|---|---|---|---|---|
| 1 | 10/2a | 4.49 | 4.58 | 4.37 | 4.49 |
| 2 | 2b/10 | 7.06 | 7.04 | 7.51 | 7.21 |
| 3 | 2d/2b | 5.27 | 5.32 | 4.95 | 5.18 |
| 4 | 2d/2c | 3.97 | 3.35 | 3.98 | 3.74 |
| 5 | 2e/2d | 1.88 | 1.89 | 1.93 | 1.90 |
The reaction time did not affect the relative reactivity much. For example, k2a/k10 values of 4.80, 4.28 and 4.49 were obtained when the competitive reactions of donor 2a against donor 10 were quenched at 3, 5 and 120 minutes respectively. Next we examined the effect of promoter. Relative reactivities between donor pair of 1b and 10 were measured with two promoter systems, NIS/TfOH and p-TolSCl/AgOTf.40 The observed k1b/k10 values using the two systems were similar (3.02 for NIS/TfOH and 2.86 for p-TolSCl/AgOTf). This suggested that relative reactivities were independent of promoter, consistent with our hypothesis and observations by Wong and coworkers.15 Greater than 92% yields were obtained for the two methyl glycoside products based on consumption of 1b and 10, confirming that consumed donors were converted into products.
With the experimental conditions established, the RRVs for three series of donors (Gal 1a–1e, GlcN 2a–2e, GlcBn 3a–3e) were obtained using MeOH as the acceptor, with the reactivity of thiomannoside 11 set as 115 (Table 2). Although donors 2a–2e contain a free hydroxyl group, due to the much higher reactivity of methanol and excess methanol used,41 chemoselective glycosylation of 2a–2e with methanol was achieved with little self couplings of donors. It is evident that the para-substituents on the aglycon of thioaryl glycoside donors significantly impact anomeric reactivities with over three orders of magnitude difference obtained by a single substitution (3evs3a). This effect is much larger than that resulting from changing one protective group on the glycon ring, which is generally less than 10 fold.6 The much bigger influence of aglycon on anomeric effect is due to the direct conjugation of para-substituents with the anomeric sulfur atom.
| Y | NO2 | Br | N3 | NHAc | OMe |
|---|---|---|---|---|---|
|
0.042 (1a) | 0.89 (1b) | 3.12 (1c) | 18.1 (1d) | 21.7 (1e) |
|
0.601 (2a) | 19.2 (2b) | 27.1 (2c) | 101 (2d) | 191 (2e) |
|
3.19 (3a) | 161 (3b) | 657 (3c) | 2482 (3d) | 3772 (3e) |
For our glycosylations, Hammett plots were constructed by plotting log(RRV) values of the three series of donors against σp values of the para-substituents24,42 (Fig. 3). Excellent linear correlations were observed within each donor series, suggesting that the same glycosylation mechanism is operative over a large range of reactivities.29,43 Furthermore, the slopes of the Hammett plots (ρ values) were all negative, indicating that the reactive intermediates were cationic in nature and electron donating substituents such as methoxy expedited the reaction compared with electron withdrawing substituents such as nitro.
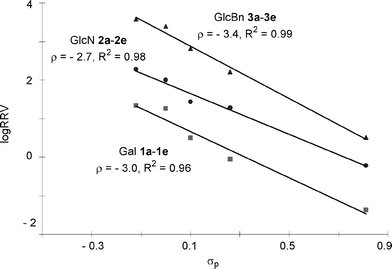 | ||
| Fig. 3 Hammett plots of log(RRV)vsσp using methanol as the acceptor. | ||
Prediction of donor RRVs by Hammett equation and correlation of 1H-NMR anomeric proton chemical shifts with σP values
Building block preparation is a time-consuming process and it requires much experience to design and fine tune the protective groups in order to achieve exact anomeric reactivities required by the armed-disarmed chemoselective glycosylations.6,15,44 It will be highly desirable if reactivities of the building blocks can be predicted prior to the actual synthesis. Anomeric reactivities have been estimated by calculating the energy difference between the ground state and the oxacarbenium ion.45 With the large number of atoms in typical fully protected glycosyl donors, global minima of the structures can be difficult to obtain by calculation. Furthermore, the need to include solvation for accurate determination of the oxacarbenium ion energy renders this approaches challenging.45 With the excellent linear correlations between log(RRV) against σp and large number of σp values available,24σp can be employed as a predictor of RRV. For example, log(RRV) of galactoside 12 was calculated to be 1.38 from the Hammett plot (Fig. 3, σp = −0.14 for methyl group), while the experimentally determined value was 1.28.6 The same was applicable to thioglycosides 13 and 14 (Table 3).| Donor | Calculated log(RRV) value | Experimental log(RRV) value |
|---|---|---|
|
1.38 | 1.28 |
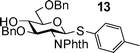
|
2.28 | 2.41 |
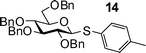
|
3.68 | 3.42 |
Since the only structural differences among our donor series are in their aglycon substituents, we envision that the chemical shifts of their anomeric protons should only be affected by the electronic properties of para-substituents on aglycons. Excellent linear correlations between chemical shifts of the anomeric protons (Table 4) and σp values were obtained in all donor series examined (Fig. 4), which supports previous observations that chemical shifts could be used to predict relative reactivities.16,23
| Y | NO2 | Br | N3 | NHAc | OMe | NH2 |
|---|---|---|---|---|---|---|
|
5.09 (1a) | 4.90 (1b) | 4.88 (1c) | 4.90 (1d) | 4.82 (1e) | 4.78 (1f) |
|
5.69 (2a) | 5.50 (2b) | 5.47 (2c) | 5.46 (2d) | 5.40 (2e) | 5.35 (2f) |
|
4.82 (3a) | 4.62 (3b) | 4.58 (3c) | 4.57 (3d) | 4.53 (3e) | 4.47 (3f) |
|
4.88 (5) | 4.65 (6) | 4.62 (9) | 4.59 (15) | 4.54 (7) | 4.51 (8) |
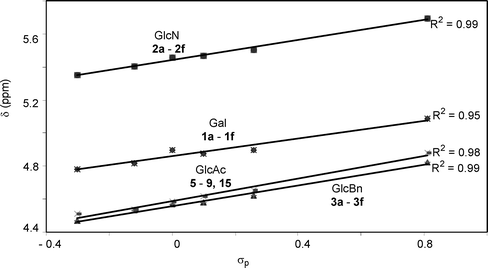 | ||
| Fig. 4 Correlations of anomeric proton chemical shifts of the four donor series with σp values. | ||
Effect of acceptor and solvent on relative reactivity values
Prior competitive glycosylation studies have been mainly focused on systematic investigations of the effect of donor structural change on glycosylation.6,16,22 It is known that acceptors can also drastically affect glycosylation.46–49 In order to quantified the effect of acceptor, we performed competitive glycosylation reactions of Gal donors 1a–1e using galactoside 16 and glucosamine 17 respectively as representatives of primary and secondary carbohydrate acceptors (Table 5).| Entry | Acceptor (reaction solvent) | Y = NO2 (1a) | Y = Br (1b) | Y = N3 (1c) | Y = NHAc (1d) | Y = OMe (1e) |
|---|---|---|---|---|---|---|
| 1 | Methanol (CH2Cl2) | 1 | 21.2 | 74.3 | 431.0 | 516.7 |
| 2 | Acceptor 16 (CH2Cl2) | 1 | 11.6 | 72.3 | 177.5 | 234.2 |
| 3 | Acceptor 17 (CH2Cl2) | 1 | 5.95 | 11.8 | 51.4 | 62.5 |
| 4 | Acceptor 16 (diethyl ether) | 1 | 7.63 | 20.0 | 34.3 | 47.1 |
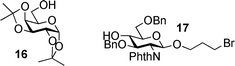
Excellent linear correlations were obtained in the Hammett plots for reactions of donors 1a–1e with acceptors 16 and 17 in CH2Cl2 with ρ values of −2.7 and −2.0 respectively (Fig. 5). Interestingly, compared with methanol acceptor for the same donor series, the reactivity differential became smaller (k1e/k1a 62.5 with acceptor 17vs 516.7 with methanol acceptor) and the reaction rate was less sensitive to changes in para-substituents of the aglycon as reflected by the smaller absolute ρ value.
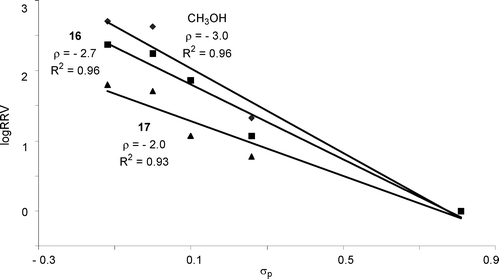 | ||
| Fig. 5 Hammett plots of log(RRV) of donors 1a–1evsσp (methanol, galactoside 16 and glucosamine 17 as the respective acceptor and CH2Cl2 as the glycosylation solvent). | ||
In the programmable reactivity based chemoselective glycosylation, RRVmethanol values were utilized as a quantitative measure to guide building block selection.6,9,10,14,15,18,19 However, in some cases, glycosylation yields were lower than those indicated based on RRVmethanol differentials,15,44 which could be due to the decrease of chemoselectivity resulting from glycosylating a less reactive carbohydrate acceptor.
The glycosylation solvent can also significantly impact the chemoselectivity. When glycosylations of acceptor 16 by donors 1a–1e were carried out in diethyl ether using p-TolSCl/AgOTf promoter system, good linear correlation between log(RRV) and σp was retained (Fig. 6 and Table 5 entry 4). However, the RRV difference in diethyl ether was smaller than that in CH2Cl2 as indicated by the smaller absolute ρ value. Oscarson and coworkers reported that using NIS/AgOTf as the promoter, better selectivity was observed in diethyl ether compared with CH2Cl2 for competitive glycosylation of two thioglycosyl building blocks.50 This discrepancy may be attributed to the low solubility of NIS in diethyl ether, thus reducing effective concentration of promoter available for glycosylation leading to higher selectivity.50,51
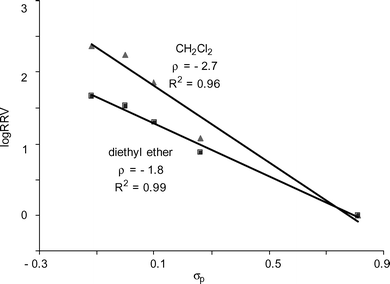 | ||
| Fig. 6 Hammett plots of log(RRV) of donors 1a–1evsσp (galactoside 16 as the acceptor; promoter p-TolSCl/AgOTf; CH2Cl2 or diethyl ether as the glycosylation solvent). | ||
Discussion
Thioglycoside glycosylation reaction can be generally viewed as a three-step process.15,45 The glycosyl donor is first activated by eletrophilic addition of the promoter onto the anomeric sulfur atom, generating a glycosyl sulfonium ion (Scheme 3, step A), which is converted to other reactive intermediates such as glycosyl triflate or dioxolenium ion (Scheme 3, step B). Nucleophilic attack of the reactive intermediate by the acceptor leads to the glycoside product (Scheme 3, step C). Crich and coworkers have demonstrated in a series of ground breaking low temperature NMR studies that glycosyl triflates were the dominant intermediates following activation of thioglucosides and thiomannosides without a participating neighboring group on O-2.52–54 Recently, our group monitored the activation process of thioglycosyl donors bearing participating neighboring group on O-2 by low temperature NMR.55 Upon addition of the promoter, the disappearance of the glycosyl donor was almost instantaneous. The glycosyl sulfonium ion was not observed during the activation process. Rather, the major intermediates were the glycosyl triflate and the dioxolenium ion formed due to neighboring group participation. Glycosylation of the acceptor by the reactive intermediate occurred at a slower rate, often requiring more than one hour to complete at low temperature with carbohydrate acceptors. Although in these NMR studies donor activation was performed in the absence of the acceptor, these results suggested that steps A and B in thioglycoside glycosylation are rapid with the glycosyl sulfonium ion being consumed as it is forming, and step C is the slow step for glycosylation. The absence of glycosyl sulfonium ion indicated that the rate of the reverse reaction for step B must be small compared to the forward reaction.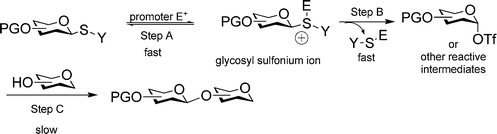 | ||
| Scheme 3 General mechanism for glycosylation of thioglycosides. | ||
Acceptors had a significant impact on the glycosylation rate as shown in Hammett analyses (Fig. 5), suggesting that acceptor attack is involved in the rate limiting step, which is consistent with the results from previous low temperature NMR studies. The dependence of ρ value on acceptor could be explained as the acceptor nucleophilicity decreased from methanol to galactoside 16, the acceptor reacted slower with the reactive intermediate, allowing its accumulation. Therefore, rate differences resulting from differential aglycons became less pronounced leading to a smaller absolute ρ value (2.7 vs 3.0, Fig. 5). Secondary hydroxyl group in glucosamine 17 was less reactive56 than the primary alcohol in 16, causing further decrease in the absolute value of ρ (2.0 vs 2.7).
In the Hammett relationship studies, ρ values were negative, demonstrating building blocks with electron donating groups were more reactive with acceptors. Since the donors used in Hammett plots differed only in their aglycon substituents, the reactive intermediates generated within the same donor series should have the same structure as aglycons have already been cleaved. Therefore, the rate constants for step C for the same donor series should be the same. The fact that building blocks with electron withdrawing aglycons reacted slower could be understood by assuming step A is a fast equilibrium. As the sulfur atoms in p-nitrophenyl glycosides are less electron rich, their activation by an electrophilic promoter will be slower than the corresponding p-methoxyphenyl glycosides. This will generate lower concentrations of the reactive intermediates from p-nitrophenyl glycosides, leading to lower overall glycosylation rates.
The absolute ρ value of Gal donor series 1a–1e was larger than that of the GlcN series 2a–2e (3.0 vs 2.7, Fig. 3), suggesting that Gal series of donors were more sensitive to the substituents. This can be rationalized by the presence of more electron withdrawing protective groups on the glycon ring in the Gal donor series, imparting more cationic character to the reactive intermediates.
Since OH is an equally potent electron donating moiety as benzyl group,15GlcN series 2a–2e contain one more electron withdrawing protective group on the glycon ring as compared to GlcBn series 3a–3e donors. Yet, the GlcN series were less sensitive to aglycon changes as shown by a smaller absolute ρ value in its Hammett plot (2.7 vs 3.4, Fig. 3). This presumably resulted from the participating neighboring group NPhth on C-2 of GlcN donors. With NPhth participation, the positive charge on the corresponding reactive intermediate from GlcN donors becomes more dispersed; hence the electron donating para-substituent on aglycon has lesser stabilizing effect leading to a smaller absolute ρ value (Fig. 7). Similar phenomena of smaller absolute ρ value due to neighboring group participation have been observed in several nucleophilic displacement reactions.57,58
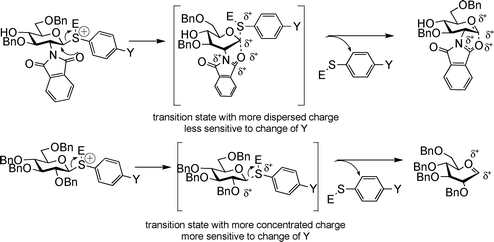 | ||
| Fig. 7 The smaller absolute ρ values for GlcN donor series can be rationalized by neighboring group participation of the phthalimido group. | ||
Conclusions
We have demonstrated that simple modification of the para-substituents of thioaryl glycosides can render over three orders of magnitude difference in anomeric reactivities of glycosyl donors. The reactivity differential was found to be dependent upon the identity of the acceptor with the less reactive carbohydrate acceptors leading to lower chemoselectivities. Excellent linear correlations were obtained in Hammett relationship studies demonstrating that the glycosylation mechanism remains unchanged over the wide range of donor reactivities and reaction conditions. The negative slopes of the Hammett plots suggest that electron donating substituents expedite the reactions and the magnitudes of slopes can be rationalized by neighboring group participation and electronic properties of the glycon protective groups. The knowledge gained from our results can provide valuable insights into further improvement of the powerful armed-disarmed chemoselective glycosylation methodology and the understanding of chemical glycosylation in general.Experimental section
General procedure for competitive glycosylation and HPLC measurements
Two donors of interest (0.05 mmol each) were dried together with a reference compound (50 mg) under high vacuum overnight. They were then dissolved in anhydrous CH2Cl2 (5 mL). Aliquots of this solution (1 mL each) were transferred to three pre-dried vials. The exact ratio of two donors in each vial was determined by HPLC analysis of an aliquot (10 μL) of the solution. Each vial was measured at least three times. Anhydrous MeOH (2.04 μL, 5 eq), acceptor 16 or acceptor 17 was then added to each vial followed by 0.5 M NIS solution in acetonitrile (20 μL) and 0.1 M TfOH in Et2O (10 μL). The reactions were left at room temperature for 2 hours and quenched by triethylamine (20 μL). The reaction mixture was diluted with CH2Cl2 (2 mL), washed with saturated aqueous sodium thiosulfate solution containing 10% sodium hydrogen bicarbonate, dried over Na2SO4 and evaporated to dryness. The solid obtained was then dissolved in CH2Cl2 (1 mL). The concentration of remaining donors in each vial were determined by HPLC (∼10 μL each injection; 3 injections for each vial). RRVs are calculated according to equation 1 as the average of all measurements. All HPLC analyses were performed using a HP 1050 series HPLC system with a SUPELCO normal phase analytical HPLC column (25 cm * 4.6 mm ID) and UV detection at 270 nm.For glycosylations using the p-TolSCl/AgOTf promoter system, all procedures are the same as above except that 0.5 M AgOTf solution in acetonitrile (40 μL) and p-TolSCl (1.5 μL) were added to each vial instead of NIS and TfOH.
Acknowledgements
We are grateful for financial support from the National Institutes of Health (R01-GM-72667). We would like to thank Prof. James E. Jackson (Michigan State Univ.) for helpful discussions and Dr. Thomas Carter (Michigan State Univ.) for graphics.References
- D. P. Galonić and D. Y. Gin, Nature, 2007, 446, 1000–1007 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS.
- Comprehensive Glycoscience, from Chemistry to Systems Biology, ed. J. P. Kamerling, Elsevier Science, 2007 Search PubMed.
- T. Buskas, S. Ingale and G. -J. Boons, Glycobiology, 2006, 16, 113R–136R CrossRef CAS.
- K. M. Koeller and C. -H. Wong, Nat. Biotech., 2000, 18, 835–841 CrossRef CAS.
- K. M. Koeller and C. -H. Wong, Chem. Rev., 2000, 100, 4465–4493 CrossRef CAS and references cited therein.
- H. Paulsen, Angew. Chem. Int. Ed. Engl., 1982, 21, 155–173 CrossRef.
- B. Fraser-Reid, U. E. Udodong, Z. F. Wu, H. Ottosson, J. R. Merritt, C. S. Rao, C. Roberts and R. Madsen, Synlett, 1992, 1992, 927–942 CrossRef.
- H. Tanaka, M. Adachi, H. Tsukamoto, T. Ikeda, H. Yamada and T. Takahashi, Org. Lett., 2002, 4, 4213–4216 CrossRef CAS.
- L. Green, B. Hinzen, S. J. Ince, P. Langer, S. V. Ley and S. L. Warriner, Synlett, 1998, 440–442 CrossRef CAS.
- H. Jona, H. Mandai, W. Chavasiri, K. Takeuchi and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2002, 75, 291–309 CrossRef CAS.
- P. Pornsuriyasak and A. V. Demchenko, Chem. Eur. J., 2006, 12, 6630–6646 CrossRef CAS and references cited therein.
- S. Raghavan and D. Kahne, J. Am. Chem. Soc., 1993, 115, 1580–1581 CrossRef CAS.
- Y. Wang, X. Huang, L. -H. Zhang and X. -S. Ye, Org. Lett., 2004, 6, 4415–4417 CrossRef CAS.
- Z. Zhang, I. R. Ollman, X. -S. Ye, R. Wischnat, T. Baasov and C. -H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS.
- N. L. Douglas, S. V. Ley, U. Lucking and S. L. Warriner, J. Chem. Soc. Perkin Trans. 1, 1998, 51–65 RSC.
- J. -C. Lee, W. A. Greenberg and C. H. Wong, Nat. Protocols, 2006, 1, 3143–3152 CAS.
- J. -C. Lee, C. -Y. Wu, J. V. Apon, G. Siuzdak and C. -H. Wong, Angew. Chem. Int. Ed., 2006, 45, 2753–2757 CrossRef CAS.
- T. K. -K. Mong, H. -K. Lee, S. G. Duron and C. -H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 797–802 CrossRef CAS.
- S. Cao, F. Hernández-Matéo and R. Roy, J. Carbohydr. Chem., 1998, 17, 609–631 CrossRef CAS.
- R. Geurtsen, D. S. Holmes and G. J. Boons, J. Org. Chem., 1997, 62, 8145–8154 CrossRef CAS.
- M. Lahmann and S. Oscarson, Can. J. Chem., 2002, 80, 889–893 CrossRef CAS.
- L. Huang, Z. Wang and X. Huang, Chem. Commun., 2004, 17, 1960–1961 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. Vallmitjana, M. Ferrer-Navarro, R. Planell, M. Abel, C. Ausin, E. Querol, A. Planas and J. -A. Perez-Pons, Biochemistry, 2001, 40, 5975–5982 CrossRef CAS.
- A. Planas, M. Abel, Ó. Millet, J. Palasi, C. Pallarés and J. -L. Viladot, Carbohydr. Res., 1998, 310, 53–64 CrossRef CAS.
- H. Nakatani, Biologia, 1998, 53, 343–347 CAS.
- S. L. Lawson, W. W. Wakarchuk and S. G. Withers, Biochemistry, 1997, 36, 2257–2265 CrossRef CAS.
- X. Guo, W. G. Laver, E. Vimr and M. L. Sinnott, J. Am. Chem. Soc., 1994, 116, 5572–5578 CrossRef CAS.
- C. K. De Bruyne, G. M. Aerts and R. L. De Gussem, Eur. J. Biochem., 1979, 102, 257–267 CrossRef CAS.
- C. S. Tsai, J. Y. Tang and S. C. Subbarao, Biochem. J., 1969, 114, 529–534 CAS.
- J. C. Gebler, D. E. Trimbur, R. A. J. Warren, R. Aebersold, M. Namchuk and S. G. Withers, Biochemistry, 1995, 34, 14547–14553 CrossRef CAS.
- J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9961–9969 CrossRef.
- L. A. J. M. Sliedregt, K. Zegelaar-Jaarsveld, G. A. van der Marel and J. H. van Boom, Synlett, 1993, 335–337 CrossRef.
- C. Denekamp and Y. Sandlers, J. Mass Spectrom., 2005, 40, 1055–1063 CrossRef CAS.
- S. -S. Weng, Y. -D. Lin and C. -T. Chen, Org. Lett., 2006, 8, 5633–5636 CrossRef CAS.
- G. H. Veeneman, S. H. van Leuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef CAS.
- D. G. Blackmond, Angew. Chem. Int. Ed., 2005, 44, 4302–4320 CrossRef CAS.
- B. Fraser-Reid, Z. Wu, U. E. Udodong and H. Ottosson, J. Org. Chem., 1990, 55, 6068–6070 CrossRef CAS.
- X. Huang, L. Huang, H. Wang and X. -S. Ye, Angew. Chem. Int. Ed., 2004, 42, 5221–5224 CrossRef.
- T. Zhu and G. J. Boons, Angew. Chem. Int. Ed., 1999, 38, 3495–3497 CrossRef CAS.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem. Int. Ed., 2005, 44, 5188–5240 CrossRef.
- S. Yamago, K. Kokubo, O. Hara, S. Masuda and J. -I. Yoshida, J. Org. Chem., 2002, 67, 8584–8592 CrossRef CAS.
- T. -K. Mong, C. -Y. Huang and C. -H. Wong, J. Org. Chem., 2003, 68, 2135–2142 CrossRef CAS.
- C. W. Andrews, R. Rodebaugh and B. Fraser-Reid, J. Org. Chem., 1996, 61, 5280–5289 CrossRef CAS.
- C. Uriel, A. Agocs, A. M. Gomez, J. C. Lopez and B. Fraser-Reid, Org. Lett., 2005, 7, 4899–4902 CrossRef CAS.
- B. Fraser-Reid, J. C. López, A. M. Gómez and C. Uriel, Eur. J. Org. Chem., 2004, 1387–1395 CrossRef CAS.
- B. Fraser-Reid, J. C. López, K. V. Radhakrishnan, M. V. Nandakumar, A. M. Gomez and C. Uriel, Chem. Commun., 2002, 2104–2105 RSC.
- N. M. Spijker and C. A. A. van Boeckel, Angew. Chem. Int. Ed., 1991, 30, 180–183 CrossRef.
- M. Lahmann and S. Oscarson, Org. Lett., 2000, 2, 3881–3882 CrossRef CAS.
- M. Fridman, D. Solomon, S. Yogev and T. Baasov, Org. Lett., 2002, 4, 281–283 CrossRef CAS.
- T. Nokami, A. Shibuya, H. Tsuyama, S. Suga, A. A. Bowers, D. Crich and J. -I. Yoshida, J. Am. Chem. Soc., 2007, 129, 10922–10928 CrossRef CAS.
- D. Crich and W. Cai, J. Org. Chem., 1999, 64, 4926–4930 CrossRef CAS.
- D. Crich and S. Sun, J. Am. Chem. Soc., 1997, 119, 11217–11223 CrossRef CAS.
- Y. Zeng, Z. Wang, D. Whitfield and X. Huang, J. Org. Chem., 2008, 73, 7952–7962 CrossRef CAS.
- D. Crich and V. Dudkin, J. Am. Chem. Soc., 2001, 123, 6819–6825 CrossRef CAS.
- K. C. Kemp and D. Metzger, J. Org. Chem., 1974, 33, 4165–4168.
- K. Bowden and J. M. Byrne, J. Chem. Soc. Perkin Trans. 2, 1997, 123–127 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Selected 1H- and 13C- NMR spectra, HPLC chromatograms. See DOI: 10.1039/b813048e |
| This journal is © The Royal Society of Chemistry 2009 |