Polymer hydrogel capsules: en route toward synthetic cellular systems
Brigitte
Städler
a,
Andrew D.
Price
a,
Rona
Chandrawati
a,
Leticia
Hosta-Rigau
abc,
Alexander N.
Zelikin
a and
Frank
Caruso
*a
aCentre for Nanoscience and Nanotechnology, Department of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Australia. E-mail: fcaruso@unimelb.edu.au; Fax: +61 3 8344 4153; Tel: +61 3 8344 3461
bInstitute for Research in Biomedicine, Barcelona Science Park, Barcelona 08028, Spain
cDepartment of Organic Chemistry, University of Barcelona, Barcelona 08028, Spain
First published on 28th August 2009
Abstract
Engineered synthetic cellular systems are expected to become a powerful biomedical platform for the development of next-generation therapeutic carrier vehicles. In this mini-review, we discuss the potential of polymer capsules derived by the layer-by-layer assembly as a platform system for the construction of artificial cells and organelles. We outline the characteristics of polymer capsules that make them unique for these applications, and we describe several successful examples of microencapsulated catalysis, including biologically relevant enzymatic reactions. We also provide examples of subcompartmentalized polymer capsules, which represent a major step toward the creation of synthetic cells.
Introduction
Understanding and mimicking cellular structure, its activity and (multi)functionality has long attracted the interest of scientists across various disciplines. However, despite intense efforts, the ability to mimic cellular functions substantially lags behind the understanding of the underlying phenomena, and even the most successful current synthetic systems are far simpler than living cells, fulfilling only limited, typically one, simple cellular function.As artificial organs and tissue engineering have literally given new life to macroscale physiological systems, artificial cells or organelles are expected to substitute for missing or lost cellular functions.1 The minimal requirements for the successful design of artificial cells or organelles are a structurally sound scaffold for the vessel wall and controllable loading of the synthetic machinery into the vessel. More advanced designs attempt to conduct continuous reactions and, consequently, the exchange of reagents/nutrients with the surrounding environment is crucial. Further advances relate to the possibility of subcompartmentalizing the vessel, in a manner very much similar to biological cells, to conduct multiple, spatially separated reactions within the same carrier.
The most successful examples of synthetic mimics of cellular systems to date are lipid vesicles and polymersomes. These two systems represent carrier vehicles and are formed by the self-assembly of amphiphilic lipids and copolymers , respectively.2 An alternative approach to the design of carrier systems is based on the layer-by-layer (LbL) assembly technology,3,4 which affords engineered hollow capsules with facile control over the size, shape and composition of the capsule membrane. In the assembly of polymer capsules, the LbL technology entails the sequential adsorption of interacting polymers (e.g., through electrostatics, hydrogen bonding and covalent bonding) onto sacrificial template particles, followed by removal of the template (Scheme 1a). A plethora of charged and noncharged polymers have been used as structural components of LbL capsules to provide custom-designed carriers for a number of applications, including drug delivery and microencapsulated catalysis. The key advantage of LbL assembly for the design and construction of synthetic mimics of cells and organelles resides in the ability to prepare engineered capsules with excellent structural stability and high permeability (to small molecules). In this mini-review, we outline recent progress in the bottom-up assembly of functional LbL-assembled carrier vehicles for the development of therapeutic artificial cells and organelles. We focus on: (i) the types of carrier capsules, the permeability properties of the capsules, and methods used to load the capsules; (ii) continuous biocatalytic reactions within LbL capsules; and (iii) recent approaches that enable the compartmentalization of LbL capsules. We present specific examples for each, highlighting progress in these areas. We also detail the remaining challenges associated with the development of LbL-based capsules as therapeutic artificial cells and organelles.
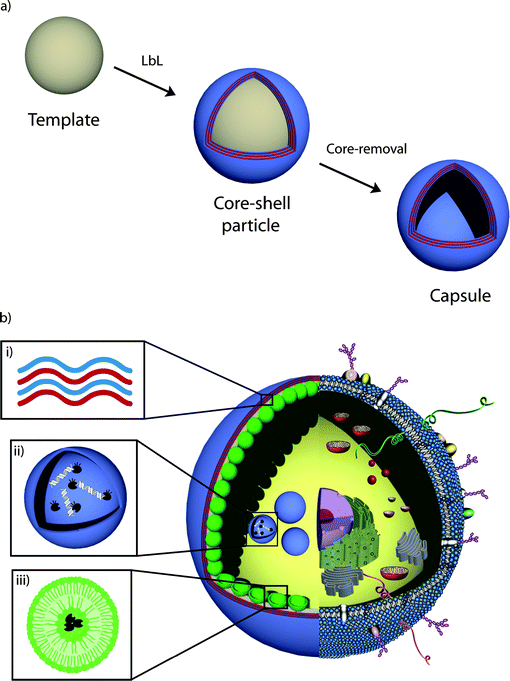 | ||
| Scheme 1 Schematic illustration of (a) LbL assembly (see text for details) to form polymer hydrogel capsules and (b) a synthetic mimic of cells and subcellular organelles derived from LbL capsules. i) The polymer hydrogel membrane provides the structural scaffold and its semipermeability allows for the exchange of nutrients between the interior and exterior environment. ii) A LbL-derived lysosome mimic for the triggered degradation of DNA. iii) Incorporated liposomes compartmentalize the capsule's interior. | ||
Polymer capsules—the structural scaffold
The use of polymer capsules as scaffolds for artificial cells or organelles defines the requirement for their size, from several micrometers (cells) to submicrometer size (organelles) (Scheme 1(b)I). Although a range of LbL capsules (including hydrolytically degradable systems) with sizes larger than ∼4 µm3 have been assembled, the submicrometer size requirement for synthetic organelles precludes the use of most LbL capsules reported to date, often due to aggregation occurring during the assembly and/or core removal steps. Two prominent systems that fulfill this requirement and which exhibit high colloidal stability are based on poly(allylamine hydrochloride) (PAH)/polystyrene sulfonate (PSS), a pair of polyelectrolytes that associate through electrostatic interactions, and poly(N-vinyl pyrrolidone) (PVPON)/poly(methacrylic acid) (PMA), a hydrogen bonded polymer pair.The PSS/PAH capsule system has been well characterized and has been shown to exhibit high colloidal stability.5 The surface of these capsules has also been exploited for postfunctionalization with low fouling polymers6 and for imparting specific recognition properties through the coupling of targeting molecules (e.g., antibodies).7 Furthermore, studies have shown that the permeability of these capsules can be tailored through the number of polymer layers deposited.8 The nonbiodegradable nature of PSS/PAH capsules, as well as the possibility to prepare capsules with sizes as low as tens of nanometers,5 make them well suited for the creation of synthetic and stable organelles.
PMA/PVPON capsules owe their structural stability to intermolecular hydrogen bonds that form and remain intact in acidic media, where the polyacid is protonated. However, these capsules spontaneously disintegrate upon ionization of PMA at solution pH values above the pKa of PMA (6.5).9 With the aim to create (bio)degradable capsules, we recently proposed an approach to crosslink the constituent polymersvia degradable covalent bonds. We hypothesized that above the pKa of PMA the stability of these capsules will be maintained solely by the degradable crosslinks and therefore the degradation profile of the capsules would closely follow the degradation of the crosslinking bonds. To this end, we synthesized thiol-modified PMA, assembled the multilayered capsules via the alternate deposition of the thiol-modified PMA with PVPON, and stabilized the capsules via the disulfide linkages.10 Above the pKa of PMA, in the absence of hydrogen bonding, PVPON was released and single-component PMA hydrogel capsules were obtained.11 We showed that the PMA/PVPON build-up on colloidal particles is linear,11 which allows simple control over the thickness of the capsule walls12 and dictates its properties, in particular permeability.13 These hydrogel capsules exhibit excellent colloidal stability in a range of conditions,14 including the presence of serum proteins, and degrade in the presence of a natural reducing agent, glutathione,11 verifying their (bio)degradable nature. These characteristics make the PMA hydrogel capsules attractive candidates for the creation of synthetic microreactors and artificial organelles.
Further challenges in the design of artificial cells and organelles relate to the loading of synthetic machinery into the reactor vessel and its retention within the carrier while permitting controlled accessibility of other solutes. LbL capsules are typically semipermeable and exhibit size exclusion behavior: while small solutes, such as buffer salts, solvent molecules and other low molecular weight molecules readily permeate through the multilayered polymer film, higher molecular weight solutes can have restricted diffusivity through the capsule wall.3,13 The cut-off value for such a “dialysis sphere” depends on the nature of the constituent polymers, the thickness and density of the polymer film (which depends on the number of deposited bilayers and the assembly conditions), and the solution conditions (pH, temperature, ionic strength). For both PSS/PAH8 and PMA13 capsules, the number of deposited layers controls the permeability of solutes. Single-component PMA capsules, being highly negatively charged, also exhibit semipermeable characteristics based on the charge of the solute,11 providing a barrier for diffusion of negatively charged polymers15 but not uncharged polymers.11 Charge exclusion was also observed for PSS/PAH capsules enriched with a negative component.16 As discussed below, the aforementioned means of control over capsule permeability allows the effective encapsulation of macromolecular cargo within these colloidal carriers. However, the dialysis membrane-like nature of highly hydrated LbL-derived polymer capsules implies that most solutes will have a non-negligible permeability through the capsule walls, potentially via a reptation mechanism for polymers17 and a relay-race mechanism for proteins.18
Encapsulation of drugs and reagents within the polymer capsules can be achieved via preloading or postloading techniques. The postloading method exploits temporary changes in the permeability of the preformed capsule membrane, which for PSS/PAH can be induced by the change in solvent pH and temperature19 or dehydration/rehydration.20 Alternatively, the cargo molecules can be associated with the template particles prior to the LbL assembly of the polymer film and become encased within the polymer capsule upon removal of the core particles. This approach, termed preloading, typically uses porous templates to adsorb the encapsulated material21 or inorganic salts with coprecipitated material22 and is widely used for the encapsulation of proteins. To exploit commercially available monodisperse templates in the micrometer and submicrometer size range, we have recently developed an approach to immobilize cargo, specifically single- and double-stranded DNA15 or polymer-peptide conjugates,13 onto the surface of amine-functionalized solid silica particles. Subsequent LbL assembly of thiol-modified PMA and PVPON followed by core removal yields monodisperse capsules with a uniform distribution of cargo (Fig. 1a,b), two factors crucial for reliable drug and reagent dosage.
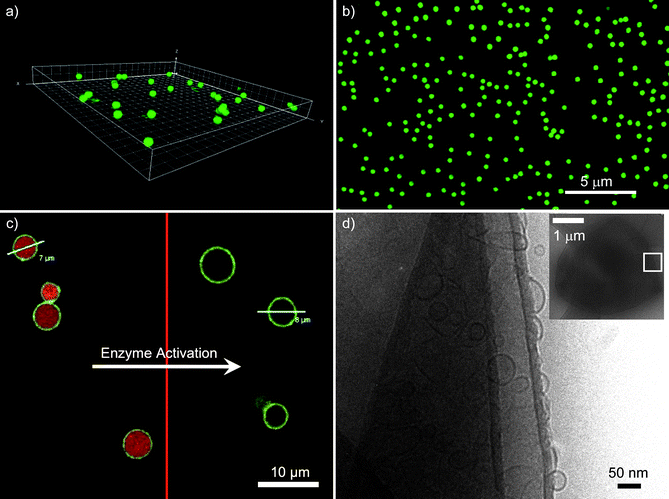 | ||
| Fig. 1 Confocal laser scanning microscopy image of the fluorescently-labeled PMA-peptide (PMA-CKKFGAEVVP (KP9)) within (a) 1 µm and (b) 0.5 µm diameter PMA capsules. Images reprinted with permission from ref. 13. (c) AF-546 labeled DNA co-encapsulated with DNase I in AF-488 labeled PMA hydrogel capsules. Triggering the enzymes causes digestion of the DNA and release of the low molecular weight products. Image reprinted with permission from ref. 37. (d) A cryo-transmission electron microscopy image of a capsosome (inset) and a close-up of the PSS/PAH polyelectrolyte shell, which contains intact liposomes. The liposome membranes are visible as black circles. Image reprinted with permission from ref. 46. | ||
Semipermeable vessels for biocatalysis
The exchange of nutrients between the internal milieu and external volume is necessary for an enclosed vessel to be considered a self-sustaining system. Unlike cellular systems, which have evolved a myriad of membrane-spanning proteins to allow controlled and active transport, many synthetic assemblies are impermeable to charged solutes. Without a means of obtaining nutrients from their surroundings, the encapsulated reaction quickly exhausts its supply of substrates. Continuous reactions, such as the synthesis of RNA within lipid vesicles, have been performed by increasing the membrane's permeability through temperature-induced transient defects,23 permeabilization with a mild detergent,24 or the assembly of leaky membranes.25 The incorporation of transmembraneproteins, although challenging, has proven to provide specific gating to liposome and polymersome assemblies.26 Alternatively, as previously mentioned, the semi-permeable nature of LbL-derived capsules coupled with the diverse techniques to achieve the loading of peptides, nucleic acids and intact proteins makes them standalone candidates for the creation of synthetic microreactors and artificial organelles.The most well-studied class of LbL-encapsulated bio-relevant transformations is the conversion of small molecules in enzymatic reactions. The encapsulated enzymes remain accessible to their substrates but are protected from high molecular weight inhibitors, with the products of the reaction able to diffuse to the exterior. Enzymes, including glucose oxidase,27 horseradish peroxidase,28 α-chymotrypsin,29catalase,30 pronase,31 or urease,32 have been encapsulated and their catalytic activity within the capsule was demonstrated. As a synthetic mimic to coupled enzymatic reactions within cellular organelles, the bienzyme catalytic system of glucose oxidase and horseradish peroxidase was encapsulated within LbL capsules.33 While recent evidence suggests the incorporation of enzymes into PSS/PAH capsules may cause a decrease in biocatalytic activity due to mass transfer restrictions and enzyme/polymer multilayer interactions,34 it is likely that this effect can be controlled via the judicious choice of the enzyme/polymer capsule system.
The synthesis and degradation of macromolecules within polymer capsules is a second set of reactions performed within LbL capsules and presents a significant step toward cell and organelle mimicry. Successful approaches include the polymerization of a phenolic polymer35 or DNA by polymerase chain reaction (PCR )36 within PSS/PAH capsules via the external introduction of the monomers. In particular, the ability to carry out PCR in micrometer-sized capsules may potentially allow for multiplexed PCR without a limitation on the number of nucleic acid amplification cycles, an inherent drawback to emulsion-based PCR technologies. As a biomedical platform, microcapsulated PCR can be envisaged as a technique for the synthesis and delivery of DNA-based therapeutics with a single vessel.
A prominent organelle of the cell, the lysosome, digests larger macromolecules by a variety of hydrolases. We have recently demonstrated the use of LbL capsules as synthetic lysosomes for the triggered digestion of DNA.37 Both DNA and the endonucleaseDNase I were encapsulated within the interior of PMA hydrogel capsules with the aid of mesoporous silica templates (Scheme 1(b)ii). The external introduction of divalent cation cofactors triggered the digestion of DNA by the endonuclease; the small products of digestion were permeable to the walls of the capsule (Fig. 1c). The use of fluorescently labeled DNA allowed its digestion to be monitored by high-throughput flow cytometry. We emphasize that the selective retention of DNA, permeability to smaller oligonucleotides, and external triggering of the reaction were made possible by the use of a semipermeable PMA hydrogel capsule.
Subcompartmentalized capsules
Biological cells are capable of performing different encapsulated (enzymatic cascade) reactions in parallel with high accuracy and specificity within their organelles. Hence, attempts to mimic eukaryotic cells require the assembly of a carrier vessel containing multiple subcompartments that can host different cargo. However, only a few groups have succeeded in engineering compartmentalized systems. Multicompartment micelles38 or trapping small lipid39–41 or polymer42,43 vesicles in a larger carrier vesicle are examples. The former assembly, liposomes trapped within a liposome, has proven to show controllable drug release39 and potential for combinatory drug delivery40 as compared to single liposomes. These assemblies have also been used to conduct triggered consecutive enzymatic reactions by making use of the higher permeability of liposomes at their phase transition temperature.41 In the latter approach (polymersomes within polymersomes), pH-sensitive channels incorporated in the membrane are proposed as selective gates to the subcompartments for a pH-dependent transport of small molecules.42 Polymersomes were also employed to perform a three-enzyme reaction, not by trapping the enzymes in subcompartments per se, but by localizing them in different parts of a polymersome, namely in its interior, within the polymer membrane and/or on its surface.43 Attempts to subdivide the interior of LbL-assembled capsules to date have involved the assembly of dual-compartmentalized capsules22,44 or the embedding of smaller capsules into a polyelectrolyte multilayer-stabilized gel bead.45We recently explored the combination of liposomes and polymer capsules to form capsosomes (Scheme 1(b)iii). Features from both systems are preserved within capsosomes, while some of the challenges of the individual systems are overcome. The polymer hydrogel carrier capsule provides the required structural integrity, with its degradability and permeability controlled by the choice of the building blocks. The semipermeable membrane of the polymer capsule allows for the controlled exchange of (bio)molecules between the interior and exterior, an important factor to enable continuous encapsulated reactions. Liposomes, on the other hand, divide the capsule's interior into subcompartments, which are suitable to efficiently trap and protect small and/or fragile (bio)molecules.
Capsosomes are assembled via a LbL approach, commencing with the adsorption of a polymer precursor layer onto silica template particles, followed by the assembly of the first layer of liposomes. Polymer separation layers are required for the adsorption of the next layer of liposomes, followed by a polymer capping layer that not only efficiently traps the liposomes, but also allows for the subsequent assembly of the polymer carrier capsule. Upon removal of the template core, capsosomes are obtained.
We first reported the incorporation of zwitterionic liposomes into PSS/PAH multilayered films on planar surfaces,46 demonstrating that the liposomes can be efficiently entrapped within the multilayer film without being ruptured or displaced. Assembly of the same system on colloidal particles yielded the formation of the first generation of capsosomes—structurally intact liposomes incorporated within a nondegradable polymer carrier capsule (Fig. 1d). With the goal to move beyond electrostatically driven buildup and aiming at a generalizable capsosome assembly, we introduced a novel noncovalent concept to anchor liposomes to polymers. Cholesterol, a native membrane constituent, was conjugated to two different polymers, poly(L-lysine) and PMA.48 Employing these modified polymers as precursor and separation layers in the assembly process resulted in more efficient liposome incorporation than the use of their unmodified counterparts. This anchoring concept is widely applicable since the incorporation of cholesterol into the membrane of the liposomes is a spontaneous process and largely independent of the lipid composition or buffer conditions. Moreover, the use of cholesterol-modified PMA as a capping layer allowed for the subsequent hydrogen-bonding driven assembly of PVPON and thiol-modified PMA to construct (bio)degradable carrier capsules. Upon crosslinking of the thiols in the film with either an oxidizing reagent or via the infiltration of a polymeric crosslinker,47 intact capsosomes consisting of a PMA hydrogel capsule with incorporated liposomes were obtained. Such capsosomes allow for the retention of cargo (the liposomes themselves and the entrapped enzymes within them) for up to two weeks.48 With the goal to confirm the presence and activity of a model enzyme, β-lactamase, within the liposomal subcompartments of the capsosomes and to quantify the number of subcompartments, we performed a colorimetric enzymatic assay ; the conversion of the yellow substrate nitrocefin to its red hydrolysed product.49 We demonstrated that as long as the enzymes remained trapped within the liposomal subcompartments, no color change was observed, but upon their lysis, the enzymes obtain access to their substrates and the conversion occurred. Using this quantitative assay , we calculated that a 3 µm capsosome confines ∼8 × 103 liposomes. Taken together, the above results establish a platform that accommodates key aspects involved in the assembly of therapeutic artificial cells and organelles.
Conclusions/outlook
In this mini-review, we have discussed the achievements in the design of LbL-derived polymer capsules, including their assembly and permeability properties, and loading techniques, in light of their use as synthetic mimics of cells and subcellular organelles. We expect that advances in the field of microencapsulated catalysis and further improvements in subcompartmentalization techniques will lead to enhanced opportunities in the assembly of artificial cells and organelles. The presented proof-of-concept examples demonstrate the potential of LbL capsules for these applications, yet most of the examples employ model enzymes with limited medical relevance. Future challenges lie in the use of specific enzymes associated with certain diseases, and conducting cascade multi-enzyme reactions within the synthetic subcompartmentalized systems. Further challenges to be addressed include self-reproduction/division, self-repair, targeting, recognition and induced self-destruction. The polymer hydrogel capsules provide an exciting platform for advancing such challenges.Acknowledgements
This work was supported by the Australian Research Council under the Federation Fellowship and Discovery Project schemes.Notes and references
- S. Mann, Angew. Chem., Int. Ed., 2008, 47, 5306 CrossRef CAS; A. Pohorille and D. Deamer, Trends Biotechnol., 2002, 20, 123 CrossRef CAS; Y. Zhang, W. C. Ruder and P. R. Le Duc, Trends Biotechnol., 2008, 26, 14 CrossRef CAS; R. V. Sole, Int. J. Biochem. Cell Biol., 2009, 41, 274 CrossRef CAS.
- D. Deamer, Trends Biotechnol., 2005, 23, 336 CrossRef CAS; D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838 CrossRef CAS; C. Lo Presti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576 RSC; P.-A. Monnard and D. W. Deamer, The Anatomical Record, 2002, 268, 196 Search PubMed; T. Oberholzer and P. L. Luisi, J. Biol. Phys., 2002, 28, 733 CrossRef CAS; S. M. Christensen and D. Stamou, Soft Matter, 2007, 3, 828 RSC; D. M. Vriezema, M. C. Aragones, J. Elemans, J. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445 CrossRef CAS.
- B. G. De Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2007, 36, 636 RSC.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707 RSC; E. Kharlampieva, V. Kozlovskaya and S. A. Sukhishvili, Adv. Mater., 2009, 21, 3053 CrossRef CAS.
- D. I. Gittins and F. Caruso, Adv. Mater., 2000, 12, 1947 CrossRef CAS; D. I. Gittins and F. Caruso, J. Phys. Chem. B, 2001, 105, 6846 CrossRef CAS; G. Schneider and G. Decher, Langmuir, 2008, 24, 1778 CrossRef CAS.
- R. Heuberger, G. Sukhorukov, J. Vörös, M. Textor and H. Möhwald, Adv. Funct. Mater., 2005, 15, 357 CrossRef CAS.
- C. Cortez, E. Tomaskovic-Crook, A. P. R. Johnston, A. M. Scott, E. C. Nice, J. K. Heath and F. Caruso, ACS Nano, 2007, 1, 93 CrossRef CAS.
- A. S. Angelatos, A. P. R. Johnston, Y. J. Wang and F. Caruso, Langmuir, 2007, 23, 4554 CrossRef CAS; A. A. Antipov, G. B. Sukhorukov, E. Donath and H. Mohwald, J. Phys. Chem. B, 2001, 105, 2281 CrossRef CAS.
- S. A. Sukhishvili and S. Granick, Macromolecules, 2002, 35, 301 CrossRef CAS.
- A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27 CrossRef CAS.
- A. N. Zelikin, Q. Li and F. Caruso, Chem. Mater., 2008, 20, 2655 CrossRef CAS.
- A. L. Becker, A. N. Zelikin, A. P. R. Johnston and F. Caruso, Langmuir, 2009 DOI:10.1021/la901687a.
- S.-F. Chong, A. Sexton, R. De Rose, S. J. Kent, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5178 CrossRef CAS.
- R. De Rose, A. N. Zelikin, A. P. R. Johnston, A. Sexton, S. F. Chong, C. Cortez, W. Mulholland, F. Caruso and S. J. Kent, Adv. Mater., 2008, 20, 4698 CrossRef CAS; S. Sivakumar, V. Bansal, C. Cortez, S. F. Chong, A. N. Zelikin and F. Caruso, Adv. Mater., 2009, 21, 1820 CrossRef CAS.
- A. N. Zelikin, A. L. Becker, A. P. R. Johnston, K. L. Wark, F. Turatti and F. Caruso, ACS Nano, 2007, 1, 63 CrossRef CAS.
- W. J. Tong, W. F. Dong, C. Y. Gao and H. Mohwald, J. Phys. Chem. B, 2005, 109, 13159 CrossRef CAS.
- P. G. De Gennes, J. Chem. Phys., 1971, 55, 572 CrossRef.
- V. A. Kabanov, V. B. Skobeleva, V. B. Rogacheva and A. B. Zezin, J. Phys. Chem. B, 2004, 108, 1485 CrossRef CAS.
- A. A. Antipov and G. B. Sukhorukov, Adv. Colloid Interface Sci., 2004, 111, 49 CrossRef CAS.
- O. Kreft, R. Georgieva, H. Baumler, M. Steup, B. Muller-Rober, G. B. Sukhorukov and H. Mohwald, Macromol. Rapid Commun., 2006, 27, 435 CrossRef CAS.
- Y. J. Wang and F. Caruso, Chem. Mater., 2006, 18, 4089 CrossRef CAS; Y. Wang, A. D. Price and F. Caruso, J. Mater. Chem., 2009 10.1039/b901742a.
- O. Kreft, M. Prevot, H. Mohwald and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2007, 46, 5605 CrossRef.
- P.-A. Monnard, A. Luptak and D. W. Deamer, Philos. Trans. R. Soc. London, Ser. B, 2007, 362, 1741 CrossRef CAS.
- M. Treyer, P. Walde and T. Oberholzer, Langmuir, 2002, 18, 1043 CrossRef CAS.
- A. C. Chakrabarti, R. R. Breaker, G. F. Joyce and D. W. Deamer, J. Mol. Evol., 1994, 39, 555 CrossRef CAS; S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobe, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122 CrossRef CAS.
- H. J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538 CrossRef CAS; V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669 CrossRef CAS; G. Steinberg-Yfrach, J. L. Rigaud, E. N. Durantini, A. L. Moore, D. Gust and T. A. Moore, Nature, 1998, 392, 479 CrossRef CAS.
- R. Srivastava, J. Q. Brown, H. Zhu and M. J. McShane, Macromol. Biosci., 2005, 5, 717 CrossRef CAS; Z.-l. Zhi and D. T. Haynie, Chem. Commun., 2006, 147 RSC.
- S. V. Rao, K. W. Anderson and L. G. Bachas, Biotechnol. Bioeng., 1999, 65, 389 CrossRef CAS.
- A. I. Petrov, D. V. Volodkin and G. B. Sukhorukov, Biotechnol. Prog., 2005, 21, 918 CrossRef CAS; O. P. Tiourina, A. A. Antipov, G. B. Sukhorukov, N. I. Larionova, Y. Lvov and H. Mohwald, Macromol. Biosci., 2001, 1, 209 CrossRef CAS.
- F. Caruso, D. Trau, H. Mohwald and R. Renneberg, Langmuir, 2000, 16, 1485 CrossRef CAS; Y. Wang and F. Caruso, Chem. Commun., 2004, 1528 RSC.
- T. Borodina, E. Markvicheva, S. Kunizhev, H. Moehwald, G. B. Sukhorukov and O. Kreft, Macromol. Rapid Commun., 2007, 28, 1894 CrossRef CAS.
- A. Antipov, D. Shchukin, Y. Fedutik, I. Zanaveskina, V. Klechkovskaya, G. Sukhorukov and H. Mohwald, Macromol. Rapid Commun., 2003, 24, 274 CrossRef CAS; A. Yu, I. Gentle, G. Lu and F. Caruso, Chem. Commun., 2006, 2150 RSC.
- O. Kreft, M. Prevot, H. Moehwald and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2007, 46, 5605 CrossRef; W. C. Mak, J. Bai, X. Y. Chang and D. Trau, Langmuir, 2009, 25, 769 CrossRef CAS; P. Pescador, I. Katakis, J. L. Toca-Herrera and E. Donath, Langmuir, 2008, 24, 14108 CrossRef CAS; E. W. Stein, D. V. Volodkin, M. J. McShane and G. B. Sukhorukov, Biomacromolecules, 2006, 7, 710 CrossRef CAS.
- L. O. Wiemann, A. Buthe, M. Klein, A. van den Wittenboer, L. Daehne and M. B. Ansorge-Schumacher, Langmuir, 2009, 25, 618 CrossRef CAS.
- R. Ghan, T. Shutava, A. Patel, V. T. John and Y. Lvov, Macromolecules, 2004, 37, 4519 CrossRef CAS.
- W. C. Mak, K. Y. Cheung and D. Trau, Adv. Funct. Mater., 2008, 18, 2930 CrossRef CAS.
- A. D. Price, A. N. Zelikin, Y. Wang and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 329 CrossRef CAS.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS.
- E. T. Kisak, B. Coldren, C. A. Evans, C. Boyer and J. A. Zasadzinski, Curr. Med. Chem., 2004, 11, 199 CrossRef CAS; C. Boyer and J. A. Zasadzinski, ACS Nano, 2007, 1, 176 CrossRef CAS; V. Mishra, S. Mahor, A. Rawat, P. Dubey, P. N. Gupta, P. Singh and S. P. Vyas, Vaccine, 2006, 24, 5559 CrossRef CAS.
- W. T. Al-Jamal and K. Kostarelos, Int. J. Pharm., 2007, 331, 182 CrossRef CAS.
- P. Y. Bolinger, D. Stamou and H. Vogel, Angew. Chem., Int. Ed., 2008, 47, 5544 CrossRef CAS.
- H. C. Chiu, Y. W. Lin, Y. F. Huang, C. K. Chuang and C. S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875 CrossRef CAS.
- S. F. M. van Dongen, M. Nallani, J. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem.-Eur. J., 2009, 15, 1107 CAS.
- O. Kreft, A. G. Skirtach, G. B. Sukhorukov and H. Möhwald, Adv. Mater., 2007, 19, 3142 CrossRef CAS.
- B. G. De Geest, S. De Koker, K. Immesoete, J. Demeester, S. C. De Smedt and W. E. Hennink, Adv. Mater., 2008, 20, 3687 CrossRef CAS.
- B. Städler, R. Chandrawati, K. Goldie and F. Caruso, Langmuir, 2009, 25, 6725 CrossRef CAS.
- S.-F. Chong, R. Chandrawati, B. Städler, J. Park, J. Cho, Y. Wang, Z. Jia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009 DOI:10.1021/smll.20090090.
- R. Chandrawati, B. Städler, A. Postma, L. A. Connal, S.-F. Chong, A. N. Zelikin and F. Caruso, Biomaterials, 2009 DOI:10.1016/j.biomaterials.2009.07.040.
- B. Städler, R. Chandrawati, A. D. Price, S.-F. Chong, K. Breheney, A. Postma, L. A. Connal, A. N. Zelikin and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 4359 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |