DOI:
10.1039/B9NR00112C
(Feature Article)
Nanoscale, 2009,
1, 61-67
Received
12th June 2009
, Accepted 19th August 2009
First published on 4th September 2009
Abstract
Gold nanoparticles (AuNPs) are promising nanocarriers for therapeutics due to their facile synthesis, ease of functionalization, biocompatibility, and inherent non-toxicity. The unique chemical and physical properties of AuNP monolayers provide versatility in delivery method and tunability of surface properties. Here, we discuss several strategies to utilize the properties of AuNPs for drug delivery.
1. Introduction
Considerable effort has been focused on the creation of drug delivery systems (DDSs) for improving cancer chemotherapy, with the goals of enhancing therapeutic selectivity and efficacy.1 One area where DDSs have proved highly beneficial is in targeted delivery, where marked improvements in chemotherapy have been achieved through passive and/or active targeting approaches with nanocarriers including liposomes,2polymer micelles and vesicles,3dendrimers,4 and metal nanoparticles.5
Gold nanoparticles (AuNPs) have recently emerged as highly promising DDSs.6AuNPs have multiple aspects that make them well-suited for use in delivery applications. First, AuNPs of many different sizes (1–150 nm) can be fabricated in a controlled fashion with control over size dispersity,7 whereas polymer-based carriers can be heterogeneous in structure due to high polydispersity.5 Furthermore, AuNPs have high surface-area-to-volume ratios, and, as a result, a high density of ligands can be appended for targeting or drug-loading purposes. For example, a 2 nm diameter core AuNP can load ∼100 ligands per particle.8 Secondly, functional diversity can be readily achieved by creation of multifunctional monolayers using techniques such as ligand place-exchange reactions,9 allowing multiple functional moieties such as drugs and targeting agents to be placed onto the particle surface with fewer synthetic challenges than many other delivery vehicles.5 Finally, the gold core is essentially inert, non-toxic, and biocompatible, making it an ideal starting point for carrier construction.10
The ease of functionalization of AuNPs has enabled their use in a variety of delivery strategies Scheme 1). In one approach, prodrugs can be covalently conjugated to AuNPsvia cleavable linkers. Alternatively, hydrophobic drugs can be non-covalently loaded onto AuNPs, allowing conjugation without structural modification of the drug payload. Once loaded, AuNP payloads can be released by either internal (e.g.glutathione)6a or external (e.g. light)11 stimuli. The versatility of the AuNP monolayer is central to both release mechanisms, providing tunability for endogenous release mechanisms and a functional platform for external release strategies.
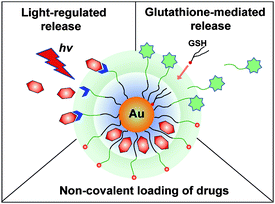 |
| | Scheme 1
AuNPs as a multimodal drug delivery system. | |
Several reviews regarding Au NPs for biomedical applications have recently been published, generally describing biosensing, diagnostics, and therapy.12 In this review we will discuss various strategies which have been employed in creating AuNPs as DDS platforms, focusing on the role of the monolayer structure on DDS function.
One of the key advantages of AuNPs is our ability to fabricate these nanomaterials with a wide variety of core sizes. Some general synthetic procedures of core–shell AuNPs are summarized in Table 1. The one-pot protocol developed by Brust and Schiffrin et al. in 1994 (Scheme 2) is of particular interest.13 A wide variety of monolayer-protected AuNPs can be formed rapidly and in a scalable fashion using this approach. In this method, an AuCl4− salt is reduced with NaBH4 in presence of the desired ligands. The core size of these particles can be varied from 1.5 nm to ∼6 nm by varying the ligand–gold stoichiometry. Larger particles can be fabricated through either ripening approaches14 or viacitrate reduction of gold salts.15 To enhance the functional versatility of AuNPs, mixed monolayer-protected AuNPs can be synthesized directly with proper ligands or through post-functionalization. The most commonly used method for creation of mixed monolayer-protected AuNPs is through use of the place-exchange reaction first introduced by Murray (Scheme 2).9 In this protocol, external thiols displace the existing ligands of AuNPs in an equilibrium process. Several research groups have fabricated delivery systems based on AuNPs bearing functional moieties that are anchored with thiol linkers.6 Control of ligand structure can be used to make the particles more suitable for delivery applications, e.g. through use of biocompatible oligo(ethylene glycol) (OEG) and poly(ethylene glycol) (PEG) moieties.16
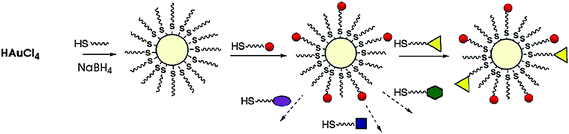 |
| | Scheme 2 Formation of AuNPs using the Schiffrin reaction and a mixed monolayer of AuNPs using the Murray place-exchange reaction. | |
Table 1 Synthetic methods and capping agents for AuNPs of varying core sizes
The first requirement for developing a DDS is an understanding of the interaction of the carrier with cells. Cellular uptake and intracellular fate of AuNPs depend on surface functionality and size.17 Charge is also an important determinant,18 Rotello and Vachet have demonstrated that cellular uptake of functionalized AuNPs is dependent on both charge and hydrophobicity.19 Stellacci et al. have reported that cell membrane penetration by NPs was also dependent on ligand shell morphology,20 with “striped” (structured ligand shell) NPs apparently capable of passing directly through the plasma membrane of the cells without creating pores on the cell membrane that can cause cytotoxicity (Fig. 1). Furthermore, organelle-specific localization of particles can be easily achieved by decorating the surface with a targeting moiety. For example, Feldheim et al. have demonstrated nuclear targeting of particles modified with a nuclear localization sequence (NLS).21 Recently, Brust et al. have used transmission electron microscopy22 to show that specific cellular targets such as the nucleus and other organelles can be targeted by modifying the particle surface with cell penetrating peptides (CPPs) [e.g. the oligopeptides TAT (AGRKKRRQRRR) and Pntn (GRQIKIWFQNRRMKWKK)] and the nuclear localization sequence (NLS: GGFSTSLRARKA) (Fig. 2).
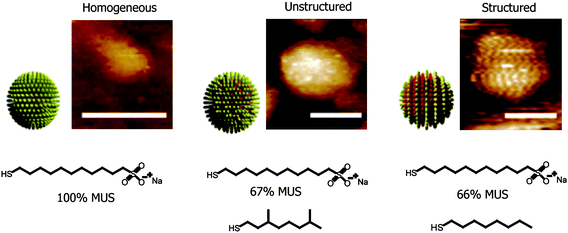 |
| | Fig. 1 Schematic diagrams of the ligand shell structure of the nanoparticles and representative STM images (scale bars 5 nm). Reprinted with permission from ref. 20 (Copyright 2008 Nature Publishing Group). | |
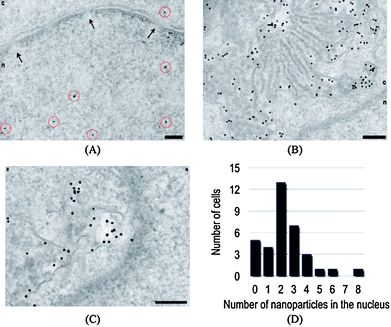 |
| | Fig. 2 Nuclear targeting (A) by PEG-modified nanoparticles functionalized with a combination of CPPs (2% TAT and 2% Pntn) and 2% NLS. Nanoparticles are highlighted by red circles. The nuclear envelope with nuclear pores (arrows) is clearly shown in this image. The nucleus is denoted ‘n’, and the cytosol ‘c’. Unusual perinuclear membranous structures (B and C) that are highly loaded with nanoparticles are typically also observed under these conditions. Nuclear targeting is enhanced in comparison with experiments in the absence of CPPs (D). Scale bars are 200 nm. Reprinted with permission from ref. 22 (Copyright 2008 The American Chemical Society). | |
Particle size provides the second key determinant of particle uptake. Chan et al. have demonstrated that cellular uptake depends on the size of nanoparticles.23AuNPs (2–100 nm) coated with Herceptin were fabricated and tested for ErbB2 receptor-mediated internalization in breast cells. They found that most efficient cellular uptake occurred with the 20–50 nm particles and programmed cell death (apoptosis) was enhanced by particles in the 40–50 nm range. The authors also have investigated the effect of AuNP size (10–100 nm) on passive targeting of tumors in vivo.24 The larger nanoparticles remained near the vasculature, whereas smaller nanoparticles rapidly diffused from blood vessels to the tumor matrix (Fig. 3).
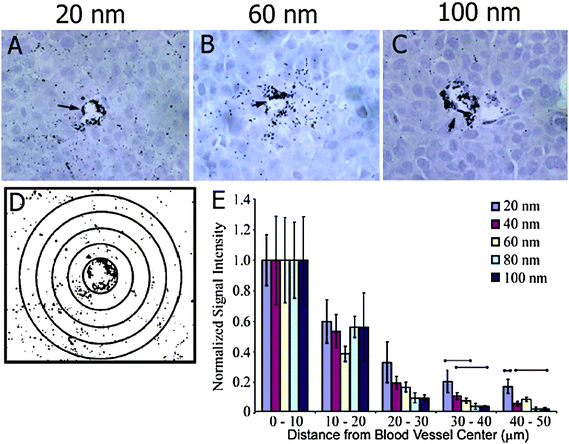 |
| | Fig. 3 Particle-size-dependent permeation of the tumor interstitial space. (A–C) Histological samples were obtained for 20, 60, and 100 nm particle sizes at 8 h post-injection (HPI). (D) ImageJ software was used to generate contrast-enhanced images for densitometry analysis. (E) Densitometry signal was quantified at 10 mm distances away from blood vessel centers 8 HPI and was normalized to the signal at 0–10 µm. Reprinted with permission from ref. 24 (Copyright 2009 The American Chemical Society). | |
4. Drug attachment and release strategies using gold nanoparticles
Both the transport and release of drugs play critical roles in providing effective delivery systems. In general drugs can be loaded onto nanocarriers by either covalent conjugation or non-covalent interactions.25 The non-covalent approach employs active drugs while the covalent attachment generally requires intercellular processing of a prodrug.26 The ease of controlling the functionality and structure of AuNP monolayers makes them excellent platforms for DDS creation.
4.1
Glutathione-mediated release in covalent and non-covalent conjugation strategies
DDS systems based on glutathione (GSH)-mediated payload release exploit the dramatic higher intracellularGSH concentration (1–10 mM)27 relative to extracellularthiol levels (GSH 2 μM, cysteine 8 μM). 28 The high levels of intracellularGSH can be used to release prodrugs (payloads) on AuNPs through either place-exchange reactions at the core or viadisulfide exchange. Importantly, the monolayer of nanoparticles can provide steric shielding against exchange with surface cysteines of proteins in the bloodstream,29 enabling their potential use in vivo.
In early studies, Rotello et al. developed a nanoparticle-based delivery system featuring glutathione release.6a The particles (core diameter: ∼2 nm) featured a mixed monolayer composed of cationic ligands (TTMA) and fluorogenic ligands (HSBDP) (Fig. 4). The cationic surface of the nanoparticles facilitated their penetration through cell membranes, and the payload release was triggered by intracellularglutathione (GSH). In this delivery system, the Bodipy moiety of the HSDBP ligand was non-fluorescent when attached to the particle due to fluorescence quenching by the Au core.30 The fluorescence was recovered upon GSH treatment in a cuvette, or with intracellularthiols in human liver cells (Hep G2). GSH-controlled release of the dye was verified by treating mouse embryonic fibroblast cells (MEF, having >50% lower intracellularGSH levels than HepG2) with varying concentration of glutathione monoester (GSH-OEt), transiently increasing the glutathione level inside the cells. In these studies, a dose-dependent increase in fluorescence was observed with increasing GSH-OEt concentration. In a similar fashion, AuNP-mediated DNA transfection efficiency was regulated by controlling the intracellularglutathione level.31
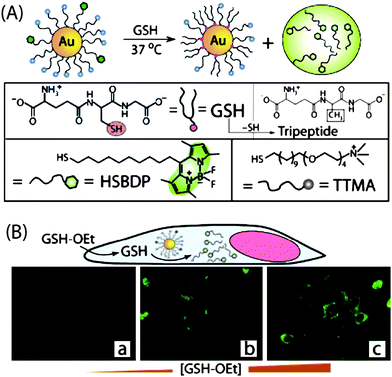 |
| | Fig. 4 (A) Schematic illustration of GSH-mediated surface monolayer exchange reaction/payload release. (B) Fluorescence images of MEF cells displaying GSH-controlled release of the fluorophore after incubation with 0, 5, and 20 mM GSH-OEt. | |
The ligand-displacement rate by intracellularthiols (e.g. DHLA and GSH) depends strongly on the monolayer structure and surface charge,32 allowing controlled release of payload. As an example, Kotov et al. have demonstrated that AuNPs bearing 6-mercaptopurine-9-β-D-ribofuranoside significantly enhanced the anti-proliferative effect against K-562 leukemia cells compared to the same drug in free form.33 This improvement was attributed to enhanced intracellular transport followed by the subsequent GSH-mediated release in cytoplasm and lysosomes.
In a strategy that combines glutathione-mediated release with non-covalent loading of drugs (vide infra), Kim et al. reported cyclodextrin-functionalized AuNPs as carriers of anti-cancer drugs.34 The AuNPs used the cyclodextrin moieties to encapsulate drugs, anti-epidermal growth factor receptor (anti-EGFR) antibody as a targeting moiety, and poly(ethyleneglycol) (PEG) as an anti-fouling shell (Fig. 5). The anticancer drug β-lapachone, was efficiently encapsulated into the hydrophobic cavity of cyclodextrin on AuNPs and then released by intracellularGSH. The introduction of an anti-EGFR antibody onto the AuNPs was shown to both enhance the uptake of AuNPs and increase the degree of apoptosis.
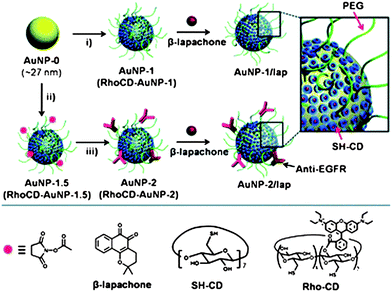 |
| | Fig. 5 Schematic illustration of the functionalization of AuNP carriers with β-lapachone, using: i) SH-CD and mPEG-SH for AuNP-1 (RhoCD and mPEG-SH for RhoCD-AuNP-1); ii) SH-CD, mPEG-SH, and NHS-PEG-SH for AuNP-1.5 (RhoCD, mPEG-SH, NHS-PEG-SH for RhoCD-AuNP-1.5) and iii) anti-EGFR. Reprinted with permission from ref. 34 (Copyright 2009 The Royal Society of Chemistry). | |
4.2 Monolayer encapsulation of therapeutics
Non-covalent incorporation of drugs into AuNP monolayers provides an alternative delivery strategy that allows direct use of unmodified drugs, thus avoiding prodrug processing issues. Drug encapsulation with AuNPs relies on the use of ligands that generate a hydrophobic interior to the monolayer. Structurally, the radial nature of the monolayer results in a decrease in ligand density as one goes further from the core of small AuNP cores (< 6 nm).35 Consequently “hydrophobic pockets” are created inside the monolayer of the AuNP into which hydrophobic materials can be partitioned. Pasquato et al. demonstrated the encapsulation of radical probes in AuNP monolayers, using EPR spectroscopy to monitor the partition of lipophilic probes between a monolayer of AuNPs and bulk water.36 As expected, smaller particles featuring more strongly radial monolayers favor guest encapsulation (Fig. 6).
![(A) Schematic representation of AuNP and the nitroxide probe inclusion. (B) Plot of the ratio between the concentration of 2 partitioned in the monolayer and that of the free species (● 16 nm; ▼ 3.4 nm; ■ 5.3 nm) as a function of [HS-C8-TEG] bound to the gold. Reprinted with permission from ref. 36b (Copyright 2005 The American Chemical Society).](/image/article/2009/NR/b9nr00112c/b9nr00112c-f6.gif) |
| | Fig. 6 (A) Schematic representation of AuNP and the nitroxide probe inclusion. (B) Plot of the ratio between the concentration of 2 partitioned in the monolayer and that of the free species (● 16 nm; ▼ 3.4 nm; ■ 5.3 nm) as a function of [HS-C8-TEG] bound to the gold. Reprinted with permission from ref. 36b (Copyright 2005 The American Chemical Society). | |
The concept of monolayer encapsulation has recently been applied to drug delivery. Rotello et al. have developed a biocompatible AuNP carrier that employs hydrophobic pockets to encapsulate drugs and deliver them into cancer cells.37 The particles (∼2.5 nm core) featured a hydrophobic alkanethiol interior and a hydrophilic shell composed of a tetra(ethylene glycol) (TEG) unit terminated with a zwitterionic headgroup designed to minimize nonspecific binding with biomacromolecules38 and other cell surface functionalities (Fig. 7). Hydrophobic payloads were kinetically entrapped in the monolayer, with the resulting host–guest materials stable in buffer and serum. The entrapped payloads were released into cells by membrane-mediated diffusion, as demonstrated by both fluorescence microscopy (using a fluorophore payload) and through drug efficacy with therapeutic guests. No particle uptake was observed with these systems using ICP-MS, making these systems excellent candidates for passive targeting using the enhanced permeability and retention effect.39
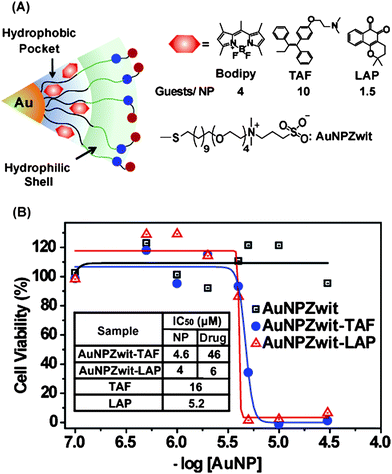 |
| | Fig. 7 (A) Structure of particles and guest compounds: Bodipy, TAF, and LAP, the number of encapsulated guests per particle (B) Cytotoxicity of AuNPZwit complexes measured by Alamar blueassay after 24 h incubation with MCF-7 cells. IC50 of AuNP (NP), equivalent drugs (Drug), and free drugs are shown in the table. | |
Photodynamic therapy (PDT) is a promising strategy that uses reactive oxygen species (ROS) to induce apoptosis or necrosis. Burda et al. reported the use of PEGylated AuNP-Pc4 (Si-phthalocyanine) conjugates as efficient platforms for PDT (Fig. 8a).40 The phthalocyaninephotosensitizing agent was encapsulated by the PEG ligands through van der Waals interactions, with the PEG ligand inhibiting colloid aggregation and providing significant resistance to non-specific binding with biomacromolecules. The release of Pc4 from AuNPsin vitro in a two-phase solution system and in vivo in cancer-bearing mice (Fig. 8b) indicated that the delivery process was highly efficient, with Pc4 preferentially accumulated in tumor sites. With the AuNP-Pc 4 conjugates, the drug delivery time required for PDT was reduced from 2 days using the free drug to 2 h, using the conjugate (Fig. 8).
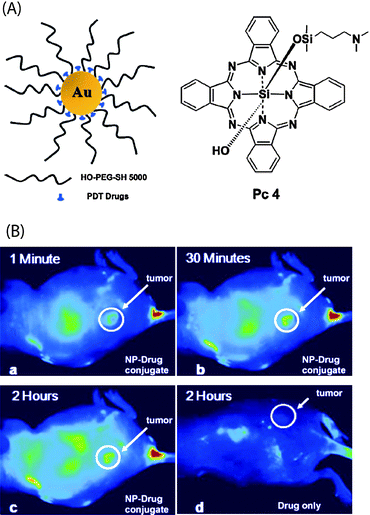 |
| | Fig. 8 (A) Structure of the water-soluble AuNPs as a PDT drug delivery agents, Pc 4 structure (B) Fluorescence images of a tumor-bearing mouse after being injected with AuNP-Pc 4 conjugates in normal saline (0.9% NaCl, pH 7.2), (a) 1 min, (b) 30 min, and (c) 120 min after intravenous tail injection. Any bright signal is due to Pc 4 fluorescence. For comparison, a mouse that got only a Pc 4 formulation without the AuNP vector injected is shown in panel (d). No circulation of the drug in the body or into the tumor was detectable 2 h after injection without the AuNPs as drug vector. Reprinted with permission from ref. 40 (Copyright 2008 The American Chemical Society). | |
4.3. Light-regulated release
The ability to regulate drug release is an important property for drug delivery systems.5d While endogenous approaches such as GSH-mediated release provide very useful strategies for delivery, externally controlled release provides a complementary tool for site- and time-specific control of payload release.41 Recently, caged drugs have been developed where the activity of the drug was suppressed by attaching it to a blocking element through a photoremovable protecting group.42 Rotello et al. have applied this strategy to AuNP delivery vehicles, utilizing a photo-cleavable o-nitrobenzyl ester moiety that dissociates upon light irradiation to alter the surface potential from positive to negative, thereby releasing adsorbed DNA.11 In more recent studies, the authors demonstrated light-controlled release of anticancer drug (5-fluorouracil) from nanoparticles (Fig. 9).43 The AuNPs (Au_PCFU, Au core: ∼2 nm) featured a mixed monolayer of zwitterionic and photocleavable ligands. The zwitterionic ligand provided solubility and prevented cellular uptake. The photocleavable ligand linked the fluorouracil to the particle through an orthonitrobenzyl group that could be effectively cleaved using near-UV irradiation (365 nm). An IC50 value of 0.7 μM was observed upon irradiation for Au_PCFU on a per particle basis, whereas no significant cell death was observed in cells treated with only light or only Au_PCFU.
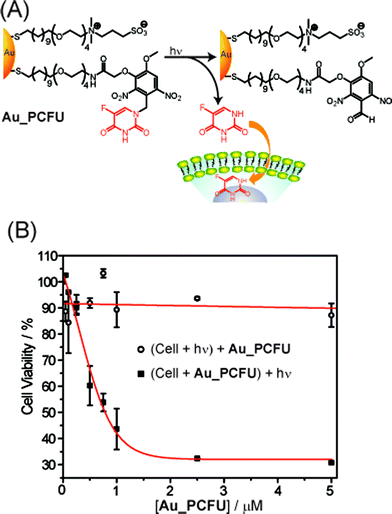 |
| | Fig. 9 (A) Photochemical reaction (365 nm) of Au_PCFU and delivery of payload to cell. (B) Cytotoxicity of different concentrations of Au_PCFU under uncaging and control conditions. The IC50 value was 0.7 µM per particle, 11.9 µM per drug. | |
In related research, Nakanishi et al. have reported a photoresponsive nanocarrier of amines, including cell-signaling agents. (Fig. 10).44 In this approach a carbamate linkage could be dissociated via the photocleavage reaction of the 2-nitrobenzyl group upon near-UV irradiation. The caging process was very effective: histamine had no biological activity while it was attached to AuNPs but became active when it was released from the particles upon photo-irradiation.
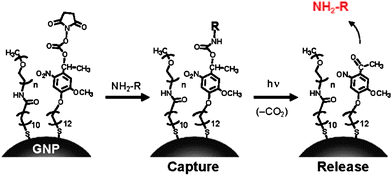 |
| | Fig. 10 Capture and release of amines on AuNPs having a photocleavable succinimidyl ester. Reprinted with permission from ref. 44 (Copyright 2009 The American Chemical Society). | |
4.4. Other attachment/release strategies
A variety of additional delivery strategies have been developed using AuNP platforms. Schoenfisch et al. have demonstrated that nitric oxide (NO) can be efficiently released at acidic pH from AuNPs,45 providing a potential means of controlling multiple cellular processes including angiogenesis, vasodilation, and the immune response.46 Hwu et al. have used a phosphate linker to conjugate paclitaxel to AuNP and Fe3O4 particles.47 The drug could be released from the particles using phosphodiesterase.
Conclusions and outlook
Gold nanoparticles provide a promising scaffold for drug delivery. The combination of tunable monolayer properties and stability, low inherent toxicity, functional versatility, and controlled release of payloads provides a wealth of options for the design of DDSs. Concurrent with the creation of new vehicles, however, a number of important issues require further examination. These issues include the size and surface dependent cytotoxicity, immune response, and biodistribution/pharmacokinetics of AuNPs. These investigations will serve not only to help in the design of effective DDSs, but will also provide fundamental insight into the interactions of nanomaterials with biological systems.
Acknowledgements
Support by the NIH (GM077173) is gratefully acknowledged.
References
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS.
-
(a) M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS;
(b) L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 56, 1649–1659 CrossRef CAS.
-
(a) C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS;
(b) H. L. Crampton and E. E. Simanek, Polym. Int., 2007, 56, 489–496 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 Search PubMed.
-
(a) R. Hong, G. Han, J. M. Fernandez, B. J. Kim, N. S. Forbes and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 1078–1079 CrossRef CAS;
(b) G. F. Paciotti, D. G. I. Kingston and L. Tamarkin, Drug Dev. Res., 2006, 67, 47–54 CrossRef CAS;
(c) J. D. Gibson, B. P. Khanal and E. R. Zubarev, J. Am. Chem. Soc., 2007, 129, 11653–11661 CrossRef CAS.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- A. C. Templeton, M. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
-
(a) E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS;
(b) M. Tsoli, H. Kuhn, W. Brandau, H. Esche and G. Schmid, Small, 2005, 1, 841–844 CrossRef CAS;
(c) R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289–1306 CrossRef CAS.
- G. Han, C. C. You, B. J. Kim, R. S. Turingan, N. S. Forbes, C. T. Martin and V. M. Rotello, Angew. Chem., Int. Ed., 2006, 45, 3165–3169 CrossRef CAS.
-
(a) N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS;
(b) E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC;
(c) M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS;
(d) C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS;
(e) M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- T. Teranishi, S. Hasegawa, T. Shimizu and M. Miyake, Adv. Mater., 2001, 13, 1699–1701 CrossRef CAS.
-
(a) K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735–743 CrossRef CAS;
(b) G. Frens, Nature Phys. Sci., 1973, 241, 20–22;
(c) J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–57 RSC.
-
(a) H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS;
(b) S. Takae, Y. Akiyama, H. Otsuka, T. Nakamura, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2005, 6, 818–824 CrossRef CAS;
(c) H. Otsuka, Y. Akiyama, Y. Nagasaki and K. Kataoka, J. Am. Chem. Soc., 2001, 123, 8226–8230 CrossRef CAS;
(d) T. Ishii, H. Otsuka, K. Kataoka and Y. Nagasaki, Langmuir, 2004, 20, 561–564 CrossRef CAS;
(e) H. Khalil, D. Mahajan, M. Rafailovich, M. Gelfer and K. Pandya, Langmuir, 2004, 20, 6896–6903 CrossRef CAS;
(f) R. G. Shimmin, A. B. Schoch and P. V. Braun, Langmuir, 2004, 20, 5613–5620 CrossRef CAS;
(g) J. C. Olivier, R. Huertas, H. J. Lee, F. Calon and W. M. Pardridge, Pharm. Res., 2002, 19, 1137–1143 CrossRef CAS.
- M. Ferrari, Nat. Nanotechnol., 2008, 3, 131–132 Search PubMed.
- E. C. Cho, J. W. Xie, P. A. Wurm and Y. N. Xia, Nano Lett., 2009, 9, 1080–1084 CrossRef CAS.
- Z.-J. Zhu, P. S. Ghosh, O. R. Miranda, R. W. Vachet and V. M. Rotello, J. Am. Chem. Soc., 2008, 130, 14139–14143 CrossRef CAS.
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H.-S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS.
- A. G. Tkachenko, H. Xie, D. Coleman, W. Glomm, J. Ryan, M. F. Anderson, S. Franzen and D. L. Feldheim, J. Am. Chem. Soc., 2003, 125, 4700–4701 CrossRef CAS.
- P. Nativo, I. A. Prior and M. Brust, ACS Nano, 2008, 2, 1639–1644 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 Search PubMed.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- M. T. Morgan, Y. Nakanishi, D. J. Kroll, A. P. Griset, M. A. Carnahan, M. Wathier, N. H. Oberlies, G. Manikumar, M. C. Wani and M. W. Grinstaff, Cancer Res., 2006, 66, 11913–11921 CrossRef CAS.
-
(a) M. E. Anderson, Chem.-Biol. Interact., 1998, 111–112, 1–14 CrossRef CAS;
(b) H. Sies, Free Radical Biol. Med., 1999, 27, 916–921 CrossRef CAS.
-
(a) D. P. Jones, J. L. Carlson, V. C. Mody, J. Y. Cai, M. J. Lynn and P. Sternberg, Free Radical Biol. Med., 2000, 28, 625–635 CrossRef CAS;
(b) D. P. Jones, J. L. Carlson, P. S. Samiec, P. Sternberg, V. C. Mody, R. L. Reed and L. A. S. Brown, Clin. Chim. Acta, 1998, 275, 175–184 CrossRef CAS.
-
(a) P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef;
(b) C. M. McIntosh, E. A. Esposito, A. K. Boal, J. M. Simard, C. T. Martin and V. M. Rotello, J. Am. Chem. Soc., 2001, 123, 7626–7629 CrossRef CAS;
(c) A. Verma, J. M. Simard, J. W. E. Worrall and V. M. Rotello, J. Am. Chem. Soc., 2004, 126, 13987–13991 CrossRef CAS.
-
(a) K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS;
(b) E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. C. J. M. van Veggel, D. N. Reinhoudt, M. Moller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 203002 CrossRef CAS.
- P. S. Ghosh, C. K. Kim, G. Han, N. S. Forbes and V. M. Rotello, ACS Nano, 2008, 2, 2213–2218 CrossRef CAS.
-
(a) A. Chompoosor, G. Han and V. M. Rotello, Bioconjugate Chem., 2008, 19, 1342–1345 CrossRef CAS;
(b) S. S. Agasti, C. C. You, P. Arumugam and V. M. Rotello, J. Mater. Chem., 2008, 18, 70–73 RSC.
- P. Podsiadlo, V. A. Sinani, J. H. Bahng, N. W. S. Kam, J. Lee and N. A. Kotov, Langmuir, 2008, 24, 568–574 CrossRef CAS.
- C. Park, H. Youn, H. Kim, T. Noh, Y. H. Kook, E. T. Oh, H. J. Park and C. Kim, J. Mater. Chem., 2009, 19, 2310–2315 RSC.
-
(a) M. J. Hostetler, J. J. Stokes and R. W. Murray, Langmuir, 1996, 12, 3604–3612 CrossRef CAS;
(b) J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
-
(a) M. Lucarini, P. Franchi, G. F. Pedulli, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2004, 126, 9326–9329 CrossRef CAS;
(b) M. Lucarini, P. Franchi, G. F. Pedulli, C. Gentilini, S. Polizzi, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2005, 127, 16384–16385 CrossRef CAS.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z.-J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS.
-
(a) L. L. Rouhana, J. A. Jaber and J. B. Schlenoff, Langmuir, 2007, 23, 12799–12801 CrossRef CAS;
(b) Q. Jin, J. P. Xu, J. Ji and J. C. Shen, Chem. Commun., 2008, 3058–3060 RSC.
- A. D'Emanuele and D. Attwood, Adv. Drug Delivery Rev., 2005, 57, 2147–2162 CrossRef CAS.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. W. Fei and C. Burda, J. Am. Chem. Soc., 2008, 130, 10643–10647 CrossRef CAS.
-
(a) M. Rooseboom, J. N. M. Commandeur and N. P. E. Vermeulen, Pharmacol. Rev., 2004, 56, 53–102 CrossRef CAS;
(b) S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS;
(c) H. J. Kim, H. Matsuda, H. S. Zhou and I. Honma, Adv. Mater., 2006, 18, 3083 CrossRef CAS;
(d) I. C. Kwon, Y. H. Bae and S. W. Kim, Nature, 1991, 354, 291–293 CAS;
(e) H. Park, J. Yang, S. Seo, K. Kim, J. Suh, D. Kim, S. Haam and K. H. Yoo, Small, 2008, 4, 192–196 CrossRef CAS;
(f) C. Park, J. Lim, M. Yun and C. Kim, Angew. Chem., Int. Ed., 2008, 47, 2959–2963 CrossRef CAS;
(g) N. W. S. Kam, Z. Liu and H. J. Dai, J. Am. Chem. Soc., 2005, 127, 12492–12493 CrossRef CAS.
-
(a) C. P. Mccoy, C. Rooney, C. R. Edwards, D. S. Jones and S. P. Gorman, J. Am. Chem. Soc., 2007, 129, 9572 CrossRef CAS;
(b) G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS.
- S. S. Agasti, A. Chompoosor, C. C. You, P. Ghosh, C. K. Kim and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 5728–5729 CrossRef CAS.
- J. Nakanishi, H. Nakayama, T. Shimizu, H. Ishida, Y. Kikuchi, K. Yamaguchi and Y. Horiike, J. Am. Chem. Soc., 2009, 131, 3822–3823 CrossRef CAS.
- M. A. Polizzi, N. A. Stasko and M. H. Schoenfisch, Langmuir, 2007, 23, 4938–4943 CrossRef CAS.
-
(a) S. Mocellin, V. Bronte and D. Nitti, Med. Res. Rev., 2007, 27, 317–352 CrossRef CAS;
(b) Q. Yang, S. H. Wang, P. W. Fan, L. F. Wang, Y. Di, K. F. Lin and F. S. Xiao, Chem. Mater., 2005, 17, 5999–6003 CrossRef CAS.
- J. R. Hwu, Y. S. Lin, T. Josephrajan, M. H. Hsu, F. Y. Cheng, C. S. Yeh, W. C. Su and D. B. Shieh, J. Am. Chem. Soc., 2009, 131, 66–67 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.