Self-healing at the nanoscale
Vincenzo
Amendola
* and
Moreno
Meneghetti
*
Department of Chemical Sciences, Nanophotonic Laboratory, University of Padova, Via Marzolo 1, I-35131 Padova, Italy. E-mail: vincenzo.amendola@unipd.it; moreno.meneghetti@unipd.it
First published on 28th August 2009
Abstract
The design of self-healing materials is a very important but challenging topic in nanotechnology. Self-healing strategies, also inspired by natural processes, allow the fabrication of auto-repairing systems, and in recent years, materials engineering at the nanoscale has allowed further advances in this emerging field. In this mini review, we recall some interesting self-healing systems found in natural processes and others created by man-made activity with special emphasis on the role played in this field by nanostructures. Finally, the self-healing of gold nanoparticles during laser irradiation is considered in more detail since it is a rare example of a functional nanomaterial with self-repairing properties.
1. Introduction
Nanoscience and nanotechnology are moving towards new frontiers and the design and fabrication of self-healing materials is one of the future challenges. According to the definition, self-healing materials have the ability of autonomous damage recovery. In several cases, however, the self-healing is induced by an external stimulus, like temperature, and the materials with this ability are defined as non-autonomic self-healing systems.1Self-healing is useful, in general, for the prolongation of material life, but it is especially valuable when human intervention is arduous, like in space applications, or in the presence of harsh physical and chemical conditions. Self-healing is also needed to preserve material properties when kinetic and thermodynamic conditions favour a high density of defects, like in the case of nanostructures, which often show excellent functional properties but are also subject to fast degradation because of the higher number of interface atoms than in ordinary materials. However, nanosystems can be fabricated by using several functional nanostructures among which one can also include some components with a self-healing task. This can be a simpler strategy than designing a more robust nanosystem.2 Just 50 years ago, during the annual meeting of the American Physical Society, Richard Feynman gave his famed talk “There's plenty of room at the bottom” and diffused the concept of “a hundred tiny hands” capable of self-replication down to the atomic scale.3 Self-healing and self-replication are two connected abilities in living organisms, and the realization of self-healing nanomaterials can be an important step for the realization of self-replicating systems.2,3
In this paper we provide an overview of self-healing at the nanoscale, organized in three main sections. In the first section we recall some interesting strategies of self-healing processes in natural systems which show sophisticated, elegant and efficient repairing mechanisms. Biological self-healing processes are the most efficient and complex among them. Frequently, they have been a source of inspiration for the fabrication of self-healing materials. In the second section we consider man-made self-healing systems which can be grouped, according to their repairing strategy, into the following main classes: auto-assembling materials, shape-memory materials and materials capable of responsive chemical reactions or reversible bonding. We evidence in particular the advantages brought by nanotechnology in this field, for instance by means of nanoparticles or nanostructured patterns. At present, many self-healing nanostructured materials can be considered as proof of concepts and various theoretical calculations and computer simulations have given useful indications for their design. In some cases, the proposed fabrications of self-healing materials are still waiting for an experimental confirmation.
In the third section we deal with a system consisting of a solution of gold nanoparticles (AuNPs) which is capable of self-repairing during high-intensity laser irradiation. Metal nanoparticles are good nonlinear optical materials but, when irradiated with laser pulses, they quickly lose their properties. In contrast, the self-healing of gold nanoparticles was observed when they were blended with a Zn-phthalocyanine. In this case thousands of laser pulses can be used without degradation of their multiphoton absorption properties. In a panorama where nanomaterials for structural self-healing processes are usually found, the blend of gold nanoparticles and Zn-phthalocyanines represents a rare case of a functional nanomaterial able of dynamic self-repairing.
2. Self-healing strategies in natural processes
Reparation at material interfaces can be considered one of the most common processes among natural healing mechanisms and probably those with lower complexity. The decrease of free surface energy is the reason for spontaneous atomic rearrangement at interfaces, after which damage recovery and/or protection of the underlying layer can follow. In almost all solid materials, two fractured surfaces have the potential for sticking together spontaneously if they meet at the nanoscale after the crack.4,5 From our everyday experience, this seldom happens because the two faces only meet at a few points due to irregular interfaces or because chemical reactions took place soon after the crack.4 If two nanoscale surfaces, not necessarily with the same composition, are placed in contact with the help of pressure and temperature, chemical bonds at the interface can take place and a whole piece can form.4 This technique is extensively applied to glasses, semiconductors, ceramics and metals with polished surfaces.4 Dynamics simulations showed that the mechanism of autonomous bond restoration is really efficient for nanoscale flaws, thanks to thermal fluctuations of atoms in the fractured surfaces.4,5 In nanoparticles, flaw self-healing is more rapid than in macroscopic materials because of the presence of soft boundaries, which facilitate lattice adaptation in order to aid recovery of the crack and minimize the interface energy (Fig. 1a).5 Such a mechanism takes place in natural materials with nanostructured components like the abalone shell, which is composed of multilayers of hard aragonite lamellae and soft protein lamellae (Fig. 1b).5,6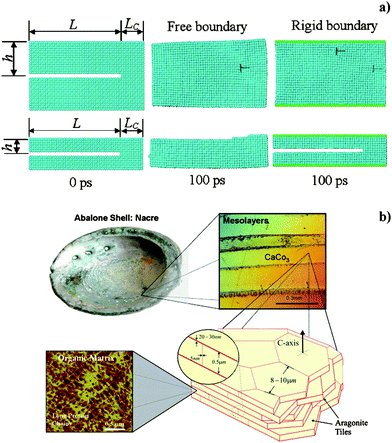 | ||
| Fig. 1 a) Snapshots obtained by molecular dynamics simulation at 300 K at time 0 fs and 100 fs for copper nanoclusters with two different sizes (height of top and bottom cluster is 30 and 14 rows of atoms respectively), in the hypothesis of free and rigid boundaries (central and right columns), with flaws corresponding to the missing two rows of atoms. Reproduced with permission from ref. 5. Copyright (2003) The American Physical Society. b) Overall view of the hierarchical structure of an abalone shell, showing mesolayers, mineral tiles, and tile pull-out in a fracture region. Reproduced with permission from ref. 6. Copyright (2008) Elsevier Science. | ||
Another self-healing process is present in many metals exposed to air, for instance Al, Cr, Ti, Zr, and Cu. On their surfaces a passivating oxide layer forms in order to decrease the surface free energy.7 This layer is quite impermeable and protects the underlying metal from further oxidation. In the case of mechanical removal of a part of this layer, air oxidation of the exposed metal surface readily restores the protective film. Diffusive processes promoted by oxygen and hydrogen adsorption allow the perfect matching and homogenization of the new oxide layer with the old one.7
Sometimes the reduction of surface energy at interfaces produces surface reconstruction instead of passivation. Computer calculations8,9 pointed out how CdSe and CdTe semiconductor quantum dots undergo a surface reconstruction in the presence of ligands on their surface. Reconstruction allows the preservation of inner crystalline structure and composition and has, therefore, a great importance in the control of the opening of the optical gap in these nanoparticles.8,9
Many other natural self-healing mechanisms of great efficiency and simplicity are based on self-assembly. Self-assembly occurs when an ensemble of objects, the building blocks of the system, spontaneously organize in an ordered structure by means of physical interactions.10 Phospholipid membranes,11,12 micelles13,14 and thiol monolayers11,15 have been deeply investigated in past years and show clear self-healing behaviour in the case of mechanical damage, as a consequence of their self-assembly abilities. The minimization of surface free energy is the driving force for the process, though the kinetics are controlled by a diffusion-limited phenomenon which requires thermal energy to occur (Fig. 2).12,13 Self-assembled monolayers have found application as corrosion inhibitors and lubricants, but only in those systems where the rate of film unravelling was lower or equal to the rate of self-repairing.14 The repairing efficiency is facilitated when the system has some redundancy, namely when excess building blocks are available for self-healing.14Self-assembly at liquid–liquid or solid–liquid interfaces, of monolayers of a wide series of materials, like carbon nanotubes,16 rigid macromolecules17 or metal nanoparticles,18 were also found to have self-healing abilities in response to scratches or defects.
 | ||
| Fig. 2 Snapshots of vesicle formation at three increasing times for a cluster composed of amphiphilic molecules, obtained by Brownian dynamics simulation. The left column shows the outer appearance of the micelle, while the right column shows a central slice of the same micelle. Reproduced with permission from ref. 13. Copyright (2001) The American Physical Society. | ||
On the scale of natural self-healing systems with increasing complexity, there is no doubt that living organisms hold the highest levels. Living organisms use the informations stored in their DNA for self-assembly and self-replication from the molecular scale up to whole organisms, through proteins, cell organelles, cells, tissues and organs.19 Self-healing is just a consequence of the availability in each single cell of the instructions for the development of the whole organism contained in the DNA. It is worth recalling that cells have microscopic size and that their components have typical dimensions of hundreds of nanometres or less. Processes inside the cell are, therefore, very interesting for understanding how self-healing can occur at the nanoscale. Repairing mechanisms are common processes in biological systems, because they represent a normal turnover of components consumed during everyday life, more often than the response to accidental external damage.19 Therefore, despite their complexity, the strategies for self-repairing can be extremely useful for inspiring the development of biomimetic self-healing materials and various general concepts can be pointed out by considering some examples of biological repairing mechanisms.
The 11-cis retinal molecule, which is bound to a protein called opsin hosted on the membrane of rod and cones cells on the surface of the retina, is the object of an interesting ‘self-healing’ mechanism called the visual retinoid cycle.20 Several millions of 11-cis retinal molecules change their conformation into all-transretinal during the vision process. This is a consequence of an energy transfer of the absorbed photons from a sensitizer to the molecules. After the isomerization, the all-transretinal no longer fits into the opsin binding site, which induces a conformational change of the protein resulting in the expulsion of the retinal from the protein and the separation of the opsin from the cell membrane site.20 On the one hand, this process is the first step of a cascade of biochemical reactions which produce the electric signals required for vision. On the other hand, 11-cis retinal needs to be recovered in order to repeat the same process. The visual retinoid cycle consists of the migration of the all-transretinal from the cone or the rod to the retinal pigment epithelium, where it is first esterified and then hydrolyzed and isomerized again to 11-cis retinal by an enzyme called RPE65. Then it travels back to cones or rods, where it binds again a free ospin (Fig. 3).20,21 Some of the aspects of the whole cycle, which occurs at the nanoscale, can be pointed out: i) various functional nanostructures are assembled together; ii) the part to be ‘repaired’ autonomously moves toward the repairing site as a consequence of its new state; iii) the part is ‘repaired’ because there are specific nanostructures with repairing functions, i.e. the enzymes; iv) the whole system has a functional redundancy and continues to operate if a part is in the repairing stage.
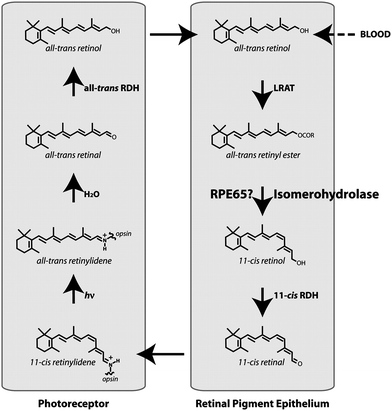 | ||
| Fig. 3 Scheme of the visual retinoid cycle. Reproduced with permission from ref. 21. Copyright (2005) National Academy of Sciences, USA | ||
A similar healing strategy can be found in photosynthesis. The fundamental process of water splitting in photosynthesis is catalysed by the photosystem II complex, which is made of more than 25 proteins and is hosted in chloroplast membranes.22 In case of high light intensity, photoinhibition of the photosynthesis occurs due to light-induced oxidative damage in photosystem II. The damage takes place prevalently in the D1 reaction center, which is one of the 25 proteins in photosystem II.22,23 Damage of the D1 polypeptide produces a cascade of events consisting of the step-by-step release of specific biomarkers for the activation of a set of enzymes responsible for: i) the cleavage of D1 from the photosystem II, ii) its disassembly iii) restoration and iv) re-assembly in the photosystem II.22,23 In the case of very high light intensities, the symmetrical reaction center, called D2, can also undergo damage and can be repaired in an analogous way.22 Hence, the healing process requires, in this case, that a set of specific functional components act on the part to be repaired after its disassembly.
In the case of irreversible damage to the whole photosystem II complex or to the rhodopsin (the complex of 11-cis retinal and opsin) for the vision cycle, another level of self-healing comes into play, namely the cells can still recover the missing parts by recalling the instructions contained in the DNA.24
DNA is the most precious resource of all cells, but is itself subject to frequent damage like oxidation, alkylation, hydrolysis and crosslinking of bases, formation of adducts , mismatch of bases and breaks in DNA strands.24,25 The main sources of damage are endogenous cellular processes, due to oxidative stress, but also external damaging factors can be relevant.24 Consequently, evolution provided each cell with a full set of mechanisms for DNA healing.24,25 The complexity of DNA repairing mechanisms increases with the organism complexity.24,25 Mammalians have multiple repairing mechanisms, some are diversified for damage typology and some are redundant and act in parallel for the same type of damage, probably for a compensatory strategy.24,25 An interesting characteristic of the DNA healing processes is their first step, where a class of proteins with the role of damage sensors bind to DNA looking for errors.24 When an error is detected, a so-called checkpoint starts, consisting of the temporary arrest of the cell cycle and the activation of the specific repairing pathway.24,25 A protein called p53 has the role of managing cell functionality after the damage detection.24,25 Some damage can be repaired by direct restoration of the bases, as occurs in some living organisms in the case of ultraviolet-induced cyclobutane pyrimidine dimerization. In this case, an enzyme called photolyase binds to the DNA portion containing the pyrimidine dimer and repairs the damage by photoreactivation (Fig. 4).24,26 In many other cases, the damage-repairing mechanisms consist of the replacement of damaged single bases or nucleotides with new undamaged ones, operated by polymerase or glycosylase enzymes.24,25 For relevant damages, break, crosslink or recombinatorial repairing are necessary, although these are error-prone procedures and can produce mutations in the restored genome .24,25 Interestingly, when DNA repairing is impossible, the p53 center can induce the definitive cell cycle stop, defined senescence, or can induce cell apoptosis.24,25 While this is not a repairing mechanism at the cellular level, it represents an excellent repairing mechanism for the whole living organism.
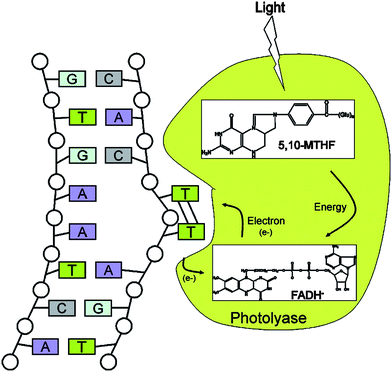 | ||
| Fig. 4 Scheme of direct repair of ultraviolet-induced cyclobutane pyrimidine dimerization by photoreactivation. Photolyase binds to DNA containing a pyrimidine dimer in a light-independent reaction and flips the dimer out into the active-site pocket. Catalysis is initiated by light. The photoantenna cofactor, methenyltetrahydrofolate (5,10-MTHF), absorbs a photon and transfers the excitation energy to the catalytic flavincofactor, FADH−. Then, the excited state FADH− transfers an electron to the pyrimidine dimer, splitting the dimer into two pyrimidines. The electron returns to the flavin radical to regenerate FADH−, and the enzyme dissociates from the repaired DNA. Reproduced with permission from ref. 24. Copyright (2004) Annual Reviews. | ||
A self-healing strategy used by biological tissues is that of a direct replacing of their building blocks, namely the cells. A flux of biomarkers , for instance growth factors, triggers the whole process of tissue regeneration.27,28 Cutaneous wound healing is a good example of the complexity of tissue self-reparation. After the formation of a clot which re-establishes the haemostasis, wound healing has three main phases, inflammation, tissue formation and tissue remodelling, partially overlapped in time (Fig. 5).28,29 Each step triggers the next one and is stopped when its goal is reached. Initially, a set of biological signals induces the accumulation of neutrophils and macrophages in order to clean the wounded area of microbes. In the intermediate phase, epidermal cells nearby begin to proliferate and undergo structural modifications which allow their migration to the wounded region. Collagen is degraded around the wounded area for facilitating cell migration. In the third phase, the new tissue and a capillary network gradually advance in the wound. Epidermal cells revert their structural modifications and firmly adhere to the tissue, while fibroblasts produce collagen until the replenishment of the wound area. When the scar is formed, non epidermal cells in the wound undergo controlled apoptosis and collagen production slows down.
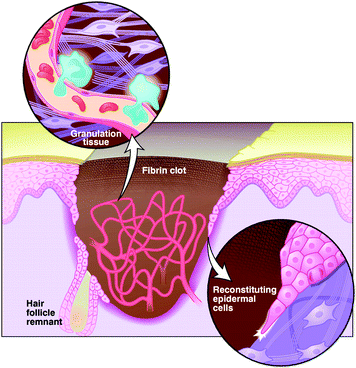 | ||
| Fig. 5 Cartoon to illustrate the main phases in the healing of a skin wound. The tearing is temporarily plugged with a fibrin clot, which is soon infiltrated by inflammatory cells. Later, a dense capillary network and a temporary granulation tissue form. An epidermal layer is reconstituted from the edges of the wound and from the remnants of the pristine cute, hair-like follicles, by the migration of epidermal cells and by production of collagen. Reproduced with permission from ref. 29. Copyright (1997) The American Association for the Advancement of Science. | ||
One should recall that another general strategy based on adult stem cells exists in complex organisms for the replacement of building blocks. Stem cells are defined as cells that have clonogenic and self-renewing capabilities and that can differentiate into multiple cell lineages.30 In the presence of stress or biomarker signalling damage, it is supposed that stem cells can move to the critical point, differentiate and participate in tissue repairing.30 Such behaviour is observed and intensely studied in organs with different regenerative capability, for instance in liver, brain or cartilage.30–34
The suggestions coming from such complex processes of building-block replacement can be used at different levels for designing materials with self-healing properties. In any case, they show also that the replacement of a single building block is obtained by a system with many functional components which act at the nanoscale.
The above biological examples show that very efficient self-healing mechanisms are dynamic processes with many actors playing interconnected roles and with transport of active components. In particular, one can recognize the importance of biomarkers which activate specific repairing processes27 and of a hierarchical organization both for structures and functions.19 It is worth recalling that living organisms are the result of evolution and, therefore, each function is part of a complex ensemble and cannot be considered as an isolated process. However, some of the characteristics of self-healing biological processes, like the multifunctionality, the self-replication ability, the self-assembly ability and the markers for the activation of specific processes should be in the mind of researchers who are designing new biomimetic materials. In any case, the nanoscale at which the living organisms activate self-healing processes makes it easier to transport some mechanisms to the nanomaterials. One can also point out that the use of nanostructures for the healing of the human body is at an advanced stage in both fields of nanomedicine and nanostructured biomaterials for tissue repairing.35 One wonders if in the near future self-healing designed materials could be used in these fields.
3. Artificial self-healing systems
Self-healing in man-made materials is still in its infancy. A heterogeneous series of self-repairing systems has been created, although the complexity of repairing mechanisms are, at the moment, far from their biological analogues. In many cases, nanoscale engineering has provided new solutions for prolonged durability and resistance against structural degradation. In this section we will not review all the self-healing systems fabricated to date, rather we will describe some of the most interesting self-healing approaches, which can be grouped into the following main classes: auto-assembling materials, shape-memory materials and materials capable of responsive chemical reactions or reversible bonding.Auto-assembly
The simplest technique for obtaining self-healing consists of the design of a material with auto-assembly properties. Sometimes, redundancy of the assembled components is present in order to help the repairing mechanism or to ensure system functionality while healing of the damaged parts occurs.Tsukruk and coworkers reported nanocomposite membranes obtained by spin-assisted layer-by-layer assembly of polymeric monolayers with a metal nanoparticle intralayer, with thickness of 35–55 nm and diameter of hundreds of micrometres.36 The membranes were composed of a central layer containing 13 nm gold nanoparticles sandwiched between nine bilayers of two alternating polyelectrolytes: poly(allylamine hydrochloride) and poly(sodium 4-styrenesulfonate). After plastic deformation, the membranes showed complete and autonomous recovery of their properties on a time scale of hours, which was possible by means of a reversible polymer–nanoparticle self-organization process (Fig. 6). The multilayered structure of the membrane, the in-plane spread of polymer chains typical of polyelectrolyte multilayers obtained by spinning, the increased mobility of polymer chains due to the intralayer nanoparticles and the presence of reversible bonds between polyelectrolyte chains gave the membrane its self-assembly properties and efficient repairing ability.36
 | ||
| Fig. 6 a–e) Self healing of the polyelectrolyte–gold nanoparticle composite membrane after the application of an external pressure of 4 kPa, as shown by snapshots taken every 2 s after pressure release. f) The relative membrane cross-sections obtained every second with arrows showing the direction of the recovery of the central portion. Reproduced with permission from ref. 36. Copyright (2004) Nature Publishing Group. | ||
Nanoparticles can be also used as mobile carriers with healing functionalities both in solid and liquid environments, exploiting intermolecular interactions and maximization of entropy changes.37,38 As shown first with computer simulations by Balazs and coworkers39 and later experimentally by Gupta and coworkers,40nanoparticles embedded in a polymeric matrix can autonomously migrate to the damaged area after a fracture occurs (Fig. 7a). A similar mechanism of nanoparticle migration from a matrix of melted polymer into a notch in the underlying substrate has been predicted by molecular dynamics simulations (Fig. 7b).41,42 In both cases, the driving force for particle expulsion from the polymeric matrix is the gain in conformational entropy of polymer chains. Important parameters for the process are particles size and surface functionalization. In the experiments of Gupta's group,40 CdSe/ZnSe nanoparticles coated with tri-n-octylphosphine oxide or poly(ethylene oxide) with different average sizes between 3 and 40 nm were embedded in a poly(methyl methacrylate) matrix deposited on a silica film. During the rupture of the silica film, only CdSe/ZnSe nanoparticles with average size of 5 nm and coated with poly(ethylene oxide) percolated to the crack boundaries in the silica layer from the poly(methyl methacrylate) matrix. Particles with lower size did not migrate due to the small effect on conformational entropy of polymeric chains, while particles with larger size travelled for a short length and did not reach the fractured region. Particles coated with tri-n-octylphosphine oxide did not migrate due their stronger interactions with the polymer matrix. Also, the shape of particles can influence the percolation behaviour and this is currently under investigation. Although the effectiveness of this approach has not yet been demonstrated for self-healing, it has the potential for autonomous repeated multiscale repairing. Most importantly, spontaneous percolation of nanoparticles during a fracture can be used for the recovery of functional properties like electrical conductivity or light emission, as well as mechanical performance.37,40
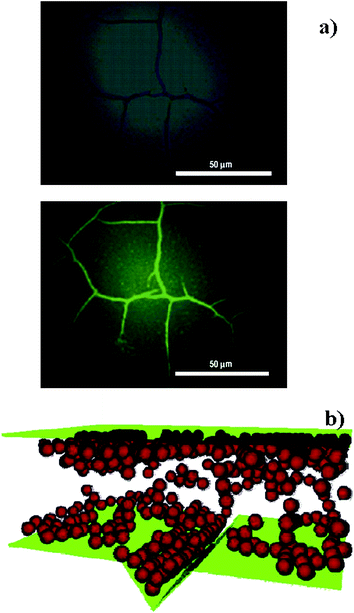 | ||
| Fig. 7 a) Bright-field (top) and fluorescence (bottom) microscopy images of a cracked silica surface on a poly(methyl methacrylate) layer containing 3 nm CdSe/ZnSe nanoparticles coated with poly(ethylene oxide). Reproduced with permission from ref. 40. Copyright (2007) Nature Publishing Group. b) The geometry adopted for molecular dynamics simulations of nanoparticle migration from a matrix of melted polymer into a notch in the underlying substrate. Nanoparticles are in red, the container walls are in green and polymer molecules are not shown for clarity. Reproduced with permission from ref. 42. Copyright (2005) American Chemical Society. | ||
Computer simulations can help in designing nanostructures with self-healing properties. The behaviour of Janus nanoparticles composed of hydrophobic and hydrophilic portions, has been recently studied when embedded in a lipid bilayer membrane.43 The results indicated that, during an external compressive stress on the membrane surface in an aqueous environment, Janus nanoparticles can self-assemble on the edge of a pore, with hydrophilic ends facing the inner of the pore (Fig. 8). This assembly lasts for a long time and allows further repeatable pore opening and closing with lower stress intensity than for the first cycle. This happens without affecting membrane integrity and represents a sensible enhancement of the healing ability with respect to a bare lipid bilayer membrane. Pore opening can be also triggered by other parameters like pH or temperature.43
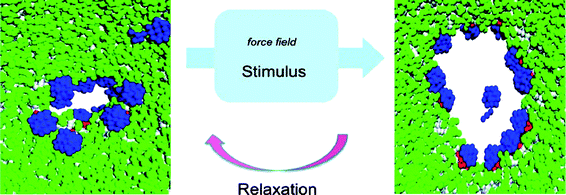 | ||
| Fig. 8 Janus nanoparticles (in blue) in a bilayer phospholipid membrane (in green) can originate pores with the ability of reversible opening by external stimuli. Reproduced with permission from ref. 43. Copyright (2008) American Chemical Society. | ||
One observes that auto-assembly is a simple mechanism that can operate at different scales. At the macroscale, some examples which show self-healing properties have also been reported, e.g. a set of hourglass shaped polyurethane beads patterned with copper patches, inspired by the backbone,44 or electromechanical systems.45 These two examples are proof of concepts that the same mechanisms observed for self-repairing at the molecular scale can also be successfully applied to macroscopic objects.
Shape memory
Shape-memory materials have the ability of restoring their initial form after a plastic deformation (Fig. 9).46 The shape recovery requires an external trigger which, in most cases, is a temperature increase above a transition threshold.46,47 Though shape-memory materials require an external trigger, they represent an original, non-biomimetic approach to the design of self-healing materials. The most popular shape-memory material is an equiatomic NiTi alloy, nitinol, which is able to recover a strain of about 10% after the phase transition.46 Other than metal alloys, ceramics, polymers and hydrogels with shape-memory behaviour are available.46,47 Shape memory basically exploits a phase transition, for instance from martensite to austenite for NiTi, from tetragonal to monoclinic for ZrO2, from glassy state to rubber-elastic state for polymers and from hydrophilic to hydrophobic state for hydrogels.46,47 The form of these materials can be programmed at high temperature, and the modifications at room temperatures can be ‘repaired’ by heating again above the transition temperature. In general, shape-memory materials can recover their form after a bulk deformation or their surface integrity after slight bumps, indentations or scratches.46,47 A typical application of metal shape-memory alloys in self-healing systems is for self-repairing joints.48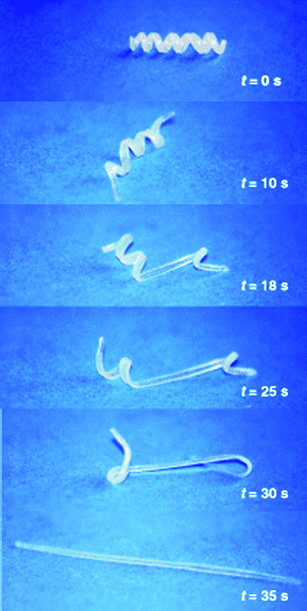 | ||
| Fig. 9 Transition from the temporary shape (spiral) to the memorized shape (linear rod) in a (ε-caprolactone) dimethacrylate-butylacrylate copolymer with transition temperature of 46 °C. Reproduced with permission from ref. 46. Copyright (2002) Wiley-VCH. | ||
Hydrogels, like those composed of poly(N-isopropylacrylamide) are usually disregarded for macroscopic self-healing systems because of their poor mechanical properties and durability.46 However, the ability to program the form and trigger the shape transition by external temperature, pH, electrical fields, light and other external parameters is very promising for applications at the nanoscale,49 where functional properties can take priority over structural performances (Fig. 10).50,51
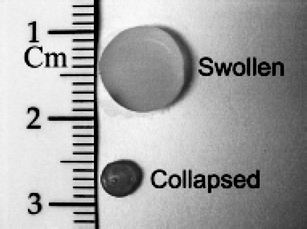 | ||
| Fig. 10 Hydrogels composed of a copolymer of (N-isopropylacrylamide) and acrylamide with embedded silica@gold nanoshells, in the swollen and collapsed states. Reproduced with permission from ref. 51. Copyright (2001) Springer Science. | ||
In recent years, molecular dynamics simulations suggested that shape-memory materials with enhanced performance may be found at the nanoscale.52–55 Simulations performed on gold, copper, nickel and nickel aluminium nanowires with diameter less than 5 nm and on ZnO nanowires with diameter less than 20 nm indicated the presence of shape-memory effects after tensile stress, due to surface-stress-mediated phase transitions.52–55 Simulations predicted a recovery ability up to 50% of the wire length in the best cases.53,55 These effects are not observed in bulk forms, since they are a consequence of nanoscale surface stress. To date, experimental evidence of such behaviour has not been found, due to the difficulties in the manipulation of metal nanowires thinner than 5 nm and probably also due to the interference of surface roughness.55
Responsive chemical reactions and reversible bonds
A large class of self-healing materials exploits chemical reactions for the restoration of structural properties or for the protection of surfaces. These chemical reactions can be initiated by an external stimulus or can start autonomously when a chemical bond is broken. Some systems also exploit the intrinsic reversibility of physical bonds for damage recovery. In some other cases, the reactions requires the addition of specific reagents from reservoirs stored in the material.Ismagilov and coworkers developed a biomimetic microfluidic system able of haemostasis using three chemical reactions in dynamic equilibrium.56 The first two reactions consisted of the second-order autocatalytic production of H3O+ ions and their first-order consumption. The equilibrium between H3O+ production and consumption is guaranteed by solution flow in the channels. When the flow decreases for the presence of a hole in the microfluidic device, H3O+ concentration increases above the equilibrium threshold and initiates the third chemical reaction, consisting of the gelling of sodium alginate at low pH. The precipitation of alginic acid gel obstructs the hole and stops the solution leak, similar to the formation of a clot in damaged blood vessels. The whole system is based on the threshold response of the three coupled chemical reactions, in close analogy to haemostasis.56
Self-healing organic–inorganic multilayers have been designed by Balkus and coworkers, based on the hydrolyzation of metal oxide precursors like TiCl4 in the presence of water and oxygen.57 Metal and oxide layers are frequently used for the protection of organic electronics from air humidity and oxygen, instead of more permeable polymers. Nonetheless, both metal and oxide layers easily suffer from microcracks due to their low flexibility. Balkus and coworkers fabricated multilayers of oxide films alternating with films of polymer fibers. They prepared microporous polymeric fibers with TiCl4 microparticles encapsulated by electrospinning. In case of a break in the external oxide layer, TiCl4 readily reacts with atmospheric water and oxygen giving a swelled titanium oxide clot (Fig. 11). The good permeability to air of the polymeric matrix is functional for the hydrolysis reaction and allows the formation of TiO2 islands large enough to replenish the crack void. In this system, the self-healing reaction is autonomously triggered by the damage.
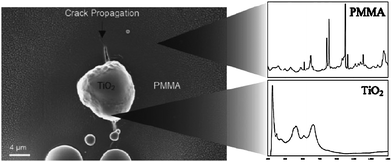 | ||
| Fig. 11 SEM micrograph (left) and corresponding Raman spectra (right) of a poly(methyl methacrylate) surface showing that a microcrack was healed by a TiO2 clot formed by reaction of air with TiCl4 contained in the inner of the polymeric matrix. Reproduced with permission from ref. 57. Copyright (2008) Wiley-VCH. | ||
Damage-responsive chemical reactions are also used in carbon composites which can resist oxidation at high temperature. Composites like B4C–SiC/C resist air oxidation by forming a borosilicate oxide layer with very low permeability to oxygen.58 The composition of the oxide layer depends on composite stoichiometry and on temperature: boron oxide dominates below 800 °C, silica dominates above 1200 °C, while at intermediate temperatures a mixture of the two oxides is present.
A well known class of autonomous self-healing chemical reactions is used for corrosion protection of metals, like in stainless steel. Stainless steel is an alloy of iron and chromium. A passivation layer of Cr2O3 protects the underlying iron-rich metal phase from corrosion. In the case of scratches, the passivation layer is readily restored if the chromium percentage in the alloy is higher than 13%.59 Restriction of the use of chromates in the field of anticorrosion coatings has opened the research toward environmentally friendly alternatives.60 For instance, the use of cerium(III) nitrate together with sodium phosphate, calcium nitrate or magnesium nitrate have been proposed. Ce(III) nitrate and other salts can be deposited as a bare layer or they can be included in organic or organosilanic polymeric films.61–63 In all cases, the suppression of zinc or aluminium corrosion was observed after a scratch in the coating. Unfortunately, the regenerative ability of these layers is limited.60,64 A different approach consists of the controlled release of corrosion inhibitors on the damaged region of protective layers.60,64,65 Three different strategies have been proposed by nanoscientists for the controlled release of corrosion inhibitors. The first one is the use of porous coatings impregnated with anticorrosion agents like benzotriazole.64 In the case of scratches, the diffusion of inhibitor molecules from the porous layer can protect the metal surface. However, a high concentration of inhibitors is not allowed because it has a negative effect on film stability, durability and impermeability.60,64,65 A double layer composed of a nanoporous titania film impregnated with benzotriazole, deposited by direct contact with the metal surface and sealed by a sol–gel impermeable film allowed better results.64 In any case, the anticorrosion effect lasts for a limited time because of inhibitor consumption or removal.60,65 The second strategy consists of the use of nanocontainers like halloysite nanotubes or cyclodextrins for the slow release of corrosion inhibitors.60,66,67 In particular, halloysite nanotubes with average length of 300–800 nm, outer diameter of about 50 nm and inner diameter of about 15 nm were revealed as promising nanostructures for large-scale production, due to their low cost and easy availability in natural deposits.67 The third, more elegant alternative consists of the encapsulation of benzotriazole within polyelectrolyte layers obtained by layer-by-layer assembly.65,68–70 The release of the corrosion inhibitors is triggered by the corrosion process, because corrosion is accompanied by local changes in the pH. Polyelectrolyte coatings are sensitive to pH changes in such a way that they can expand the polymeric net and allow benzotriazole diffusion out of the layers. When the corrosion process has been arrested, the pH is restored to the initial value and the leakage of inhibitors out of the polyelectrolyte coating is stopped. Moreover, different electrolytes respond to different pH, hence allowing the release of inhibitors in the pH range of a given corrosion process. This technique was successfully experimented both by direct deposition of films on metal surface and by deposition of a nanocomposite made of polyelectrolyte nanocapsules embedded in a polymeric matrix.65,68–70 Capsules were obtained using silica nanoparticles as scaffolds for the layer-by-layer assembly of polyelectrolytes impregnated with benzotriazole.65,68–70 The advantage of using nanoparticles is the simultaneous loading of the same coating of different pH-responsive containers which can act, for instance, as both corrosion inhibitors and damage sensors (Fig. 12).60,65,68
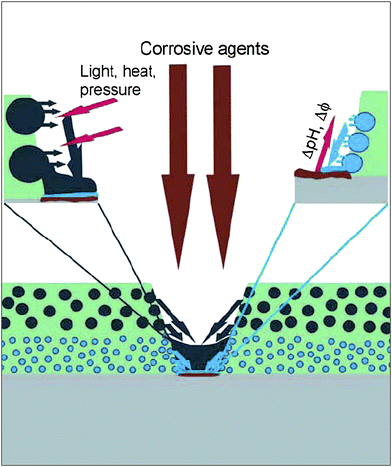 | ||
| Fig. 12 Cartoon representing the opportunities offered by coatings containing multifunctional nanoparticles. The release of healing agents can be triggered by the corrosion process, through a local pH change, or can be externally triggered by light, heat or mechanical stress. Reproduced with permission from ref. 68. Copyright (2007) Wiley-VCH. | ||
Polymers are a class of materials where reversible or responsive chemical reactions for self-healing have been widely studied.71–74 On the one hand, the chemistry of polymers is well known and offers endless opportunities, on the other hand, polymers can undergo fatigue and degradation by oxidation, heat and light, more easily than inorganic materials.
In case of polycarbonate and polyphenylene ether, bonds breaking due to ageing can be successfully restored by the catalytic activity of suitable additives.74 As shown in Fig. 13a, hydrolysis of carbonate bonds in polycarbonate produces phenoxy-terminated chains, which can react with phenyl ends to form new carbonate bonds in presence of weak alkalis like Na2CO3. Polyphenylene ether chains are easily broken by heat or light, but the ether is reformed in presence of copper(II) ions and oxygen (Fig. 13b). These mechanisms are useful for autonomous self-healing of polymers in response to nanovoids and microcracks produced by fatigue, which are at the origin of larger fractures.72,74
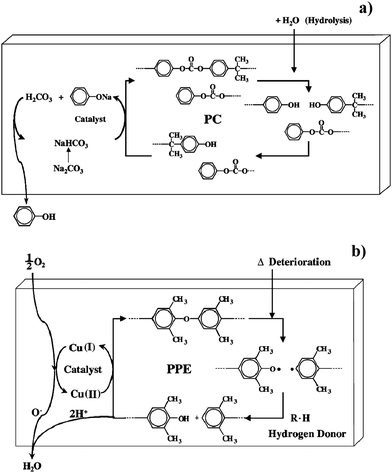 | ||
| Fig. 13 Self-repairing scheme for polycarbonate (a) and polyphenylene ether (b). Reproduced with permission from ref. 74. Copyright (2003) Elsevier Science B.V. | ||
Another approach for obtaining innovative self-healing materials is supramolecular chemistry.71–73 Leibler's group proposed a rubber based on oligomers with complementary non-covalent bonds capable of self-healing in the case of real material break or cut.75 The rubber was composed of a mixture of hydrogen-bonding groups: amidoethyl imidazolidone, di(amidoethyl) urea, diamido tetraethyl triurea plus dodecane as plasticizer. By bringing together the two ends of the rubber after the fracture, the material self-heals at room temperature. This is basically the effect of the long lifetime of hydrogen bonds when they are created in excess at the fractured surface. Further thermal curing allows the recovery of 100% of mechanical properties.
According to recent calculations of Balazs and coworkers, a nanocomposite made of nanoparticles with both permanent and reversible bonds can strongly enhance the mechanical properties of the material, thanks to autonomous breaking and restoration of labile bonds.76 Disulfide bridges are examples of such bonds. These nanomaterials can be considered nanogels where reversible bonds have a primary role for material self-healing after mechanical solicitations, and not only for increasing material tensile strength and fracture threshold. While the mechanical properties of nanogels have not been reported until now, recent advances in polymeric nanoparticles open good perspectives for these materials. Some natural materials like wood, bone and seashells exploit a similar mechanism of bond breaking and reforming to allow plastic deformations without fractures.19,76
The groups of White and Sottos fabricated interesting self-healing polymers by direct engineering of their microstructures.71,77 The main idea consists of the presence of containers encapsulating monomers and in polymerization catalysts within the polymer matrix (Fig. 14).71,77 When a crack occurs in the polymer, monomers can diffuse into the fractured area, where they meet the catalyst embedded in the polymer matrix and initiate polymerization. This strategy has been applied first to dicyclopentadiene monomers contained in urea–formaldehyde microcapsules polymerized by a ruthenium-based catalyst in epoxypolymers.71 In further experiments the catalyst was included in wax microspheres for achieving an homogenous dispersion and for protection from oxygen and other interfering chemicals.78 This approach worked successfully for both fracture and fatigue damage repairing. Up to 75–50% of the mechanical properties can be recovered after the crack. The main limitation of this strategy is that crack healing is possible only once at the same point, which also becomes the weakest part of the polymer due to its lower mechanical strength.71,77,79 The application of this approach to all polymers is not straightforward, due to issues about reagent compatibility, stability and time durability.71,80
 | ||
| Fig. 14 a–c) Cartoons representing the self-healing composite consisting of microencapsulated catalyst (yellow) and phase-separated healing-agent droplets (white) dispersed in a matrix (green); a crack propagating into the matrix releases catalyst and healing agent into the crack plane, with consequent crack filling. d) Scanning electron microscopy images of the fracture surface of such a polymer, showing an empty microcapsule and voids left by the phase-separated healing agent. e) A representative integer microcapsule. Reproduced with permission from ref. 80. Copyright (2006) Wiley-VCH. | ||
A more complex approach was developed for multiple fracture healing, by a three-dimensional microchannel network inside a polymer matrix.79,81,82 Microchannels allow a continuous flow of the healing reagents and can be designed for the transport of two separate components like an epoxy resin and its curing agent. The release of the healing chemicals is possible through a fracture of the vascular system in the cracked region of the polymer, as with microcapsules. As a proof of general applicability, the approaches based on containers of healing agents embedded in a matrix and triggered by damage have also been studied for concrete, glasses and ceramics.72,73,83
The size of capsules and channels in self-healing polymers is very important for the recovery of performance.71,84–86 The density of healing containers and the amount of healing agent released both depend on container size. The best healing is obtained when the crack volume is totally filled with the released monomers. Consequently, the size of the containers and their density should be optimized for the crack volume. For instance, material fatigue and mechanical stress mostly induce formation of microvoids or nanovoids and defects. These types of damage are deep in the material and cannot be usually repaired or even detected.71,72 In these cases, nanoscale healing by homogeneously dispersed, high-density porous nanofibers and nanocapsules may be more effective than microcapsules and microchannels in order to restore mechanical performance and for prevention and retardation of damage.71,84–86 Similarly, submicron healing containers are required for thin films , coatings and adhesives.71,85
Materials which autonomously release multicomponent healing agents can be considered as biomimetic systems. In fact, the presence of multiple functional units, which come into play when a self-healing activity is required, is typical of most biological repairing mechanisms, although they show a far greater complexity. This suggests, however, that more complex man-made materials can be obtained including more interconnected players in the game.
4. Self-healing of gold nanoparticles during laser irradiation: an example of a self-healing functional material
In the self-healing systems described in the previous section, nanomaterials were important mainly for the recovery of structural properties. Some perspectives about self-healing of functional properties exist, like Janus nanoparticles for smart membranes, but the experimental work still has to be done. Indeed, a large part of the interest in nanotechnology consists of the functional properties arising at the nanoscale. In the case of nanomaterials exposed to critical conditions like intense light irradiation, heat, or oxidative environments, these interesting functional properties usually undergo fast degradation. The nonlinear optical properties of noble metal nanoparticles are a clear example of such a problem.Gold and silver nanoparticles have high linear and nonlinear susceptibilities, due to the presence of highly polarizable electrons, namely one sp-band conduction electron and ten d-band electrons for each atom in the nanoparticle.87 Due to plasmon–polariton modes, the free electrons can be collectively excited at optical wavelengths.87 The plasmon resonance extinction cross-section in noble metal nanoparticles is of the order of 10−17 cm2 per single atom, i.e. of the order of 10−12 cm2 for a 20 nm nanoparticle.87,88 Since the plasmon relaxation dynamics are on the time scale of 5–10 fs, the full-width-at-half-maximum of plasmon resonance is of the order of tens of nanometres.87,89 The interaction with electromagnetic fields at visible wavelengths is, therefore, a near-resonance or in-resonance excitation. On the one hand, near-resonance excitation further increases the nonlinear optical susceptibility.90 On the other hand, plasmon excitation induces sensible heating of nanoparticles.91,92 Due to the ultrahigh extinction cross-section and ultrafast relaxation dynamics, noble metal nanoparticles are probably among the best available converters of visible light into heat. This property generates, however, the related problems of photomelting and photofragmentation of the nanoparticles at high laser intensity.
In recent years, a series of interesting nonlinear optical phenomena like optical limiting (OL),93–102 second- and third-harmonic generation,103–109 optical Kerr effect,102,109–113 and two-photon-induced fluorescence112,113 have been reported for metal nanoparticles. In particular, gold and silver nanoparticles showed better OL performances than those of the best organic optical limiters, with ns,94,95,97–101,103,114 ps,94,96,115 and fs93 near-resonance laser pulses. The main OL mechanism has been attributed to free-carrier multiphoton absorption processes93,98–101 with subsequent particle fragmentation due to electron photoejection.116,117 Interparticle electronic coupling was also addressed as a possible OL mechanism when nanoparticles are aggregated.98,114 Nonlinear scattering is usually excluded because many investigations did not show differences between open-aperture and closed-aperture measurements.93,98–101,114 A consolidated result is that nanoparticles with sizes of tens of nanometres are better optical limiters than smaller nanoparticles.94,95,97–101,114,118 Unfortunately, gold and silver nanoparticles have a low threshold for irreversible damage due to photoinduced melting or fragmentation.91,92,94,99,100,115,119 In particular, the experiments showed that large nanoparticles lose their OL properties after a few laser pulses due to photoinduced fragmentation, which is very rapid and has a lower threshold for big particles.91,92,94,99,100,115,119 In a few cases, gold or silver nanoparticles were blended with other optical limiters with complementary nonlinear absorption mechanisms and activation thresholds, with the aim to enhance OL performances. Dendrimer-encapsulated nanoparticles and nanoparticles conjugated to fullero-pyrrolidine-pyridine derivatives or to a fulleropyrrolidine moiety showed better OL performances than the isolated moieties.95,99,101,120–123 However, these studies did not address the main problem associated with nonlinear absorption of plasmonic nanostructures, namely their fast photoinduced degradation.
In our recent work,124 we reported about a blend of AuNPs and Zn-phthalocyanines which showed enhanced and long lasting optical limiting performances with 532 nm and 9 ns laser pulses. The solution was prepared with AuNPs with average size of 16 nm obtained by laser ablation synthesis in solution (LASiS) in tetrahydrofuran (THF). The LASiS technique does not require any chemicals or stabilizing agents around the nanoparticle surfaces, because a colloidal solution of charged nanoparticles is obtained.91,125,126 The Zn–2(3)-tetra-[2-(tert-butoxy)-ethyloxy]-phthalocyanine (TEOPcZn) was added to the AuNP solution. TEOPcZn has good solubility and does not aggregate in THF thanks to the four peripheral substituents.
The blend showed better performance than isolated moieties both during a single OL measurement (Fig. 15), during multiple cycles of measurements and even after irradiation with hundreds of laser pulses at high fluence. In general, the blend showed OL performances superior to fullerene solutions with the same linear transmittance. We performed a crossed analysis of OL measurements with UV–visible spectroscopy and transmission electron microscopy (TEM). The results evidenced a clear correlation between the average volume of nanoparticles and their nonlinear absorption performances, when comparing initial and final sizes of blended and non-blended AuNPs. In particular, the long lasting OL performances of the blend were associated with the presence of a large average volume of AuNPs at the end of the measurement (Fig. 16). As recalled above, particle volume has a dramatic influence on the nonlinear absorption properties of AuNP solutions.98–100,115,115,116,127AuNPs showed complete and rapid size reduction in the absence of TEOPcZn. In contrast, the size distribution of blended AuNPs indicated the presence of both big, i.e. more than 10 nm, and small particles after the same measurements. Also a small fraction of nanoparticles with size larger than that observed before irradiation was found in size histograms. This result suggested that both a fragmentation and a regrowth process of AuNPs occurred in the blend with TEOPcZn, namely that the presence of the phthalocyanines promoted the recovery of photofragmented AuNPs.
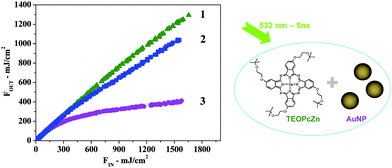 | ||
| Fig. 15 Optical limiting curves reporting the transmitted laser fluence vs. the incident fluence for 532 nm (9 ns) laser pulses. 1. TEOPcZn solution; 2. AuNP solution; 3. AuNP–TEOPcZn blend. The blend is by far the best optical limiter. From ref. 124. | ||
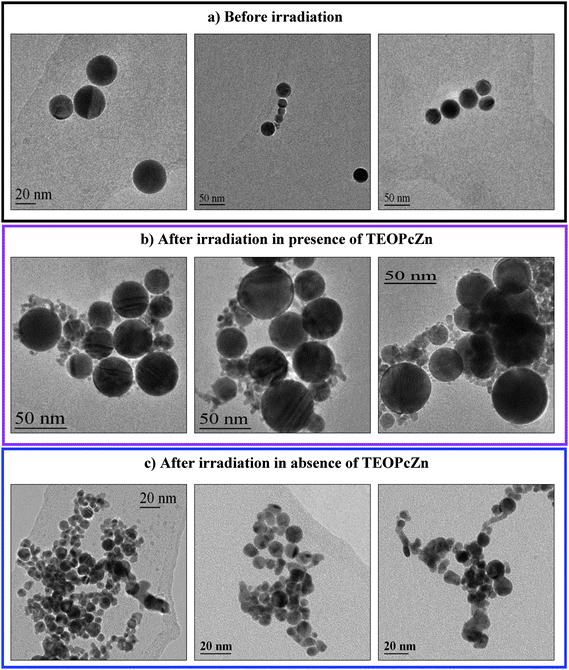 | ||
| Fig. 16 Representative TEM images of AuNPs before (a) and after irradiation with 350 laser pulses at 532 nm (1100 mJ cm−2) in the presence (b) and in the absence (c) of TEOPcZn. From ref. 124. | ||
Interestingly, the addition of TEOPcZn to a solution of AuNPs halfway through a multiple-pulse irradiation experiment produced a rapid recovery of the OL properties after a few laser pulses. This can be clearly observed in Fig. 17, where the nonlinear transmittances at 532 nm during repeated irradiation with 1100 mJ cm−2 (9 ns) laser pulses are reported for the solutions of blended AuNPs, bare AuNPs and bare AuNPs to which TEOPcZn was added after 400 pulses. At the end of the experiment, AuNPs blended with TEOPcZn after 400 pulses possessed an average size larger than before the blending. Actually, the addition immediately produced the assembly of photofragmented AuNPs in small aggregates, as monitored by UV–visible spectroscopy and simulated by Discrete Dipole Approximation calculations. According to experiments where a controlled growth of AuNPs was obtained,91,119 these aggregates can be transformed into larger particles by incoming laser pulses through a photomelting process.
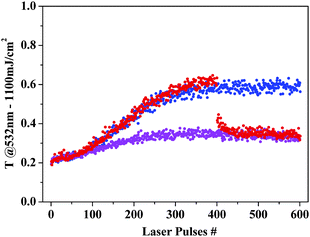 | ||
| Fig. 17 Nonlinear transmittances at 532 nm for multiple irradiation with 1100 mJ cm−2 (9 ns) laser pulses are reported for the blended AuNPs (violet), bare AuNPs (blue) and bare AuNPs to which TEOPcZn was added after 400 pulses (red). Other experimental details in ref. 124. | ||
The stability of a colloidal system composed of AuNPs obtained by LASiS without any stabilizing agent depends on the nanoparticle’s zeta-potential, which we found to be positive for the particles obtained in THF. The addition of TEOPcZn produced an aggregation of photofragmented nanoparticles, showing that phthalocyanines affect the zeta-potential of AuNPs. At the same time, we observed a constant degradation of TEOPcZn during irradiation experiments of the blend, in the form of a loss of optical density in the TEOPcZnabsorption spectra. This finding is compatible with the oxidation of phthalocyanines, as verified by spectroelectrochemical measurements.124 According to these observations, one can deduce that the aggregation of AuNPs in the presence of TEOPcZn is induced by a charge-transfer process from the molecules to the nanoparticles. Such a phenomenon can be found in the literature and may be further facilitated in this case because the AuNPs are not coated with a shell of interfering stabilizers.91,128
From the above data we conclude that enhanced optical limiting and OL durability are the result of the self-healing of AuNPs in the presence of TEOPcZn, by the following mechanism: i) initially AuNPs irradiated with 532 nm and 9 ns laser pulses show excellent OL performances but also suffer from fragmentation into smaller particles; ii) in the absence of TEOPcZn, these smaller nanoparticles repel each other due to their surface charge, but in the presence of TEOPcZn, the aggregation of these small AuNPs is favoured by a reduction of the surface charge of the nanoparticles produced by the oxidation of TEOPcZn; iii) after the aggregation process, further absorption of laser light produces melting of the aggregates into single spherical nanoparticles as evidenced by the TEM images; iv) These large nanoparticles are ready to absorb light again with a higher efficiency than the parent smaller nanoparticles (Fig. 18).
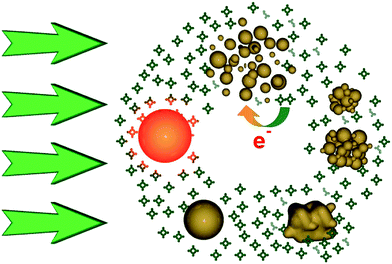 | ||
| Fig. 18 Sketch of the self-healing mechanism for AuNPs in the presence of TEOPcZn. From left in clockwise order: AuNPs absorb photons; AuNPs heat up over the explosion threshold; AuNPs fragment into smaller particles with positive charge; some of the positive charges are neutralized by the oxidation of TEOPcZn; less charge on AuNPs weakens the stability of colloidal particles and promotes their aggregation; aggregates are photomelted into new spherical AuNPs ready to efficiently limit light again. From ref. 124. | ||
The properties of the AuNP and TEOPcZn solutions show that the functional properties of a nanomaterial can self-heal in a dynamic process where the triggering of the healing process is given by the same transformation of the AuNPs. Such behaviour could be of inspiration in other cases where a functional activity is degraded and the transformed nanostructure can itself trigger its healing.
5. Conclusions
The design of self-healing nanosystems with enhanced performances and time duration is one of the new challenges in nanoscience and nanotechnology. Self-healing is particularly important for nanostructured systems which are usually prone to a high concentration of defects because of their high number of surface atoms. Natural systems, in particular biological systems, can be a useful source of inspiration for the design of self-repairing materials. However, evident differences exist, at the moment, between natural and man-made materials, like the dynamics and complexity of the healing processes, their hierarchical levels and the functional redundancy which makes possible the activity of the system, while the self-healing process is operating. Man-made self-healing materials are, at the moment, more simple, and basically can be classified as either auto-assembling materials, shape-memory materials and materials capable of responsive chemical reactions or reversible bonding.Some aspects that are important for biological repairing processes and should also be considered for self-repairing materials are: the specific damage recognition; the selective cleavage of the damaged components; the presence of a hierarchical structure with multiscale repairing mechanisms; the healing ability as a result of the self-reproduction ability, i.e. the availability of complete self-assembly instructions.
In recent years, interesting perspectives have opened for the design of innovative self-healing nanosystems. Computer simulations have provided useful indications for directing the efforts of scientists toward the fabrication of repairing systems. The engineering of membranes with nanostructures like, for instance, Janus nanoparticles or organic–inorganic nanoparticles, can open the way to a series of stimuli-responsive functional devices with self-healing abilities.129 The occurrence of entropy-driven transport phenomena at the nanoscale can be exploited for obtaining nanoparticles capable of travelling through a given matrix, when activated by external stimuli, in order to carry out healing functionalities.37 The excellent shape-memory and pseudoelastic properties predicted for nanowires, if experimentally confirmed, could offer many opportunities for the fabrication of self-healing nanosystems.55 The development of chemical reactions activated by the presence of functional groups, which respond to mechanical stress, mechanophores, could help in developing smart systems capable of autonomous self-healing in response to damage exactly at the point where the solicitation occurs.37,130 Light- or laser-triggered healing processes have been revealed as new promising approaches for dynamic repairing of functional nanostructures.124 The creation of hybrid systems composed of inorganic and proteic moieties could be the pathway for the creation of nanomaterials which can participate in complex biological processes, like biomarker activation and enzyme activity, aiming at biomimetic complex self-healing strategies.2,131,132 Integration with molecular motors could offer other concrete opportunities for dynamic healing mechanisms.132,133
The comparison of man-made self-healing systems with, in particular, biological systems shows that this field is in its infancy and many advances can be foreseen in the near future.
Acknowledgements
Authors acknowledge useful conversations on the repairing mechanism of photosystem II with Prof. Donatella Carbonera and DNA with Dr. Alessandro Sinigaglia. The University of Padova is acknowledged for funding (PRAT prot. CPDA063353, post-doc grant prot. CPDR074533).References
- S. K. Ghosh, Self-healing Materials: Fundamentals, Design Strategies, and Applications, Wiley-VCH, Weinheim, 2008 Search PubMed.
- K. E. Drexler, Nanosystems: molecular machinery, manufacturing, and computation, John Wiley & Sons, New York, 1992 Search PubMed.
- R. P. Feynman, 1959, Annual Meeting of the American Physical Society, California Institute of Technology, Pasadena Search PubMed.
- H. H. Yu and Z. Suo, J. Mech. Phys. Solids, 1998, 46, 829–844 CrossRef CAS.
- Y. Guo and W. Guo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 085411 CrossRef.
- M. A. Meyers, A. Y. M. Lin, P. Y. Chen and J. Muyco, J. Mech. Behav. Biomed. Mater., 2008, 1, 76–85 Search PubMed.
- G. Hultquist, B. Tveten, E. Hörnlund, M. Limbäck and R. Haugsrud, Oxid. Met., 2001, 56, 313–346 CrossRef CAS.
- A. Puzder, A. J. Williamson, F. Gygi and G. Galli, Phys. Rev. Lett., 2004, 92, 217401–217401 CrossRef.
- S. K. Bhattacharya and A. Kshirsagar, Eur. Phys. J. D, 2008, 48, 355–364 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- T. B. Creczynski-Pasa, M. A. D. Millone, M. L. Munford, V. R. Lima, T. O. Vieira, G. A. Benitez, A. A. Pasa, R. C. Salvarezza and M. E. Vela, Phys. Chem. Chem. Phys., 2009, 11, 1077–1084 RSC.
- B. R. A. Neves, M. E. Salmon, E. B. T. Jr and P. E. Russell, Nanotechnology, 2001, 12, 285–289 CrossRef CAS.
- H. Noguchi and M. Takasu, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 041913 CrossRef CAS.
- H. C. Schniepp, D. A. Saville and I. A. Aksay, J. Am. Chem. Soc., 2006, 128, 12378–12379 CrossRef CAS.
- J. P. Bucher, L. Santesson and K. Kern, Langmuir, 1994, 10, 979–983 CrossRef CAS.
- Y. Zhang, Y. Shen, D. Kuehner, S. Wu, Z. Su, S. Ye and L. Niu, Chem. Commun., 2008, 4273–4275 RSC.
- P. Samorí, K. MüLLEN and J. P. ROBE, Adv. Mater., 2004, 16, 1761–1765 CrossRef CAS.
- O. Balmes, J. O. Bovin and J. O. Malm, J. Nanosci. Nanotech., 2006, 6, 130–134 CAS.
- P. Fratzl, J. R. Soc. Interface, 2007, 4, 637 CrossRef CAS.
- R. R. Rando, Chem. Rev., 2001, 101, 1881–1896 CrossRef CAS.
- G. Moiseyev, Y. Chen, Y. Takahashi, B. X. Wu and J. Ma, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12413–12418 CrossRef CAS.
- J. Barber and B. Andersson, Trends Biochem. Sci., 1992, 17, 61–66 CrossRef CAS.
- E. Bergo, A. Segalla, G. M. Giacometti, D. Tarantino, C. Soave, F. Andreucci and R. Barbato, J. Exp. Bot., 2003, 54, 1665–1673 CrossRef CAS.
- A. Sancar, L. A. Lindsey-Boltz, K. Unsal-Kaçmaz and S. Linn, Annu. Rev. Biochem., 2004, 73, 39–85 CrossRef CAS.
- R. Hakem, EMBO J., 2008, 27, 589 CrossRef CAS.
- R. P. Sinha and D. P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 RSC.
- P. R. LeDuc and D. N. Robinson, Adv. Mater., 2007, 19, 3761–3770 CrossRef CAS.
- A. J. Singer and R. A. F. Clark, N. Engl. J. Med., 1999, 341, 738–746 CrossRef CAS.
- P. Martin, Science, 1997, 276, 75 CrossRef CAS.
- M. Korbling and Z. Estrov, N. Engl. J. Med., 2003, 349, 570–582 CrossRef.
- A. Björklund and O. Lindvall, Nature, 2000, 405, 892 CrossRef CAS.
- Z. Kokaia and O. Lindvall, Curr. Opin. Neurobiol., 2003, 13, 127–132 CrossRef CAS.
- A. I. Caplan, M. Elyaderani, Y. Mochizuki, S. Wakitani and V. M. Goldberg, Clin. Orthop., 1997, 254–269 CrossRef CAS.
- D. F. Mosher, Nat. Med., 2001, 7, 290–292 CrossRef CAS.
- C. M. Niemeyer and C. A. Mirkin, Nanobiotechnology: concepts, applications and perspectives, Wiley-VCH, Weinheim, 2006 Search PubMed.
- C. Jiang, S. Markutsya, Y. Pikus and V. V. Tsukruk, Nat. Mater., 2004, 3, 721–728 CrossRef CAS.
- A. C. Balazs, Mater. Today, 2007, 10, 18–23 CrossRef CAS.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS.
- J. Y. Lee, G. A. Buxton and A. C. Balazs, J. Chem. Phys., 2004, 121, 5531–5540 CrossRef CAS.
- S. Gupta, Q. Zhang, T. Emrick, A. C. Balazs and T. P. Russell, Nat. Mater., 2006, 5, 229–233 CrossRef CAS.
- S. Tyagi, J. Y. Lee, G. A. Buxton and A. C. Balazs, Macromolecules, 2004, 37, 9160–9168 CrossRef CAS.
- K. A. Smith, S. Tyagi and A. C. Balazs, Macromolecules, 2005, 38, 10138–10147 CrossRef CAS.
- A. Alexeev, W. E. Uspal and A. C. Balazs, ACS Nano, 2008, 2, 1117–1122 CrossRef CAS.
- M. Boncheva and G. M. Whitesides, Angew. Chem., Int. Ed., 2003, 42, 2644–2647 CrossRef CAS.
- S. Murata, E. Yoshida, H. Kurokawa, K. Tomita and S. Kokaji, Auton. Rob., 2001, 10, 7–21 Search PubMed.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- W. Ni, Y. T. Cheng and D. S. Grummon, Appl. Phys. Lett., 2002, 80, 3310 CrossRef CAS.
- G. Park, D. E. Muntges and D. J. Inman, JOM, 2003, 55, 33–37 CrossRef.
- W. T. S. Huck, Mater. Today, 2008, 11, 24–32 CrossRef CAS.
- S. Salmaso, P. Caliceti, V. Amendola, M. Meneghetti, G. Pasparakis and C. Alexander, J. Mater. Chem., 2009, 19, 1608–1615 RSC.
- S. R. Sershen, S. L. Westcott, J. L. West and N. J. Halas, Appl. Phys. B: Lasers Opt., 2001, 73, 379–381 CrossRef CAS.
- J. Diao, K. Gall and M. L. Dunn, Nat. Mater., 2003, 2, 656–660 CrossRef CAS.
- W. Liang, M. Zhou and F. Ke, Nano Lett., 2005, 5, 2039–2043 CrossRef CAS.
- H. S. Park, K. Gall and J. A. Zimmerman, Phys. Rev. Lett., 2005, 95, 255504 CrossRef.
- H. Huang, H. Van and G. Swygenhoven, MRS Bull, 2009, 34.
- M. K. Runyon, B. L. Johnson-Kerner and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 1531–1536 CrossRef CAS.
- H. A. Liu, B. E. Gnade and K. J. Balkus Jr, Adv. Funct. Mater., 2008, 18, 3620–3629 CrossRef CAS.
- Q. Guo, J. Song, L. Liu and B. Zhang, Carbon, 1999, 37, 33–40 CrossRef CAS.
- A. J. Sedriks, Corrosion of Stainless Steels, John Wiley & Sons, New York, 1986 Search PubMed.
- D. V. Andreeva and D. G. Shchukin, Mater. Today, 2008, 11, 24–30 CrossRef CAS.
- K. Aramaki, Corros. Sci., 2003, 45, 2361–2376 CrossRef CAS.
- K. Aramaki, Corros. Sci., 2002, 44, 1621–1632 CrossRef CAS.
- K. Aramaki, Corros. Sci., 2002, 44, 2621–2634 CAS.
- S. V. Lamaka, M. L. Zheludkevich, K. A. Yasakau, R. Serra, S. K. Poznyak and M. G. S. Ferreira, Prog. Org. Coat., 2007, 58, 127–135 CrossRef CAS.
- D. O. Grigoriev, K. Köhler, E. Skorb, D. G. Shchukin and H. Möhwald, Soft Matter, 2009, 5, 1426–1432 RSC.
- Y. Lvov, R. Price, B. Gaber and I. Ichinose, Colloids Surf., A, 2002, 198–200, 375–382 CrossRef CAS.
- Y. M. Lvov, D. G. Shchukin, H. Mohwald and R. R. Price, ACS Nano, 2008, 2, 814–820 CrossRef CAS.
- D. G. Shchukin and H. Mohwald, Small, 2007, 3, 926 CrossRef CAS 943.
- D. G. Shchukin, M. Zheludkevich, K. Yasakau, S. Lamaka, M. G. S. Ferreira and H. Möhwald, Adv. Mater., 2006, 18, 1672–1678 CrossRef CAS.
- M. L. Zheludkevich, D. G. Shchukin, K. A. Yasakau, H. Mohwald and M. G. S. Ferreira, Chem. Mater., 2007, 19, 402–411 CrossRef CAS.
- J. P. Youngblood and N. R. Sottos, MRS Bull, 2008, 33.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- K. Takeda, M. Tanahashi and H. Unno, Sci. Technol. Adv. Mater., 2003, 4, 435–444 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- G. V. Kolmakov, K. Matyjaszewski and A. C. Balazs, ACS Nano, 2009, 3, 885–892 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS.
- J. D. Rule, E. N. Brown, N. R. Sottos, S. R. White and J. S. Moore, Adv. Mater., 2005, 17, 205–208 CrossRef CAS.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997–1000 CrossRef CAS.
- K. S. Toohey, C. J. Hansen, J. A. Lewis, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2009, 19, 1399–1405 CrossRef CAS.
- J. W. C. Pang and I. P. Bond, Composites, Part A, 2005, 36, 183–188.
- C. M. Dry, Cem. Concr. Res., 2000, 30, 1969–1977 CrossRef CAS.
- J. D. Rule, N. R. Sottos and S. R. White, Polymer, 2007, 48, 3520–3529 CrossRef CAS.
- B. J. Blaiszik, N. R. Sottos and S. R. White, Compos. Sci. Technol., 2008, 68, 978–986 CrossRef CAS.
- A. Dementsov and V. Privman, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 021104 CrossRef.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, Berlin, 1995 Search PubMed.
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CrossRef CAS.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef CAS.
- R. W. Boyd, Nonlinear optics, Elsevier, Oxford, 2008 Search PubMed.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 CrossRef CAS.
- H. I. Elim, J. Yang, J. Lee, J. Mi and W. Jib, Appl. Phys. Lett., 2006, 88, 083107 CrossRef.
- L. François, M. Mostafavi, J. Belloni, J. Delouis, J. Delaire and P. Feneyrou, J. Phys. Chem. B, 2000, 104, 6133–6137 CrossRef CAS.
- R. G. Ispasoiu, L. Balogh, O. P. Varnavski, D. A. Tomalia and T. G. III, J. Am. Chem. Soc., 2000, 122, 11005–11006 CrossRef CAS.
- R. Philip, G. R. Kumar, N. Sandhyarani and T. Pradeep, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 13160–13166 CrossRef CAS.
- S. Porel, S. Singh, S. S. Harsha, D. N. Rao and T. P. Radhakrishnan, Chem. Mater., 2005, 17, 9–12 CrossRef CAS.
- W. Sun, Q. Dai, J. G. Worden and Q. Huo, J. Phys. Chem. B, 2005, 109, 20854–20857 CrossRef CAS.
- Y. P. Sun and J. E. Riggs, Int. Rev. Phys. Chem., 1999, 18, 43–90 CrossRef CAS.
- R. T. Tom, A. S. Nair, N. Singh, M. Aslam, C. L. Nagendra, R. Philip, K. Vijayamohanan and T. Pradeep, Langmuir, 2003, 19, 3439–3445 CrossRef CAS.
- R. West, Y. Wang and T. Goodson-I II, J. Phys. Chem. B, 2003, 107, 3419–3426 CrossRef CAS.
- Y. Wang, X. Xie and T. Goodson, Nano Lett., 2005, 5, 2379–2384 CrossRef CAS.
- S. I. Bozhevolnyi, J. Beermann and V. Coello, Phys. Rev. Lett., 2003, 90, 197403 CrossRef.
- C. Hubert, L. Billot, P. Adam, R. Bachelot, P. Royer, J. Grand, D. Gindre, K. D. Dorkenoo and A. Fort, Appl. Phys. Lett., 2007, 90, 181105 CrossRef.
- M. D. McMahon, R. Lopez, R. F. Haglund, E. A. Ray and P. H. Bunton, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 041401 CrossRef.
- S. Tai, Y. Wu, D. Shieh, L. Chen, K. Lin, C. Yu, S. Chu, C. Chang, X. Shi, Y. Wen, K. Lin, T. Liu and C. Sun, Adv. Mater., 2007, 19, 4520–4523 CrossRef CAS.
- M. Lippitz, M. A. v. Dijk and M. Orrit, Nano Lett., 2005, 5, 799–802 CrossRef CAS.
- R. Jin, J. E. Jureller, H. Y. Kim and N. F. Scherer, J. Am. Chem. Soc., 2005, 127, 12482–12483 CrossRef CAS.
- J. L. Gu, J. L. Shi, G. J. You, L. M. Xiong, S. X. Qian and H. R. Chen, Adv. Mater., 2005, 17, 557–560 CrossRef CAS.
- M. Hu, H. Chen, C. Shen, L. Hong, B. Huang, K. Chen and L. Chen, Nat. Mater., 2006, 5, 102–106 CrossRef CAS.
- W. Schrof, S. Rozouvan, E. V. Keuren, D. Horn, J. Schmitt and G. Decher, Adv. Mater., 1998, 10, 338–341 CrossRef CAS.
- W. Wenseleers, F. Stellacci, T. Meyer-Friedrichsen, T. Mangel, C. A. Bauer, S. J. K. Pond, S. R. Marder and J. W. Perry, J. Phys. Chem. B, 2002, 106, 6853–6863 CrossRef CAS.
- D. Yelin, D. Oron, S. Thiberge, E. Moses and Y. Silberberg, Opt. Express, 2003, 11, 1385–1391 Search PubMed.
- G. Wang and W. Sun, J. Phys. Chem. B, 2006, 110, 20901–20905 CrossRef CAS.
- J. François, L. M. Mostafavi, J. Belloni and J. Delaire, Phys. Chem. Chem. Phys., 2001, 3, 4965–4971 RSC.
- P. V. Kamat, M. Flumiani and G. V. Hartland, J. Phys. Chem. B, 1998, 102, 3123–3128 CrossRef CAS.
- N. Chandrasekharan, P. V. Kamat, J. Hu and G. Jones, J. Phys. Chem. B, 2000, 104, 11103–11109 CrossRef CAS.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS.
- V. Amendola and M. Meneghetti, J. Mater. Chem., 2007, 17, 4705–4710 RSC.
- T. I. Goodson, O. Varnavsky and Y. Wang, Int. Rev. Phys. Chem., 2004, 23, 109–150 CrossRef CAS.
- S. Qu, C. Du, Y. Song, Y. Wang, Y. Gao, S. Liu, Y. Li and D. Zhu, Chem. Phys. Lett., 2002, 356, 403–408 CrossRef CAS.
- H. Fang, C. Du, S. Qu, Y. Li, Y. Song, H. Li, H. Liu and D. Zhu, Chem. Phys. Lett., 2002, 364, 290–296 CrossRef CAS.
- V. Amendola, G. Mattei, C. Cusan, M. Prato and M. Meneghetti, Synth. Met., 2005, 155, 283–286 CrossRef CAS.
- V. Amendola, D. Dini, S. Polizzi, J. Shen, K. M. Kadish, M. J. F. Calvete, M. Hanack and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 8688–8695 CrossRef CAS.
- V. Amendola, S. Polizzi and M. Meneghetti, Langmuir, 2007, 23, 6766–6770 CrossRef CAS.
- V. Amendola, S. Polizzi and M. Meneghetti, J. Phys. Chem. B, 2006, 110, 7232–7237 CrossRef CAS.
- H. Fujiwara, S. Yanagida and P. V. Kamat, J. Phys. Chem. B, 1999, 103, 2589–2591 CrossRef CAS.
- T. Hirsch, A. Shaporenko, V. M. Mirsky and M. Zharnikov, Langmuir, 2007, 23, 4373–7 CrossRef CAS.
- M. Yoshida and J. Lahann, ACS Nano, 2008, 2, 1101–1107 CrossRef CAS.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS.
- K. E. Drexler, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 5275–5278 CAS.
- H. Hess, G. D. Bachand and V. Vogel, Chem.–Eur. J., 2004, 10, 2110–2116 CrossRef CAS.
- Y. Astier, O. Uzun and F. Stellacci, Small, 2009, 5, 1273–1278 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |