Morphology control of cobalt oxidenanocrystals for promoting their catalytic performance
Xiaowei
Xie
and
Wenjie
Shen
*
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: shen98@dicp.ac.cn; Fax: +86-411-84694447; Tel: +86-411-84379085
First published on 15th September 2009
Abstract
The design and fabrication of nanomaterials is a crucial issue in heterogeneous catalysis to achieve excellent performance. Traditionally, the main theme is to reduce the size of particles as small as possible mainly to increase the activity, so-called size-dependent catalytic chemistry. In recent years, the rapid developments in novel morphological and structural nanomaterials have enabled the fabrication of catalytic materials with exposing more reactive crystal planes, favoring a deep understanding of the active sites. Here, we highlight the recent progress in catalytic materials with unique performance caused by the morphology, by taking Co3O4 nanomaterials as an example. Firstly, we briefly summarize the important synthetic strategies and characteristics of morphology-controlled Co3O4 nanomaterials and their precursors like cobalt hydroxides, including zero- to two-dimensional and hierarchical nanostructures. Then, morphology/plane-dependent catalysis of these cobalt oxides is demonstrated, focusing on CH4combustion and COoxidation in order to elaborate the intrinsic nature of morphology and surface plane. Finally, we outline our personal understanding and perspectives on the morphology-dependent nanocatalysis with metal and metal oxides. These morphology-controlled nanomaterials with more reactive crystal planes exposed are expected to be highly efficient for practical applications based on the deep understanding of the catalytically active sites.
 Ms Xiaowaei Xie | Ms Xiaowei Xie is a PhD candidate at Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Her research topic is fabrication and characterization of morphology-controlled Co3O4 nanocrystals and their applications in CO oxidation at low temperatures dating from 2004. Her experience includes design and synthesis of metal oxide nanomaterials, structure analysis and catalytic reaction chemistry. |
 Dr Wenjie Shen | Dr Wenjie Shen is a Professor at Dalian Institute of Chemical Physics, Chinese Academy of Sciences. His scientific interests focus on the synthesis and assembly of metal and metal oxide nanomaterials with controllable size and morphology from the precursors to the activated catalysts, the structural analyses of catalytic materials, and the reaction chemistry (mechanism and kinetics) of alcohols and hydrocarbons. |
Introduction
Nanocatalysis is an explosively growing topic due to its considerable contributions both to the material sciences and to the practical heterogeneous catalysis. Since the catalytic performance strongly depends on the size and shape at nanometer scale, rational design and skilful synthesis of highly active catalysts are becoming the top priorities nowadays. Conventionally, active catalysts consist of small nanometer-sized particles because the number of corner and edge atoms for adsorption and activation of the reactants increases with decreasing the crystal domains.1–4 In this context, the catalytic performance is extremely sensitive to the particle size because the surface structure and electronic properties change considerably at this nanometer-size level.5,6 On the other hand, surface science enabled the study of the activity and selectivity of the catalysts by the characteristics at the atom-level structure of the planes that they expose.7–9 The most familiar example is the surface structure sensitivity of iron-catalyzed ammonia synthesis where the Fe (111) face is the most active plane and the activity ratio for Fe (111), Fe (100) and Fe (110) planes is 418:25:1.10,11 In addition to influencing the activity, the crystal plane of the metal also affects the selectivity. The Pt (100) plane only produces cyclohexane when used for benzenehydrogenation, but both cyclohexene and cyclohexane are formed on the Pt (111) plane.12 It is obvious that both the activity and the selectivity of a catalyst can be tailored by tuning its size and morphology.These interesting surface studies are mainly conducted using probe model reactions on single-crystal surfaces under high vacuum conditions. However, industrial heterogeneous catalysis usually occurs at ambient or elevated temperatures and pressures using complex materials as catalysts. Although the concept of morphology-dependent catalysis makes is possible to correlate the catalytic performance with the morphology/plane of the crystal under realistic reaction conditions, it is rather challenging to synthesize catalytic nanomaterials that are enclosed by specific surface planes, hopefully the most reactive facets. During the past decade, the rapid development in materials science offers an opportunity to prepare metal and/or metal oxide crystals with specific planes exposed. Several recent works have readily demonstrated the importance of well-defined morphology/plane in enhancing catalytic reactivity.13–20 Tian et al.13 have recently reported that Pt particles enclosed by high-index planes like {730}, {210}, and {520} exhibit a much higher catalytic activity for electro-oxidation of organic fuels such as formic acid and ethanol than that of traditional particles exposing stable planes, such as {111}, {100}, and even {110}. Narayanan et al.14,15 compared the reaction kinetics of the electron-transfer reaction between hexacyanoferrate (III) ions and thiosulfate ions in a colloidal solution containing tetrahedral, cubic, and near-spherical platinum nanoparticles, and found that the catalytic activity is greatly dependent on the shape of the Pt particles because of the varied fractions of surface atoms on the edges and corners. Li and co-workers16 investigated the activity of truncated triangular, cubic, and near spherical silver nanoparticles for the oxidation of styrene. The silver nanocubes exposing more reactive {100} planes show a much higher activity than the near-spherical nanoparticles and nanoplates.
Concerning metal oxides, a typical example comes from the shape-dependent oxygen storage capacity of ceria nanocrystals. Yan and co-workers17 have found that oxygen storage could occur both at the surface and in the bulk of ceria nanorods and nanocubes but is restricted only to the surface of nanopolyhedra. When used for COoxidation, the {100}- and {110}-dominated surface structures are more reactive than the {110}-dominated ones because of the facilitated migration of lattice oxygen from the bulk to the surface. Similarly, Li and co-workers18 have also observed that CeO2nanorods exposing the {100} and {110} planes are more reactive for COoxidation than the traditional CeO2nanoparticles predominantly exposing the {111} planes, although the latter has a larger surface area than the former. Zheng et al.19 reported that quasicubic α-Fe2O3 enclosed by six {110} planes could catalyze COoxidation with almost 100% conversion at 230 °C, which is a much lower temperature than those of flowerlike, hollow, and other irregular structures exposing various crystal planes. This clearly confirms that the external morphology, especially the exposed crystal planes of α-Fe2O3, affects the catalytic activity more significantly than the commonly considered parameters, such as surface area and hollow structure. Choudary et al.20 have observed that hexagonal MgO crystals with a lower surface area but exposing more (100) planes are highly active for benzylation of aromatics because of the optimal adsorption, molecular trafficking, and desorption energy on the (100) plane. Obviously, the most pronounced catalytic reactivity correlates closely with the large number of active centers provided by the special crystal planes through morphology control, instead of the particle size as conventionally regarded. Therefore, morphology/plane-dependent nanocatalysis should be explored at the atomic scale or at least nanoscale under practical conditions, facilitating the design and fabrication of highly efficient catalysts in terms of activity and selectivity. The key issue is to create more active planes in the nanostructures and/or to enhance the density of active sites on the surface of a catalyst. In other words, it is highly possible to enhance the fraction of more-reactive surface planes and decrease the fraction of less-reactive planes in a nanocrystalline catalyst by tuning the shape with specific surface crystallographic facets through an appropriate synthesis strategy.
Recently, solution-phase methods have became an essential and powerful approach toward fabricating nanomaterials with the quality, quantity, and reproducibility suitable for a meaningful study of their shape–property relationships.21 In this feature article, our consideration is to highlight the recent advances in morphology-controlled synthesis of nanomaterials and the subsequent development of nanocatalysis using the accumulated knowledge, taking Co3O4 nanomaterials as an interesting example. We initially summarize the synthetic strategies for morphology-controlled Co3O4 nanomaterials, mainly focusing on solution-based approaches. Then, we present two representative reactions catalyzed by Co3O4 nanomaterials, where the role of morphology is clearly and comprehensively elaborated. Finally, we stress the importance of combined studies of surface science, materials science and heterogeneous catalysis for the development of highly efficient nanocatalysts.
Synthesis of Co3O4nanostructures
Interest in nanocrystals has been growing steadily due to their fascinating physical and chemical properties in contrast to bulk solids, and thus sustained efforts have been made to explore efficient synthetic routes to obtain well-defined nanocrystals with controlled morphology. Solution-based methods, especially hydro- and solvothermal routes and liquid-phase precipitation that favor shape control, have been developed as the most powerful strategies based on the features of low temperature, easy control, and large-scale fabrication. Co3O4nanocrystals possessing diverse morphologies can be classified as zero- (0D), one- (1D), two-dimensional (2D), and hierarchical nanostructures.Co3O4 nanoparticles and nanocubes
Zero-dimensional nanocrystals are spherical particles and well-faceted nanopolyhedra.22 They are usually enclosed by low-index planes, mainly because the crystal growing rates in the direction perpendicular to a high-index plane are much faster than those along the direction of a low-index plane, and thus the high-index planes are rapidly eliminated during particle formation.23According to the Wulff construction, face-centered cubic (fcc) metals and metal oxides are often obtained with truncated octahedral shapes.24–27Fig. 1 shows the morphology of typical Co3O4 truncated octahedra prepared by a conventional precipitation method. The nanoparticles are connected together in a disordered fashion, and each nanoparticle consists of a single crystal grain with faceted surfaces. The size of the nanoparticles is 10–30 nm. When viewed along the [100] orientation for an individual particle, two sets of {022} planes with a planar spacing of 0.286 nm and {001} facets (two (001) and two (010)) are observed. When viewed along the [110] orientation, two sets of {111} planes with a planar spacing of 0.467 nm are clearly seen, and the nanoparticle is bound by four {111} and two {001} facets. Since the [100] projection is a truncated square and the [110] projection is a truncated rhombohedron, the shape of the particle is determined to be a truncated octahedron, surrounded by eight {111} and six {001} planes.
![TEM images of Co3O4 truncated octahedra obtained by precipitating cobalt acetate tetrahydrate dissolved in ethylene glycol with sodium carbonate aqueous solution at 80 °C, followed by thermal decomposition of the precursor at 450 °C for 4 h in air. (a) Low-magnification bright-field image; (b,c) HRTEM images of the nanoparticle viewed along [100] and [110] orientations; (d) schematic representation of a truncated octahedron.](/image/article/2009/NR/b9nr00155g/b9nr00155g-f1.gif) | ||
| Fig. 1 TEM images of Co3O4 truncated octahedra obtained by precipitating cobalt acetate tetrahydrate dissolved in ethylene glycol with sodium carbonate aqueous solution at 80 °C, followed by thermal decomposition of the precursor at 450 °C for 4 h in air. (a) Low-magnification bright-field image; (b,c) HRTEM images of the nanoparticle viewed along [100] and [110] orientations; (d) schematic representation of a truncated octahedron. | ||
Nanocubes are also typical shapes for nanopolyhedra with cubic crystal structures. The growth of cube-like Co3O4 with diversified sizes tends to terminate at their low-index {100} planes (Fig. 2a) by solution-based routes.28–33 Peculiar nanoparticles with either triangular or bullet-like (Fig. 2b,c) shapes can be simply obtained by thermal decomposition of Co fatty acid.34 Single-crystal seeds can evolve into octahedra, depending on the specific growing rates along the [111] and [100] directions.35,36Co3O4 octahedra (Fig. 2d) consisting of {111} planes have also been synthesized by thermal decomposition of cobalt complexes with proper procedures that favor a fast growing rate along the [111] direction but a slow growing rate along the [100] direction.37,38
 | ||
| Fig. 2 (a) TEM image of a single-crystalline Co3O4 nanocube, inset is the corresponding ED pattern (reprinted from ref. 29. Copyright 2003, American Chemical Society); (b,c) TEM images of triangular and bullet-like Co3O4nanocrystals (reprinted from ref. 34. Copyright 2004, American Chemical Society); (d) SEM image of Co3O4 octahedra and a magnified image of a single octahedron (reprinted from ref. 37. Copyright 2009, American Chemical Society). | ||
Co3O4 nanowires and nanorods
One-dimensional nanostructures generally include wires, rods, tubes, and belts whose lateral dimensions fall anywhere in the range of 1 to 100 nm.39 The design and synthesis of 1D Co3O4nanostructures is nowadays one of the most popular topics in materials science. The priority is to develop effective techniques for inducing anisotropic growth for forming 1D structures where the synthetic parameters, such as temperature, acidity-basicity, and type of template, all play essential roles in determining the final morphology.Co3O4 nanowires/nanorods can be accomplished by growing on solid substrates, typically involving the reaction of gaseous oxygen molecules with cobalt foil.40–43 A solution-based route has been developed for the direct growth of freestanding hollow Co3O4nanowire arrays on a variety of substrates, such as transparent conducting glass, Si wafer, Cu or Ti foil, and polystyrene substrate.44,45 These single-crystalline and mesoporous nanowires tend to grow along the [111] direction (Fig. 3a–d). The solid-liquid method using SBA-15 (Fig. 3e,f) and porous anodic aluminaoxide (AAO) as hard templates represents another important step forward in the synthesis of Co3O4nanowires/nanorods.46–51 It is amazing that even viruses can be successfully used as a template to assemble Co3O4nanowires.52 By incorporating gold-binding peptides into the filament coat, hybrid Au–Co3O4nanowires have also been obtained. Moreover, thermal decomposition of organocobalt precursors and cobalt complexes produces Co3O4nanocrystals with rod-like shapes.34,53 The inverse microemulsion method and the molten salt approach are also employed to synthesize Co3O4nanorods growing along the [111] and [−1,−1,15] directions, respectively (Fig. 3g,h)54,55
![SEM images of the Co3O4nanowires grown on silicon wafer (a) and polystyrene substrate (b); (c) TEM image of Co3O4nanowires; (d) SAED pattern of a single nanowire indexed along the [11̄2] zone axis (reprinted from ref. 44. Copyright 2006, American Chemical Society); TEM images of Co3O4nanowires growing on SBA-15 (e) perpendicular and (f) parallel to the wire axis (reprinted from ref. 48. Copyright 2005, Wiley-VCH); TEM images of Co3O4nanorods prepared (g) in an inverse microemulsion (reprinted from ref. 54. Copyright 2002, Royal Society of Chemistry) and (h) by a molten salt approach (reprinted from ref. 55. Copyright 2007, Elsevier Science).](/image/article/2009/NR/b9nr00155g/b9nr00155g-f3.gif) | ||
Fig. 3
SEM images of the Co3O4nanowires grown on silicon wafer (a) and polystyrene substrate (b); (c) TEM image of Co3O4nanowires; (d) SAED pattern of a single nanowire indexed along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 2] zone axis (reprinted from ref. 44. Copyright 2006, American Chemical Society); TEM images of Co3O4nanowires growing on SBA-15 (e) perpendicular and (f) parallel to the wire axis (reprinted from ref. 48. Copyright 2005, Wiley-VCH); TEM images of Co3O4nanorods prepared (g) in an inverse microemulsion (reprinted from ref. 54. Copyright 2002, Royal Society of Chemistry) and (h) by a molten salt approach (reprinted from ref. 55. Copyright 2007, Elsevier Science). 2] zone axis (reprinted from ref. 44. Copyright 2006, American Chemical Society); TEM images of Co3O4nanowires growing on SBA-15 (e) perpendicular and (f) parallel to the wire axis (reprinted from ref. 48. Copyright 2005, Wiley-VCH); TEM images of Co3O4nanorods prepared (g) in an inverse microemulsion (reprinted from ref. 54. Copyright 2002, Royal Society of Chemistry) and (h) by a molten salt approach (reprinted from ref. 55. Copyright 2007, Elsevier Science). | ||
In particular, solution-based routes have been considered to be more promising and powerful. Hydro- and solvothermal processes could synthesize Co3O4nanorods with diverse diameters from 10–250 nm.56–58 More interestingly, the liquid-phase precipitation method is reported to be essentially constructive for preparing Co3O4nanorods with the advantages of low temperature, simple apparatus, easy control, and large scale by simply using carbonate anions as precipitation and inhibitor agents without any surfactant and/or template. Zeng and co-workers59 synthesized rod-like cobalt-hydroxide-carbonate precursors by a precipitation method and further converted them into 1D arrays of Co3O4nanoparticles growing along the [111] axis (Fig. 4a,b) by thermal calcination. During the precipitation, the carbonate anions act as an inhibitor that selectively decreases the rates of crystal growth along certain directions, resulting in the elongated nanorods of the precursor. But the challenge with this approach is that the rod shape of the precursor is transformed into 1D arrays of Co3O4nanoparticles or porous nanorods (Fig. 4c,d) due to the removal of hydroxyl and carbonate species upon thermal treatment.59–62 In order to improve the morphological stability of the nanorod-shaped precursor, we have recently synthesize Co3O4nanorods by liquid-phase precipitation in ethylene glycol using carbonate anions as a structure-directing agent.63 As shown in Fig. 5, the nanorod-shaped solid precursor, probably cobalt hydroxide carbonate, has a diameter of 10–20 nm and a length of 200–300 nm. Calcination at 450 °C in air caused a spontaneous transformation of the morphology, forming Co3O4nanorods with diameters of 5–15 nm and lengths of 200–300 nm. Most of the nanorods have straight sides and end profiles. Observation of a single Co3O4nanorod in the [10] orientation showed that the growth direction is [110] with the flat side parallel to (001). The (001) side planes were also observed in the [100] orientation, showing two sets of {022} planes with a planar spacing of 0.286 nm. When viewed close to the [110] orientation, the nanorod showed an approximately rectangular shape with the long edges parallel to (001) and the short edges parallel to (10). That is, the single crystal Co3O4nanorod has a rectangular cross section with two {001} side planes, two {10} side planes and two {110} end planes. The Co3O4nanorod grows mainly along the [110] direction and exposes preferentially the {110} plane, where the surface area of the {110} planes is up to 40% of the total surface in a single nanorod.
 | ||
| Fig. 4 (a) TEM images for cobalt-hydroxide-carbonate nanorods and a free-standing nanorod (insert); (b) TEM image of 1D array of Co3O4nanoparticles obtained by calcination of the above sample and its selected area ED pattern (insert) (reprinted from ref. 59. Copyright 2003, American Chemical Society); (c,d) high magnification TEM images of a single Co3O4nanorod, showing the porous structure and consisting of textured aggregation of nanocrystals, the insets are the corresponding electron diffraction patterns (reprinted from ref. 62. Copyright 2009, American Chemical Society). | ||
![(a,b) TEM images of the cobalt hydroxide carbonate precursor obtained by the precipitation of cobalt acetate tetrahydrate dissolved in ethylene glycol with aqueous sodium carbonate solution at 160 °C; (c-f) TEM images of the Co3O4nanorods obtained by thermal decomposition of the as-prepared precursor at 450 °C for 4 h in air ((c) low-magnification bright-field image; (d–f) HRTEM images viewed close to the [110] (d), [1̄10] (e) and [100] (f) orientations); (g) morphological illustration of the nanorod.](/image/article/2009/NR/b9nr00155g/b9nr00155g-f5.gif) | ||
| Fig. 5 (a,b) TEM images of the cobalt hydroxide carbonate precursor obtained by the precipitation of cobalt acetate tetrahydrate dissolved in ethylene glycol with aqueous sodium carbonate solution at 160 °C; (c-f) TEM images of the Co3O4nanorods obtained by thermal decomposition of the as-prepared precursor at 450 °C for 4 h in air ((c) low-magnification bright-field image; (d–f) HRTEM images viewed close to the [110] (d), [10] (e) and [100] (f) orientations); (g) morphological illustration of the nanorod. | ||
Since the discovery of carbon nanotubes,64 synthesis of metal and metal oxidenanotubes has became one of the most popular topics in materials science. Co3O4nanotubes have been successfully fabricated by template-based chemical vapor deposition, solvothermal treatment, and microemulsion methods.65–68 In addition, one-dimensional Co3O4nanotubes are also formed by thermal decomposition of cobalt complexes having strong intermolecular hydrogen bonding.69 Self-supported topotactic transformation of β-Co(OH)2 produces needle-like Co3O4nanotubes growing along the [111] direction (Fig. 6a–c).70
 | ||
| Fig. 6 (a) SEM image of Co3O4nanotubes, the inset shows a cross-sectional view of a single nanotube; (b) TEM image showing a selected area of a Co3O4nanotube for electron diffraction, and (c) SAED pattern (reprinted from ref. 70. Copyright 2008, Wiley-VCH); (d) TEM image of the as-prepared cobalt-hydroxide-carbonate nanobelts, and (e) 1D arrays of Co3O4nanoparticles transformed from the as-prepared nanobelts (reprinted from ref. 73. Copyright 2008, Elsevier Science); (f) SEM image of Co3O4 nano-needles; (g) TEM image and (h) SAED pattern of a single Co3O4 nano-needle (reprinted from ref. 74. Copyright 2008, Royal Society of Chemistry). | ||
Other 1D Co3O4nanostructures like naofibers and nanobelts have been successfully prepared through template-directing, electro-spinning, and hydrothermal methods.32,71,72 However, the precursors of cobalt hydroxide carbonate nanobelts obtained under hydrothermal conditions are easily transformed to 1D arrays of Co3O4nanoparticles (Fig. 6d,e) upon calcination mainly because of the poor morphological stability.73Thermal decomposition and re-crystallization of β-Co(OH)2 nano-needles produces mesoporous Co3O4 nano-needles that are nearly single-crystalline (Fig. 6f–h).74
2D and hierarchical Co3O4 nanomaterials
Two-dimensional nanostructures come with only one confined dimension.22Co3O4 nanowalls, nanosheets, and nanoplatelets as typical 2D nanostructures have been studied for a long time. Sow and co-workers40,75 obtained single-crystalline Co3O4 nanowalls by heating Co foil in an oxygen atmosphere (Fig. 7a,b). For the fabrication of Co3O4nanosheets or nanoplatelets, the most frequently used route is to prepare β-Co(OH)2 precursors hydrothermally, followed by thermal decomposition of these precursors at elevated temperatures.32,76–78 Notably, Li and co-workers32 synthesized uniform hexagonal Co3O4nanosheets with average thickness and edge length of ca. 50–100 nm and 3 µm, respectively. The predominantly exposed planes are {112}. β-Co(OH)2 with a uniform hexagonal shape is generally used as the precursor possibly because it is isostructural with brucite-like compounds and consists of a hexagonal packing of hydroxyl ions with CoII occupying alternate rows of octahedral sites.79,80 Occasionally, CoOOH and Co alkoxide are also used as the precursors to prepare Co3O4nanosheets or nanoplates.38,81 However, almost all Co3O4nanosheets possess porous structures or cracks mainly because of the poor morphology stability of the precursors (Fig. 7c–f).38,76–78 | ||
| Fig. 7 (a,b) SEM images of Co3O4 nanowalls (reprinted from ref. 40. Copyright 2007, Wiley-VCH); SEM images of (c) Co(OH)2 and (d) Co3O4 hexagonal sheets; (e,f) TEM images of porous Co3O4nanosheets (reprinted with permission from ref. 78. Copyright 2009, Wiley-VCH). | ||
In addition to the low-dimensional nanocrystals with tailored geometries, the subsequent utilization of these nanostructures as building blocks to construct hierarchical structures is also quite interesting. In-situ self-assembly has been shown to be an efficient “bottom-up” route in fabricating hierarchical Co3O4 structures, and Co3O4 nanoboxes, microspheres, flower-like structures, and nanocolumns have been synthesized accordingly (Fig. 8).81–89 To date, hydro- and solvothermal treatments are still the major routes to fabricate Co3O4 superstructures.83–89 Unfortunately, most of these hierarchical nanostructures consist of arrays of interconnected particles with porous structures.81–85 Although the building blocks are nanocubes, nanorods, and nanosheets with very smooth surfaces, the removal of hydroxyl and carbonate species in the precursors by thermal calcination results in porous building blocks or nanoparticles (Fig. 8c–h). In fact, the thermal stability of the building blocks affects considerably the morphology of the final hierarchical materials.
 | ||
| Fig. 8 (a,b) TEM images of Co3O4 nanoboxes (reprinted from ref. 87. Copyright 2006, Wiley-VCH); (c,d) SEM images of porous Co3O4 microspheres (reprinted from ref. 83. Copyright 2009, American Chemical Society); (e,f) SEM/TEM images of flowerlike Co3O4 architectures, inset is the corresponding SAED pattern (reprinted from ref. 82. Copyright 2008, American Chemical Society); (g,h) SEM/TEM images of Co3O4 nanocolumns and a single nanocolumn and its SAED pattern (reprinted from ref. 84. Copyright 2009, American Chemical Society). | ||
Hollow structured Co3O4 nanomaterials have attracted great attention mainly because of their high surface areas. Co3O4 hollow spheres constructed from nanoparticles or nanosheets usually have diameters of several hundred nanometers or even several microns (Fig. 9).90–99 Hollow nanowires, nanotubes, nanoboxes and nanocages have also been synthesized.44,70,87,100 Template-directing, Ostwald ripening, oriented aggregation and Kirkendall type diffusion are the common approaches in constructing these hollow structures.44,87,90–93
 | ||
| Fig. 9 (a,b) TEM images of semi-hollow Co3O4 core–shell structures with asymmetric Ostwald ripening (reprinted with permission from ref. 91. Copyright 2005, Wiley-VCH); (c,d) SEM images of hollow microspheres assembled from nanoslices (reprinted from ref. 95. Copyright 2009, Elsevier Science). | ||
In short, Co3O4nanostructures are usually synthesized through a two-step process. Initially, the precursors with different shapes and novel structures are mainly synthesized by solution-based approaches, and then are transformed to Co3O4 by thermal decomposition at elevated temperatures. It is, however, rather challenging to control or maintain the morphology from the precursors to the final Co3O4nanostructures. Therefore, more attention should be paid to the development of efficient synthetic techniques so that the Co3O4 nanomaterials would expose more reactive planes, which is essentially important in improving the catalytic performance.
Morphology-dependent nanocatalysis
Tricobalt tetraoxide is one of most intriguing magnetic p-type semiconductors and is widely used in magnetism, optics, lithium-ion batteries, chemical sensors, and heterogeneous catalysis.30,42,45,71,81,101 One of the most promising features in these applications is that the physical and chemical properties of Co3O4 are closely associated with the morphology, the so-called morphology-dependent phenomena. Co3O4 is typically used for oxidation reactions of carbon monoxide and hydrocarbons,101,102 and the reactivity is usually correlated to the sizes of the Co3O4 particles.103–105 Here, we demonstrate the importance of the morphology and crystal plane of Co3O4 nanomaterials in CH4combustion and COoxidation reactions.CH4 combustion
Li and co-workers32 have comprehensively studied the morphology and crystal plane effects of Co3O4nanosheets, nanobelts, and nanocubes on methanecombustion. The specific surface areas of nanosheets, nanobelts, and nanocubes are 17.8, 20.1, and 22.6 m2 g−1, respectively, but the catalytic properties vary greatly with the shape of the Co3O4nanocrystal. The value of T50 (the temperature at which the conversion of methane is 50%) on Co3O4 nanocubes is 343 °C, but it decreases to 319 °C on Co3O4 nanobelts and 313 °C over Co3O4nanosheets. The reaction rates are 2.72, 2.28, and 1.25 µmol g−1s−1 for nanosheets, nanobelts, and nanocubes, respectively, at 313 °C. Apparently, the reaction proceeds about two times faster over the nanosheets than on the nanocubes. Detailed structure analyses revealed that the predominantly exposed planes are {112} in nanosheets, {011} in nanobelts, and {001} in nanocubes (Fig. 10). Judging by the surface atomic arrangement, the activity of methanecombustion over these crystal planes follows the order {112}> {011} ≫ {001}. Therefore, the nanosheets having more reactive {112} planes exhibit exceptionally high activity towards methanecombustion. | ||
| Fig. 10 (a) SEM image of Co3O4nanosheets; (b) HRTEM image and structure model of a nanosheet; (c) TEM image of Co3O4 nanobelts; (d) HRTEM image and structure model of a nanobelt; (e) TEM image of Co3O4 nanocubes, and (f) HRTEM image and structural model of a nanocube (reprinted from ref. 32. Copyright 2008, American Chemical Society). | ||
CO oxidation
Very recently, we have demonstrated that Co3O4nanorods are highly active for low temperature oxidation of CO when compared to the conventional Co3O4nanoparticles,101 clearly showing a morphology-dependent effect. As shown in Fig. 1, the nanoparticles expose eight {111} and six {001} planes, while the nanorods expose preferentially the {110} plane and its surface area is about 40% of the total surface area in a single nanorod (Fig. 5).Fig. 11 shows the H2-TPR profiles of the Co3O4 materials. There is a minor reduction peak at around 100 °C for the samples, which is usually attributed to the very active species on the surface, formed during the pre-oxidation treatment.104 Quantitative evaluation of the amounts of hydrogen consumed proves that Co3O4 is reduced almost completely into metallic cobalt in both cases. The amounts of hydrogen consumed by the low temperature reduction at 300 °C are 51–55 µmol/g, which is about one-third of that for the high temperature reduction (184–186 µmol/g), confirming the transformation of Co3O4 to CoO. However, the subsequent reduction of CoO to Co is strongly dependent on the morphology of the Co3O4 materials. The Co3O4nanoparticles show one intense reduction peak at ∼360 °C, whereas the corresponding reduction peak in the Co3O4nanorods shifts towards a much higher temperature at about 500 °C. This indicates that the reduction of CoO in the Co3O4nanorods is more difficult than that in the Co3O4nanoparticles.
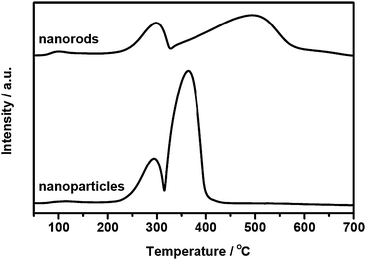 | ||
| Fig. 11 H2-TPR profiles of Co3O4nanoparticles and nanorods. The measurements were conducted using a U-shaped quartz reactor connected to a thermal conductivity detector. 15 mg of sample were pretreated with a 20% O2/N2 mixture (50 ml/min) at 450 °C for 30 min. After cooling to room temperature with a flow of N2 and introducing the reducing agent of a 10% H2/N2 mixture (20 ml/min), the sample was heated to 700 °C at a rate of 10 °C/min. | ||
Fig. 12 compares the XRD patterns recorded during hydrogen reductions of the Co3O4nanoparticles and the Co3O4nanorods. The two samples are present as Co3O4 at 200 °C and reduced rapidly to CoO at 300 °C. Again, the subsequent reduction of CoO to metallic cobalt is significantly affected by the morphology. For the Co3O4nanoparticles, the formation of Co is readily detected at 400 °C and it becomes the dominant phase at 500 °C. The diffraction peaks of CoO disappear at 550 °C, suggesting that the CoO species are fully reduced to metallic cobalt. On the other hand, the reduction of CoO in the Co3O4nanorods only starts at ∼500 °C and the complete reduction to Co is achieved at 650 °C. This phenomenon clearly indicates that the CoO species in the Co3O4nanorods is more difficult to be reduced than that in the nanoparticles, in good accordance with the TPR measurements. Therefore, it can be supposed that the morphology has significantly affected the reduction behavior of Co3O4 nanomaterials.
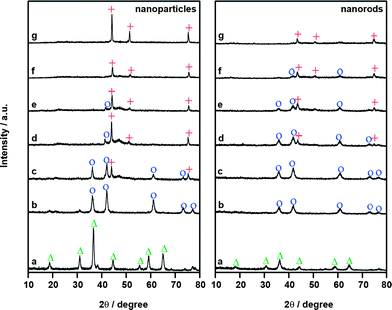 | ||
| Fig. 12 In situ XRD patterns for the reduction of Co3O4nanoparticles and nanorods. (a) 200 °C, (b) 300 °C, (c) 400 °C, (d) 500 °C, (e) 550 °C, (f) 600 °C and (g) 650 °C. △ Co3O4; ○CoO; + Co. The measurements were performed in a high temperature cell installed in the diffractometer. The sample was pressed into a pellet and mounted in the chamber. After heating to a given temperature under a flow of diluted hydrogen, the XRD patterns were then recorded. | ||
Fig. 13 compares the conversion of CO as a function of time-on-stream for COoxidation on the Co3O4 nanomaterials. The nanoparticles give an initial CO conversion of 30% which then rapidly decreases to about 10%. On the other hand, the nanorods show 100% CO conversion during the initial 6 h and then the conversion of CO only slightly decreases. After reaction for 12 h, the conversion of CO still remains at 80%. Obviously, the Co3O4nanorods are more active and stable than the Co3O4nanoparticles. This observation in morphology-dependent nanocatalysis is quite promising because the reaction temperature of −77 °C for 100% CO conversion over the Co3O4nanorods is much lower than those reported previously for cobalt oxides,106–109 other metal oxides110,111 and even noble catalysts.112,113
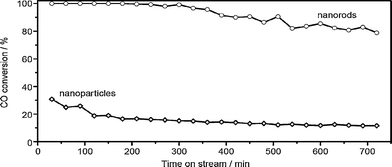 | ||
| Fig. 13 CO conversions as a function of time-on-stream over Co3O4nanoparticles and nanorods. The COoxidation reaction was performed in a continuous-flow fixed-bed quartz reactor under atmospheric pressure at −77 °C. In a typical run, 200 mg (40–60 mesh) of sample was loaded and pretreated with a 20% O2/He (50 ml/min) at 450 °C for 30 min. After cooling to −77 °C, the reaction stream of 1% CO/2.5% O2/He (50 ml/min) was introduced through a mass-flow controller. The effluent was analyzed using an on-line gas chromatograph. | ||
Table 1 lists the reaction rates of COoxidation on the Co3O4nanoparticles and nanorods in the temperature range from −56 to −86 °C. Again, the reaction rate is strongly associated with the morphology of Co3O4. The reaction rate of COoxidation on the nanorods is almost one order of magnitude larger than that over the nanoparticles. For example, the reaction rate on the nanorods is 3.91 × 10−6 mol CO g−1s−1 at −77 °C, but the corresponding value for the nanoparticles is only 4.66 × 10−7 mol CO g−1s−1. However, the estimated apparent activation energies are 21 kJ/mol on the nanoparticles and 22 kJ/mol over the nanorods. This implies that the morphology affects the reaction rate significantly but has no influence on the reaction pathway.
| Temperature (°C) | Nanorods | Nanoparticles | |
|---|---|---|---|
| Rate (mol/g s) | TOF (s−1)a | Rate (mol/g s) | |
| a The turnover frequencies (TOF) based on the Co3+ site were estimated according to the surface area of the {110} plane in the nanorods and the atomic configuration of the {110} plane. | |||
| −86 | 1.69 × 10−6 | 1.01 × 10−2 | 2.76 × 10−7 |
| −77 | 3.91 × 10−6 | 2.33 × 10−2 | 4.66 × 10−7 |
| −65 | 7.98 × 10−6 | 4.75 × 10−2 | 1.01 × 10−6 |
| −56 | 1.25 × 10−5 | 7.45 × 10−2 | 1.81 × 10−6 |
| Ea (kJ/mol) | 22 | 21 | |
| A (molecules/g s) | 8.0 × 1024 | 5.7 × 1023 | |
Here, the crystal planes show distinct catalytic activity, and the extremely high activity of the nanorods relies on its surface atomic configurations. Tricobalt tetraoxide has the spinel structure containing Co2+ in tetrahedral coordination and Co3+ in octahedral coordination. It is well known that the Co3+ species are the active sites for COoxidation, whereas the Co2+ species are almost inactive.114–116 Surface differential diffraction studies have also proved that the surface of Co3O4 is mainly composed of {111} and {110} planes, and the Co3+ species could only be present on the {110} plane.117,118 Density functional theory calculations performed on the Co3O4 surface for COoxidation indicated that the CO preferentially interacts with the surface Co3+ site which is the only favorable site for CO adsorption.119
As illustrated in Fig. 5, the Co3O4nanorods expose preferentially the reactive {110} planes, but the Co3O4nanoparticles expose mainly the {111} planes (Fig. 1). Concerning the surface atomic configuration in a unit cell of Co3O4, oxygen anions form a distorted face-centred cubic sublattice, in which Co2+ cations occupy one-eighth of the tetrahedral interstices and Co3+ cations occupy half of the octahedral interstices (Fig. 14a). The close-packed planes are {001}, {111} and {110}, and their surface atomic configurations are illustrated in Fig. 14b–d. Clearly, the {001} and {111} planes contain only Co2+ cations, while the {110} plane is composed mainly of Co3+ cations. Therefore, the dominant presence of {110} planes which are rich in catalytically active Co3+ species in the nanorods would lead to a higher activity for COoxidation.
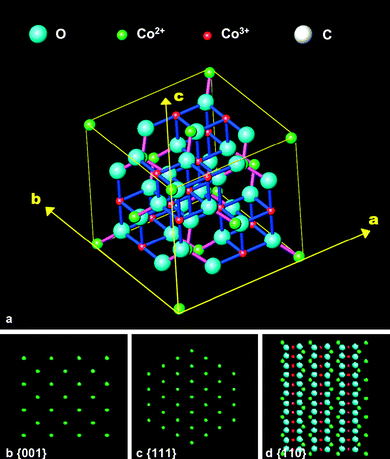 | ||
| Fig. 14 (a) Spinel structure of Co3O4 crystal, and (b–d) surface atomic configurations of the {001} (b), {111} (c), and {110} (d) planes. | ||
Table 2 summarizes the dependence of the COoxidation rate on the concentrations of CO and O2 over the Co3O4nanorods, from which the reaction orders of CO and O2 are estimated to be 0.12 and 0.28, respectively. This implies that the reaction rate is controlled by the surface reaction and it is most likely that the surfaces of the nanorods are saturated with CO and O2, leading to very weak dependences of reaction rate on CO and O2 concentrations. The estimated pre-exponential factor is 8.0 × 1024 molecules g−1s−1, but the corresponding value for the nanoparticles is only 5.7 × 1023 molecules g−1s−1. Although the crystallite size of the Co3O4nanoparticles is 16 nm with a surface area of 62 m2/g and the Co3O4nanorods give a crystallite size of 12 nm and a surface area of 163 m2/g, the significantly higher reaction rate of the nanorods is straightforwardly ascribed to the richness in active Co3+ sites on the surfaces through preferentially exposing the reactive {110} planes. As indicated by the pre-exponential factor, the nanorods have a sufficiently large number of active sites on the surface where the reaction of CO and O2 can occur rapidly once they approach the surface. Therefore, the nature of the morphology or crystal plane dependent effect is that the nanorods expose preferentially the reactive {110} plane which favors the presence of octahedrally coordinated Co3+ species, acting as the active sites for COoxidation. The large amount of active sites on the {110} planes enhances the redox properties, catalytic activity and stability in contrast to the nanoparticles. This confirms that the fabrication of more reactive crystal planes by controlling the morphology of the nanomaterials could considerably promote the catalytic performance through enriching the active sites on the surface.
| CO (vol%)a | CO conv. (%) | rCO (mol/g s) | O2 (vol%)b | CO conv. (%) | rCO (mol/g s) |
|---|---|---|---|---|---|
| a O2 = 2.50 (vol%). b CO = 1.00 (vol%). Reaction rates were measured at −77 °C. | |||||
| 0.69 | 13.1 | 3.05 × 10−6 | 0.99 | 7.6 | 3.05 × 10−6 |
| 1.03 | 9.8 | 3.91 × 10−6 | 2.56 | 9.8 | 3.91 × 10−6 |
| 1.22 | 8.2 | 3.99 × 10−6 | 3.03 | 10.9 | 4.33 × 10−6 |
| 1.51 | 7.1 | 4.09 × 10−6 | 5.11 | 11.3 | 4.67 × 10−6 |
| Reaction order: | CO = 0.12 | O2 = 0.28 | |||
These two examples convincingly evidence that morphological control of nanomaterials can lead to a large exposure of catalytically active sites. There is no doubt that this concept will be beneficial for the development of highly efficient nanocatalysts. However, the main challenge is to synthesize catalytic materials that could preferentially expose more reactive crystal planes in order to achieve the morphology/plane-dependent catalysis under practical reaction conditions.
Remarks and perspective
The merit of morphology/plane-dependent nanocatalysis is to design and fabricate materials at nanometer scale which could expose more reactive planes, hopefully a mono-plane within a uniform nanostructure. The concept of surface-structure sensitivity in surface science provides a solid base to elaborate the morphology-reactivity correlation in nanocatalysis. A recent example comes from Pt nanoparticles for benzenehydrogenation where the catalytic selectivity is strongly affected by the shape of the particles.120Cyclohexane is formed as the sole product on the cubic particles exposing the (100) planes but cyclohexane and cyclohexene are simultaneously produced on the cuboctahedral nanoparticles consisting of both (100) and (111) planes. This shape-dependent selectivity has proved their earlier surface studies.12 Moreover, Pt nanoparticles exposing {730}, {210}, and {520} high-index facets are more active for electro-oxidation of light organic fuels than the conventional particles mainly enclosed by {111}, {100}, and {110} planes, showing a morphology-dependent activity.13 Therefore, it can be expected that the synthesis of metal nanocrystals bounded by high-index facets could be a potential route for enhancing the catalytic reactivity because the high-index planes have a high density of atomic steps, ledges, and kinks serving as active sites for breaking chemical bonds.121–123Concerning metal oxides, only a few examples that evidence the morphology-dependent phenomenon are available at the moment. However, the rapidly developing nanotechnology will allow catalytic materials to be made with well-defined crystal planes. Most likely, novel and efficient synthetic approaches will concentrate more on morphology-controlled catalytic materials including metals and metal oxides which have wide applications in heterogeneous catalysis. The most important topics might be: (1) nanocrystals enclosed by high-index facets in view of catalytic activity;13 (2) nanocrystals with homogeneous surface planes with respect to catalytic selectivity;124,125 (3) nanocrystals with stable morphology under practical conditions; (4) binary metal and/or metal oxidenanocrystals;126 and (5) metal nanocrystals supported on metal oxidenanocrystals with varied morphologies.127 It could be expected that the concept of morphology/plane-dependent nanocatalysis may bridge the gap between surface science and materials science for heterogeneous catalysis, and that any new breakthrough in catalytic materials will improve the reaction efficiency greatly.
References
- R. Van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef CAS.
- S. Ladas, Surf. Sci., 1986, 175, L681–L686 CrossRef CAS.
- R. B. Greegor and F. W. Lytle, J. Catal., 1980, 63, 476–486 CrossRef CAS.
- J. J. Burton, Catal. Rev. Sci. Eng., 1974, 9, 209–222 CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- G. Ertl, Catal. Rev. Sci. Eng., 1980, 21, 201–223 CAS.
- G. Somorjai and N. Materer, Top. Catal., 1994, 1, 215–231 CrossRef CAS.
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212–9228 CrossRef CAS.
- N. D. Spencer, R. C. Schoonmaker and G. A. Somorjai, Nature, 1981, 294, 643–644 CrossRef CAS.
- N. D. Spencer, R. C. Schoonmaker and G. A. Somorjai, J. Catal., 1982, 74, 129–135 CAS.
- K. M. Bratlie, C. J. Kliewer and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 17925–17930 CrossRef CAS.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS.
- R. Xu, D. Wang, J. Zhang and Y. Li, Chem.–Asian J., 2006, 1, 888–893 CrossRef CAS.
- H. X. Mai, L. D. Sun, Y. W. Zhang, R. Si, W. Feng, H. P. Zhang, H. C. Liu and C. H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS.
- K. B. Zhou, X. Wang, X. M. Sun, Q. Peng and Y. D. Li, J. Catal., 2005, 229, 206–212 CrossRef CAS.
- Y. H. Zheng, Y. Cheng, Y. S. Wang, F. Bao, L. H. Zhou, X. F. Wei, Y. Y. Zhang and Q. Zheng, J. Phys. Chem. B, 2006, 110, 3093–3097 CrossRef CAS.
- B. M. Choudary, R. S. Mulukutla and K. J. Klabunde, J. Am. Chem. Soc., 2003, 125, 2020–2021 CrossRef CAS.
- Y. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- Z. G. Yan and C. H. Yan, J. Mater. Chem., 2008, 18, 5046–5059 RSC.
- H. E. Buckley, Crystal Growth, Wiley, New York, 1951 Search PubMed.
- G. Wulff, Z. Kristallogr., 1901, 34, 449–530 CAS.
- L. D. Marks, Rep. Prog. Phys., 1994, 57, 603–649 CrossRef CAS.
- J. A. Venables, Introduction to Surface and Thin Film Processes, Cambridge University Press, Cambridge, 2000, pp. 9–11 Search PubMed.
- Q. Yuan, H. H. Duan, L. L. Li, L. D. Sun, Y. W. Zhang and C. H. Yan, J. Colloid Interface Sci., 2009, 335, 151–167 CrossRef CAS.
- R. Xu and H. C. Zeng, J. Phys. Chem. B, 2003, 107, 926–930 CrossRef CAS.
- J. Feng and H. C. Zeng, Chem. Mater., 2003, 15, 2829–2835 CrossRef CAS.
- R. Xu and H. C. Zeng, Langmuir, 2004, 20, 9780–9790 CrossRef CAS.
- D. S. Wang, T. Xie, Q. Peng, S. Y. Zhang, J. Chen and Y. D. Li, Chem.–Eur. J., 2008, 14, 2507–2513 CrossRef CAS.
- L. H. Hu, Q. Peng and Y. D. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS.
- T. He, D. Chen, X. Jiao, Y. Wang and Y. Duan, Chem. Mater., 2005, 17, 4023–4030 CrossRef CAS.
- N. R. Jana, Y. Chen and X. Peng, Chem. Mater., 2004, 16, 3931–3935 CrossRef CAS.
- Y. J. Xiong, J. Y. Chen, B. Wiley and Y. N. Xia, J. Am. Chem. Soc., 2005, 127, 7332–7333 CrossRef CAS.
- Y. J. Xiong, H. G. Cai, B. J. Wiley, J. G. Wang, M. J. Kim and Y. N. Xia, J. Am. Chem. Soc., 2007, 129, 3665–3675 CrossRef CAS.
- T. Xu, X. Zhou, Z. Y. Jiang, Q. Kuang, Z. X. Xie and L. S. Zheng, Cryst. Growth Des., 2009, 9, 192–196 CrossRef CAS.
- B. Geng, F. Zhan, C. Fang and N. Yu, J. Mater. Chem., 2008, 18, 4977–4984 RSC.
- Y. Xia, P. Yang, Y. W. Y. Sun, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- B. Varghese, Z. Yanwu, M. V. Reddy, B. V. R. Chowdari, A. T. S. Wee, T. B. C. Vincent, C. T. Lim and C. H. Sow, Adv. Funct. Mater., 2007, 17, 1932–1939 CrossRef CAS.
- B. Varghese, Y. S. Zhang, L. Dai, V. B. C. Tan, C. T. Lim and C. H. Sow, Nano Lett., 2008, 8, 3226–3232 CrossRef CAS.
- Z. Dong, Y. Fu, Q. Han, Y. Xu and H. Zhang, J. Phys. Chem. C, 2007, 111, 18475–18478 CrossRef CAS.
- L. He, Z. C. Li and Z. J. Zhang, Nanotechnology, 2008, 19, 155606 CrossRef.
- Y. Li, B. Tan and Y. Wu, J. Am. Chem. Soc., 2006, 128, 14258–14259 CrossRef CAS.
- Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265–270 CrossRef CAS.
- K. Zhu, B. Yue, W. Zhou and H. He, Chem. Commun., 2003, 98–99 RSC.
- W. B. Yue and W. Z. Zhou, Chem. Mater., 2007, 19, 2359–2363 CrossRef CAS.
- F. Jiao, K. M. Shaju and P. G. Bruce, Angew. Chem., Int. Ed., 2005, 44, 6550–6553 CrossRef CAS.
- G. Ji, Z. Gong, W. Zhu, M. Zheng, S. Liao, K. Shen, J. Liu and J. Cao, J. Alloys Compd., 2009, 476, 579–583 CrossRef CAS.
- E. L. Salabas, A. Rumplecker, F. Kleitz, F. Radu and F. Schüth, Nano Lett., 2006, 6, 2977–2981 CrossRef CAS.
- A. Rumplecker, F. Kleitz, E. L. Salabas and F. Schuth, Chem. Mater., 2007, 19, 485–496 CrossRef CAS.
- K. T. Nam, D. W. Kim, P. J. Yoo, C. Y. Chiang, N. Meethong, P. T. Hammond, Y. M. Chiang and A. M. Belcher, Science, 2006, 312, 885–888 CrossRef CAS.
- X. Lou, J. Han, W. Chu, X. Wang and Q. Cheng, Mater. Sci. Eng., B, 2007, 137, 268–271 CrossRef CAS.
- Y. K. Liu, G. H. Wang, C. K. Xu and W. Z. Wang, Chem. Commun., 2002, 1486–1487 RSC.
- X. F. Ke, J. M. Cao, M. B. Zheng, Y. P. Chen, J. S. Liu and G. B. Ji, Mater. Lett., 2007, 61, 3901–3903 CrossRef CAS.
- Z. G. Guo and W. M. Liu, Appl. Phys. Lett., 2007, 90, 193108 CrossRef.
- S. Lian, E. Wang, L. Gao and L. Xu, Mater. Lett., 2007, 61, 3893–3896 CrossRef CAS.
- H. Zhang, J. B. Wu, C. X. Zhai, X. Y. Ma, N. Du, J. P. Tu and D. R. Yang, Nanotechnology, 2008, 19, 035711 CrossRef.
- R. Xu and H. C. Zeng, J. Phys. Chem. B, 2003, 107, 12643–12649 CrossRef CAS.
- E. Hosono, S. Fujihara, I. Honma and H. S. Zhou, J. Mater. Chem., 2005, 15, 1938–1945 RSC.
- Z. H. Wang, X. Y. Chen, M. Zhang and Y. T. Qian, Solid State Sci., 2005, 7, 13–15 CrossRef CAS.
- G. Wang, X. Shen, J. Horvat, B. Wang, H. Liu, D. Wexler and J. Yao, J. Phys. Chem. C, 2009, 113, 4357–4361 CrossRef CAS.
- X. W. Xie and W. J. Shen, unpublished work.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- W. Y. Li, L. N. Xu and J. Chen, Adv. Funct. Mater., 2005, 15, 851–857 CrossRef CAS.
- X. P. Shen, H. J. Miao, H. Zhao and Z. Xu, Appl. Phys. A: Mater. Sci. Process., 2008, 91, 47–51 CrossRef CAS.
- L. Zhuo, J. Ge, L. Cao and B. Tang, Cryst. Growth Des., 2009, 9, 1–6 CrossRef CAS.
- R. M. Wang, C. M. Liu, H. Z. Zhang, C. P. Chen, L. Guo, H. B. Xu and S. H. Yang, Appl. Phys. Lett., 2004, 85, 2080–2082 CrossRef CAS.
- X. Shi, S. Han, R. J. Sanedrin, C. Galvez, D. G. Ho, B. Hernandez, F. Zhou and M. Selke, Nano Lett., 2002, 2, 289–293 CrossRef CAS.
- X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258–262 CrossRef CAS.
- N. A. M. Barakat, M. S. Khil, F. A. Sheikh and H. Y. Kim, J. Phys. Chem. C, 2008, 112, 12225–12233 CrossRef CAS.
- B. B. Lakshmi, C. J. Patrissi and C. R. Martin, Chem. Mater., 1997, 9, 2544–2550 CrossRef CAS.
- R. J. Yu, P. F. Tao, X. S. Zhou and Y. P. Fang, J. Alloys Compd., 2008, 461, 574–578 CrossRef CAS.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, J. Mater. Chem., 2008, 18, 4397–4401 RSC.
- T. Yu, Y. W. Zhu, X. J. Xu, Z. X. Shen, P. Chen, C. T. Lim, J. T. L. Thong and C. H. Sow, Adv. Mater., 2005, 17, 1595–1599 CrossRef CAS.
- Y. L. Hou, H. Kondoh, M. Shimojo, T. Kogure and T. Ohta, J. Phys. Chem. B, 2005, 109, 19094–19098 CrossRef CAS.
- X. Liu, R. Yi, N. Zhang, R. Shi, X. Li and G. Qiu, Chem.–Asian J., 2008, 3, 732–738 CrossRef CAS.
- F. Zhan, B. Geng and Y. Guo, Chem.–Eur. J., 2009, 15, 6169–6174 CrossRef CAS.
- P. Benson, G. W. D. Briggs and W. F. K. Wynne-Jones, Electrochim. Acta, 1964, 9, 275–280 CrossRef CAS.
- C. Mockenhaupt, T. Zeiske and H. D. Lutz, J. Mol. Struct., 1998, 443, 191–196 CrossRef CAS.
- A. M. Cao, J. S. Hu, H. P. Liang, W. G. Song, L. J. Wan, X. L. He, X. G. Gao and S. H. Xia, J. Phys. Chem. B, 2006, 110, 15858–15863 CrossRef CAS.
- Y. S. Ding, L. P. Xu, C. H. Chen, X. F. Shen and S. L. Suib, J. Phys. Chem. C, 2008, 112, 8177–8183 CrossRef CAS.
- H. P. Cong and S. H. Yu, Cryst. Growth Des., 2009, 9, 210–217 CrossRef CAS.
- Y. Shao, J. Sun and L. Gao, J. Phys. Chem. C, 2009, 113, 6566–6572 CrossRef CAS.
- B. X. Li, Y. Xie, C. Z. Wu, Z. Q. Li and J. Zhang, Mater. Chem. Phys., 2006, 99, 479–486 CrossRef CAS.
- L. X. Yang, Y. J. Zhu, L. Li, L. Zhang, H. Tong, W. W. Wang, G. F. Cheng and J. F. Zhu, Eur. J. Inorg. Chem., 2006, 4787–4792 CrossRef CAS.
- T. He, D. R. Chen, X. L. Jiao and Y. L. Wang, Adv. Mater., 2006, 18, 1078–1082 CrossRef CAS.
- T. He, D. Chen, X. Jiao, Y. Xu and Y. Gu, Langmuir, 2004, 20, 8404–8408 CrossRef CAS.
- Y. Zhang, Y. Chen, T. Wang, J. Zhon and Y. Zhou, Microporous Mesoporous Mater., 2008, 114, 257–261 CrossRef CAS.
- H. Yoshikawa, K. Hayashida, Y. Kozuka, A. Horiguchi, K. Awaga, S. Bandow and S. Iijima, Appl. Phys. Lett., 2004, 85, 5287–5289 CrossRef CAS.
- B. Liu and H. C. Zeng, Small, 2005, 1, 566–571 CrossRef CAS.
- T. He, D. R. Chen, X. L. Jiao, Y. Y. Xu and Y. X. Gu, Langmuir, 2004, 20, 8404–8408 CrossRef CAS.
- J. S. Park, X. P. Shen and G. X. Wang, Sens. Actuators, B, 2009, 136, 494–498 CrossRef.
- C. H. Chen, S. F. Abbas, A. Morey, S. Sithambaram, L. P. Xu, H. F. Garces, W. A. Hines and S. L. Suib, Adv. Mater., 2008, 20, 1205–1209 CrossRef CAS.
- Y. C. Chen, L. Hu, M. Wang, Y. L. Min and Y. G. Zhang, Colloids Surf., A, 2009, 336, 64–68 CrossRef CAS.
- Y. C. Chen, Y. G. Zhang and S. Q. Fu, Mater. Lett., 2007, 61, 701–705 CrossRef CAS.
- F. F. Tao, C. L. Gao, Z. H. Wen, Q. Wang, J. H. Li and Z. Xu, J. Solid State Chem., 2009, 182, 1055–1060 CrossRef CAS.
- F. Teng, W. Q. Yao, Y. F. Zheng, Y. T. Ma, T. G. Xu, G. Z. Gao, S. H. Liang, Y. Teng and Y. F. Zhu, Talanta, 2008, 76, 1058–1064 CrossRef CAS.
- W. Zhao, Y. Liu, H. Li and X. Zhang, Mater. Lett., 2008, 62, 772–774 CrossRef CAS.
- Z. G. Guo, W. M. Liu and B. L. Su, Nanotechnology, 2008, 19, 5.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746–749 CrossRef CAS.
- T. V. Choudhary, S. Banerjee and V. R. Choudhary, Appl. Catal., A, 2002, 234, 1–23 CrossRef CAS.
- Q. Liu, L. C. Wang, M. Chen, Y. Cao, H. Y. He and K. N. Fan, J. Catal., 2009, 263, 104–113 CrossRef CAS.
- T. E. Davies, T. Garcia, B. Solsona and S. H. Taylor, Chem. Commun., 2006, 3417–3419 RSC.
- B. Solsona, T. E. Davies, T. Garcia, I. Vazquez, A. Dejoz and S. H. Taylor, Appl. Catal., B, 2008, 84, 176–184 CrossRef CAS.
- D. A. H. Cunningham, T. Kobayashi, N. Kamijo and M. Haruta, Catal. Lett., 1994, 25, 257–264 CrossRef CAS.
- Y. Ren, Z. Ma, L. Qian, S. Dai, H. He and P. G. Bruce, Catal. Lett., 2009, 131, 146–154 CrossRef CAS.
- P. Thormahlen, M. Skoglundh, E. Fridell and B. Andersson, J. Catal., 1999, 188, 300–310 CrossRef CAS.
- H. Tüysüz, M. Comotti and F. Schüth, Chem. Commun., 2008, 4022–4024 RSC.
- J. W. Saalfrank and W. F. Maier, Angew. Chem., Int. Ed., 2004, 43, 2028–2031 CrossRef CAS.
- G. Fortunato, H. R. Oswald and A. Reller, J. Mater. Chem., 2001, 11, 905–911 RSC.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CAS.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Öterlund, P. Thormälen and V. Langer, J. Catal., 2002, 211, 387–397 CrossRef CAS.
- K. Omata, T. Takada, S. Kasahara and M. Yamada, Appl. Catal., A, 1996, 146, 255–267 CrossRef CAS.
- J. P. Beaufils and Y. Barbaux, J. Appl. Crystallogr., 1982, 15, 301–307 CrossRef CAS.
- J. Ziolkowski and Y. Barbaux, J. Mol. Catal., 1991, 67, 199–215 CrossRef CAS.
- P. Broqvist, I. Panas and H. Persson, J. Catal., 2002, 210, 198–206 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS.
- G. A. Somorjai and D. W. Blakely, Nature, 1975, 258, 580–583 CrossRef CAS.
- S. G. Sun, A. C. Chen, T. S. Huang, J. B. Li and Z. W. Tian, J. Electroanal. Chem., 1992, 340, 213–226 CrossRef CAS.
- G. A. Somorjai, Chemistry in Two Dimensions: Surfaces, Cornell University Press, Ithaca, NY, 1981 Search PubMed.
- A. J. Gellman and N. Shukla, Nat. Mater., 2009, 8, 87–88 CrossRef CAS.
- I. Lee, F. Delbecq, R. Morales, M. A. Albiter and F. Zaera, Nat. Mater., 2009, 8, 132–138 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS.
- P. X. Huang, F. Wu, B. L. Zhu, X. P. Gao, H. Y. Zhu, T. Y. Yan, W. P. Huang, S. H. Wu and D. Y. Song, J. Phys. Chem. B, 2005, 109, 19169–19174 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |