Probing differential Ag+–nucleobase interactions with isothermal titration calorimetry (ITC): Towards patterned DNA metallization
Sourabh
Shukla
and
Murali
Sastry†
*
Nanoscience Group, Physical and Materials Chemistry Division, National Chemical Laboratory, Pune, 411 008, India. E-mail: msastry@tatachemicals.com
First published on 13th August 2009
Abstract
DNA has been successfully used as a scaffold for the fabrication of metallic nanowires, primarily based on the electrostatic complexation and reduction of the metal cations on the negatively charged sugar-phosphate backbone. Here, we probe the differential binding affinities of nucleobases for silver ions using sensitive isothermal titration calorimetry (ITC) measurements of the reaction enthalpies, which go in order: C > G > A ≥ T. Using the disparity between the interaction of cytosine (strong binding) and thymine (weak binding) with silver ions, we have successfully generated silver nanoparticle doublets and triplets on custom-made oligonucleotides, C30–T40–C30 and C20–T20–C20–T20–C20, respectively. Thus, a new and simple method of generating metallized DNA wires is presented, based entirely on the nucleotide sequence of DNA. The concept could be extended to other cations and complex DNA sequences in order to achieve intricately patterned DNA constructs.
Introduction
One of the primary objectives in nanotechnology research is miniaturization of future devices. The thrust therefore is on developing methodologies devoted to fabrication of active and passive components of nanoscale dimensions, and currently includes techniques such as nanolithography,1microcontact printing,2 and molecular self-assembly.3 Insofar as integrated circuits are concerned, there is a need to fabricate metallic interconnects for the components in the circuit. Among the current approaches for achieving metallic nanowires, template-mediated nanowire fabrication, wherein an array of nanoparticles is assembled either by chemical or physical means on the desired nanotemplate, has been receiving considerable attention. It is desirable that these templates possess high aspect ratios and structural flexibility. DNA is one such template that meets the above-mentioned criteria and also offers remarkable predictability and structural control through highly specific base-pairing. It has therefore been a popular choice for bottom-up assembly and conducting nanocircuits.4–7 Most strategies for achieving DNA metallization have relied on the binding of preformed metal nanoparticles to double-stranded DNA (dsDNA)7 or by chemically4,5 or photo-catalytically reducing6metal cations electrostatically bound to the negatively charged backbone (due to the phosphate groups). Such methods have been successful in achieving metallized molecular wires,4,5 rings8 and networks.9,10 However, one immediately recognizes that the non-specificity of the Columbic interaction between metal cations and the phosphate backbone leads to metallization along the entire DNA segment with little opportunity to control metallization spatially. To overcome this, sequence-specific DNA bindingproteins and sequence-specific reducing agents11 have been localized on DNA prior to the metallization step. This leads to pattern generation by either limiting or promoting the metallization process on specific loci along the DNA length. Although successful in generating patterned molecular wires on DNA, these procedures require extensive multi-step modifications of the DNA.As mentioned earlier, nitrogenous bases gives DNA an unmatched advantage over other currently available templates for metallization. Besides their role in complementary strand binding, nucleobases also govern processes such as binding of nucleoproteins , metallozymes and drug molecules in a sequence-specific manner,12 and such interactions have been studied extensively by a variety of means. The role of nucleobases in DNA–metal cation interactions has also been a subject of study for a long time. A number of these studies have described specific interactions of nucleobases, nucleosides/nucleotides with metals cations,13 and have suggested that some cations (e.g., cobalt and nickel) show an affinity towards the sugar-phosphate backbone, while others, such as silver, bind preferentially to nucleobases.13 Similarly, there have been theoretical studies on the nature of nucleobase–neutral gold cluster interactions, which explain various bonding factors influencing such complexes.14 However, to the best of our knowledge there has been no attempt to compare the strengths of interactions between nucleobases and a given cation.
Isothermal titration calorimetry (ITC) has recently emerged as a sensitive analytical tool in nanotechnology research,15 in addition to its conventional biological applications.16 In previous work, we used ITC to demonstrate that subtle structural differences between the nitrogenous bases could lead to their surprisingly different abilities to interact with metal nanoparticles.14b Here, we have employed ITC as a tool to compare the disparities in the binding strengths of nucleobases and single-stranded DNA (ssDNA) with Ag+. ITC analysis indicates significant differences between the four nucleobases in their ability to bind silver ions. We believe that these differences could be exploited to bind silver differently on DNA by selecting sequences that would either promote or inhibit the binding, resulting in selective metallization. This hypothesis is proved, in this work, by achieving sequence-dependent generation of silver particles on custom-made single-stranded oligonucleotides.
Results and discussion
ITC measures the heat of reaction for two aqueous solutions when one is titrated against the other, and the amount of heat liberated or used up during the course of a reaction is recorded as the reaction proceeds; the extent of the exothermicity or endothermicity being a function of the strength of interactions. The heat of solvation/dilution can be easily accounted for by performing control additions of equivalent amounts of solutes in the solvent. The ITC measurements for the titration of the four nucleobases against the silver ions are shown in Fig. 1. It is evident from the titration plots that the magnitude of exothermic response (enthalpies) shown by the four bases varies significantly. The extent of exothermicity recorded with the first few injections is a measure of the different strengths of interaction. For example, while cytosine showed the maximum exothermic response, thymine showed the weakest, with guanine and adenine showing intermediate values, the overall order being C > G > A > T. Further, comparative studies between cytosine (the strongest-binding nucleobase) and thymine (the weakest-binding nucleobase) were done in a 30-injection ITC measurement. This experiment also helped to establish the saturation points for the nucleobase–Ag+ interactions. The magnitude of the exothermic response was 44.48 kcal mol−1 for cytosine and 2.8 kcal mol−1 for thymine, at the time of the first injection of the respective base into the Ag+ ions (Figs. 2(a) and (b), respectively). The heat evolved during the subsequent binding events decreased, reaching saturation close to the equimolar ratio of the bases and the silver ions, beyond which there was no significant change in the heat evolved. The saturation point of the ITC curves also indicated that at the equimolar concentration of nucleobases and silver ions, there is complete binding between the two. This was further confirmed by reducing Ag+ in the presence and absence of the nucleobases. It has been previously shown by Sastry and co-workers that Ag+ can be reduced to form metallic silver by tyrosine in presence of KOH in aqueous solution.17 Therefore, in separate experiments, 10−4 moles of cytosine and thymine were added to 10−4 moles of Ag+ followed by the addition of tyrosine in alkaline media. While in the absence of any nucleobases, reduction of Ag+ leads to an intense characteristic surface plasmon resonance band at 420 nm17 (absorbance: 2.41) (Fig. 2(c), curve 1), the absorption was dampened in the presence of both cytosine (absorbance: 0.007) and thymine (absorbance: 0.064) (Fig. 2(c), curves 2 and 3, respectively). This clearly indicates strong binding of the silver ions with the bases, resulting in the formation of highly aggregated silver nanostructures on the addition of the reducing agent. Upon increasing the Ag+ concentration, an increase in the intensity of the 420 nm band was observed for both cytosine (absorbance: 0.188) and thymine (absorbance: 0.709), which still remains significantly less than the control peak, indicating the reduction of excess (unbound) Ag+ only (Fig. 2(c), curves 4 and 5). | ||
| Fig. 1 ITC response recorded by titrating the four nucleobases against Ag+ ions in a 10-injection experiment with a time interval of 5 min between successive injections. | ||
 | ||
| Fig. 2 (a) and (b) show ITC measurements recorded by titrating the nucleobasescytosine and thymine against Ag+, respectively. (c) represents the UV–vis–NIR spectra recorded upon the reduction of Ag+ without the nucleobases (black, curve 1) and in the presence of increasing concentrations of cytosine (red, curves 2 and 4) and thymine (blue, curves 3 and 5). (d) TEM images of silver nanoparticles formed in (i) the absence of any nucleobases, (ii) the presence of thymine, and (iii) the presence of cytosine. The scale bars in the TEM images represent 100 nm. | ||
The TEM images suggest that the strong interaction between cytosine and silver ions results in unusually compact structures upon the subsequent reduction of Ag+ by alkaline tyrosine [Fig. 2(d)(iii)], resulting in the absence of an intense plasmon absorption band at 420 nm. In the presence of the weakly-binding thymine, however, the loosely held Ag+ ions aggregate only slightly upon reduction to silver nanoparticles [Fig. 2(d)(ii)], resulting in a dampened plasmon absorption at 420 nm, as compared to the characteristic sharp plasmon absorption band exhibited by spherical silver nanoparticles in the absence of nucleobases [Fig. 2(d)(i)]. Such aggregation of silver nanoparticles, into clusters of irregular shapes and sizes, has been shown with strongly binding molecules such as pthalazine,18 and is characterized by the dampening of the plasmon absorption band in the UV. Similar dampening of the plasmon absorption band by the aggregation of mannose-capped silver nanoparticles upon addition of concanavalin A has been used as a bioassay .19 Thus, there is a remarkable difference between the nature of the interactions of Ag+ with cytosine and thymine. The stronger binding of silver ions by cytosine has been also documented by Dickson and co-workers who reported, using 1H NMR, that silver nanoclusters caused significant spectral shifts for cytosine compared to other nucleobases.20 This observation was the motivation behind the synthesis of silver nanoclusters using an oligo-cytosine template by the same group.21
The differential binding of Ag+ to the four nucleobases presents some exciting possibilities in terms of exploiting DNA as a template for selective metallization. However, the presence of the negatively charged sugar-phosphate backbone, in addition to the nucleobases, complicates the interaction of any charged moiety with the DNA molecule. In dsDNA, the hydrophobic core, made up of the nucleobases, remains generally inaccessible to any interacting charged species. In contrast, a short ssDNA molecule would offer an exposed sugar-phosphate backbone as well as the nucleobases for the same. Thus, any such interaction is now subject to the competitive binding affinities of the sugar-phosphate backbone and the nucleobases. The strength of binding of the sugar-phosphate backbone to Ag+ was therefore measured, similarly, by titrating the short ssDNA molecules, oligo-cytosine (C24) and oligo-thymine (T24), having similar backbone and dissimilar base composition, against Ag+ ions and measuring the exothermic response. Both ITC measurements showed exothermic responses with sigmoidal binding curves. However, the degree of exothermicity for the two oligos varied considerably. Thus, while the heat liberated for C24 was −139 kcal mol−1, for T24 this value was −25 kcal mol−1 (Figs. 3(a) and (b), respectively). These results are exciting as the two oligonucleotides have the same length and hence, similar charge densities resulting from the sugar-phosphate backbone, the only difference being the base composition. The results clearly illustrate that it is indeed the nucleobases that are interacting with the silver ions as interaction of the Ag+ ions primarily with the sugar-phosphate backbone would have generated the same extent of exothermic response in both cases. These ITC results clearly reinforce the hypothesis that nucleobases, and not the sugar-phosphate backbone, are responsible for silver ion complexation, as established previously by many other groups.13 The results also underline that there is a significant difference between the binding abilities of cytosine and thymine. Reduction of Ag+ in the presence of the two oligomers showed that the extent of binding of the silver ions with T24 and C24 varies, resulting in a less intense plasmon absorption band at 420 nm (Fig. 3(c), blue and red curves, respectively). The plasmon absorption band in the presence of C24 is further dampened compared to T24 (Fig. 3(c), red curve), highlighting the stronger binding ability of C24 over T24. TEM images reveal that the interaction of Ag+ with the strongly binding C24 results in highly aggregated structures [Fig. 3(d)(iii)] in comparison to T24 [Fig. 3(d)(ii)].
 | ||
| Fig. 3 ITC measurements recorded by titrating the short oligonucleotides (a) C24 and (b) T24 against Ag+. (c) UV–vis–NIR spectra recorded upon the reduction of Ag+ without the oligonucleotides (black), and in the presence of T24 (blue) and C24 (red). (d) Representative TEM images of silver ions reduced (i) in absence of nucleobases, (ii) in the presence of thymine, and (iii) in the presence of cytosine. Scale bars in the TEM images correspond to 50 nm. | ||
We propose that these differences in the binding ability of the nucleobases, as free molecules or as a part of the DNA structure, could be exploited for sequence-dependent metallization of DNA molecules leading to the formation of pattered nanowires. As a proof of concept experiment, two custom-made ssDNA molecules (100 bases each) were procured, consisting of cytosine and thymine as the bases but with varying sequences. The first oligonucleotide C30–T40–C30 consisted of two stretches of 30 bases each of strongly-binding cytosine flanking one stretch of 40 thymine bases in the center. The second oligonucleotide, C20–T20–C20–T20–C20, consisted of alternating cytosine and thymine stretches of 20 bases each. In two separate experiments, the oligonucleotides were incubated with silver ions at low temperatures. After an incubation period of 5 h, the solutions were centrifuged at 10![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 rpm for 15 min to remove the unbound/loosely held silver ions. This addition–centrifugation step was repeated four times to gradually build up the Ag+ concentration at the binding loci along the length of the ssDNA. The solutions were then reduced with alkaline tyrosine. TEM images showing the distinct assembly pattern of silver nanoparticles on the two oligonucleotides are shown in Figs. 4 and 5. It is evident from the TEM analysis (Fig. 4) that in the first oligonucleotide sequence (C30–T40–C30, ca. 34 nm length) silver ions are trapped on the two stretches of cytosine and are subsequently reduced to metallic silver by tyrosine in alkaline conditions. As the particles grow in size, the unoccupied thymine stretch at the center is delimited by two particles of ca.15 nm on either side. The average inter-particle separation is observed to be 12.2 ± 1.7 nm, which is consistent with the length of the T40 stretch in the middle (ca. 13.6 nm). The metallized DNA therefore, appears in the form of doublet of nanoparticles separated by a fixed distance (Fig. 4). On the other hand, for the second oligonucleotide sequence (C20–T20–C20–T20–C20, ca. 34 nm) exposure to the reducing agent following the interaction with the silver ions leads to the appearance of short linear arrangements consisting of three silver nanoparticles (Fig. 5). These linear arrangements suggest that silver ions bind to the three C20 regions in the ssDNA and subsequently form silver nanoparticles. The alternating T20 regions do not bind silver nanoparticles strongly and therefore remain non-metallized. The set of three silver nanoparticles are separated by an average distance of 6.04 ± 1.33 nm, which is comparable to the length of alternating T20 stretches (ca. 6.8 nm). Variation in the interparticle separation could result from differences in the particle size on the flanking C20 stretches (Fig. 5). Also, the diameter of the central silver nanoparticle is observed to be smaller in many of the triplets which may result form space constrains, while in certain cases, the particles in the triad seems attached to one another. Scheme 1 depicts the step-wise metallization process for the two oligonuleotides. In both cases, a few free silver nanoparticles can also be seen in the TEM images, which are formed as a result of the reduction of unbound silver ions in the solution by the reducing agents. These TEM images are comparable to those achieved by Alivisatos and co-workers22 where Watson–Crick base pairing was used to acquire different spatial arrangements of preformed gold nanocrystals. Thus, it is evident that deposition of ionic silver and its subsequent reduction to form nanoparticles is dependent on the binding ability of cytosine and thymine in the ssDNA.
000 rpm for 15 min to remove the unbound/loosely held silver ions. This addition–centrifugation step was repeated four times to gradually build up the Ag+ concentration at the binding loci along the length of the ssDNA. The solutions were then reduced with alkaline tyrosine. TEM images showing the distinct assembly pattern of silver nanoparticles on the two oligonucleotides are shown in Figs. 4 and 5. It is evident from the TEM analysis (Fig. 4) that in the first oligonucleotide sequence (C30–T40–C30, ca. 34 nm length) silver ions are trapped on the two stretches of cytosine and are subsequently reduced to metallic silver by tyrosine in alkaline conditions. As the particles grow in size, the unoccupied thymine stretch at the center is delimited by two particles of ca.15 nm on either side. The average inter-particle separation is observed to be 12.2 ± 1.7 nm, which is consistent with the length of the T40 stretch in the middle (ca. 13.6 nm). The metallized DNA therefore, appears in the form of doublet of nanoparticles separated by a fixed distance (Fig. 4). On the other hand, for the second oligonucleotide sequence (C20–T20–C20–T20–C20, ca. 34 nm) exposure to the reducing agent following the interaction with the silver ions leads to the appearance of short linear arrangements consisting of three silver nanoparticles (Fig. 5). These linear arrangements suggest that silver ions bind to the three C20 regions in the ssDNA and subsequently form silver nanoparticles. The alternating T20 regions do not bind silver nanoparticles strongly and therefore remain non-metallized. The set of three silver nanoparticles are separated by an average distance of 6.04 ± 1.33 nm, which is comparable to the length of alternating T20 stretches (ca. 6.8 nm). Variation in the interparticle separation could result from differences in the particle size on the flanking C20 stretches (Fig. 5). Also, the diameter of the central silver nanoparticle is observed to be smaller in many of the triplets which may result form space constrains, while in certain cases, the particles in the triad seems attached to one another. Scheme 1 depicts the step-wise metallization process for the two oligonuleotides. In both cases, a few free silver nanoparticles can also be seen in the TEM images, which are formed as a result of the reduction of unbound silver ions in the solution by the reducing agents. These TEM images are comparable to those achieved by Alivisatos and co-workers22 where Watson–Crick base pairing was used to acquire different spatial arrangements of preformed gold nanocrystals. Thus, it is evident that deposition of ionic silver and its subsequent reduction to form nanoparticles is dependent on the binding ability of cytosine and thymine in the ssDNA.
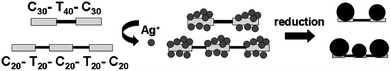 | ||
| Scheme 1 Cartoon depicting the step-wise metallization process resulting in pattered alignment of silver nanoparticles in form of doublets and triplets on the oligonucleotides C30–T40–C30 and C20–T20–C20–T20–C20. | ||
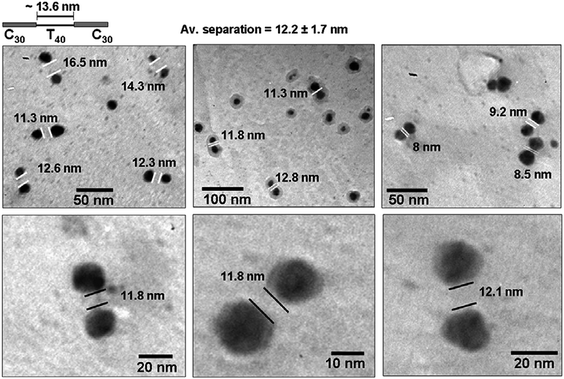 | ||
| Fig. 4 Representative TEM images of the silver nanoparticle doublets assembled on the oligonucleotide C30–T40–C30. | ||
 | ||
| Fig. 5 Representative TEM images of silver nanoparticle triplets assembled on the oligonucleotide C20–T20–C20–T20–C20. | ||
Experimental
Nucleobases and ssDNAs
The four nucleobases, adenine, cytosine, guanine and thymine, Ag2SO4, and tyrosine were obtained from Aldrich and used as received. Short oligonucleotides C24 and T24 were synthesized on a 3900 DNA synthesizer (Applied Biosystems) and their concentrations were determined by measuring the optical density at 260 nm. Custom-made oligonucleotides C30–T40–C30 and C20–T20–C20–T20–C20 were purchased from Invitrogen, US and reconstituted in TE buffer (pH 8.0).ITC measurements
ITC experiments were performed in a Micro-Cal VP-ITC instrument at 4 °C wherein 300 µL of 100 µM aqueous solution of DNA bases and 4 µM aqueous solution of ssDNA (C24, T24) at pH 7 were injected in equal steps of 10 µL into 1.47 mL of 100 µM Ag2SO4 solution.UV–Vis–NIR spectroscopy
UV–Vis–NIR spectra for all solutions were recorded on a Jasco V-570 UV–Vis–NIR spectrophotometer operated at a resolution of 1 nm.TEM analysis
Samples for TEM analysis were prepared by drop-coating the solutions on carbon coated copper grids. TEM measurements were performed on a JEOL model 1200EX instrument operated at an accelerating voltage of 80 kV.DNA metallization
For the metallization of custom-made oligos, 50 µM of the each oligonucleotide was mixed with 50 µM silver ions and incubated at 4 °C for 8 h. After the incubations, excess silver ions were removed by centrifuging the samples at 10000 rpm at 4 °C for 20 min. This cycle of silver ion addition and removal was repeated several times to gradually build up the local silver ion concentrations at binding sites. The reduction of silver ions was then brought about by addition of 5 × 10−5 M tyrosine and adjusting the pH of the samples to ∼10.
Conclusions
This study reveals the comparative strength of Ag+–nucleobase interactions using ITC. The different affinities of nitrogenous bases towards Ag+ are proved by the varying degree of aggregation of silver nanoparticles in the presence of the nucleobases, cytosine and thymine. The preferential binding of Ag+ to the nucleobases over the sugar-phosphate backbone of DNA is evident for ssDNA (T24 and C24) where the differences in binding strengths are retained, in the presence of similar backbones. It was therefore envisaged that one could control the metallization of DNA templates by varying the nucleotide sequence and promoting or limiting metal-ion deposition at specific loci. As a proof of concept, we have demonstrated controlled nanoparticle alignment on short oligoncleotides consisting of alternating silver-binding (oligo-cytosine) and nonbinding (oligo-thymine) stretches. Further studies in this field, involving different ionic species and more intricately designed DNA sequences, could therefore be used to develop patterned metal deposition on DNA scaffolds.Acknowledgements
SS thanks the Council of Scientific and Industrial Research (CSIR), Govt. of India, for financial assistance. We wish to thank Mr. A. Gaurishankar for helping with ITC measurements.References
- Z. Deng and C. Mao, Angew. Chem., Int. Ed., 2004, 43, 4068 CrossRef CAS.
- (a) C. Xu, P. Taylor, M. Ersoz, P. D. Fletcher and V. N. J. Paunov, J. Mater. Chem., 2003, 13, 3044 RSC; (b) C. Thibault, V. L. Berre, S. Casimirius, E. Trévisiol, J. François and C. Vieu, J. Nanobiotechnol., 2005, 3, 7 CrossRef CAS; (c) S. A. Lange, V. Benes, D. P. Kern, J. K. H. Horber and A. Bernard, Anal. Chem., 2004, 76, 1641 CrossRef CAS.
- Y. Ma, J. Zhang, G. Zhang and H. He, J. Am. Chem. Soc., 2004, 126, 7097 CrossRef CAS.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775 CrossRef CAS.
- C. F. Monson and A. T. Woolley, Nano Lett., 2003, 3, 359 CrossRef CAS.
- L. Berti, A. Alessandrni and P. Facci, J. Am. Chem. Soc., 2005, 127, 11216 CrossRef CAS.
- (a) M. Mertig, L. C. Ciacchi, R. Seidel and W. Pompe, Nano Lett., 2002, 2, 841 CrossRef CAS; (b) H. Nakao, H. Shiigi, Y. Yamamoto, S. Tokonami, T. Nagaoka, S. Sugiyama and T. Ohtani, Nano Lett., 2003, 3, 1391 CrossRef CAS.
- A. A. Zinchenko, K. Yoshikawa and D. Baigl, Adv. Mater., 2005, 17, 2820 CrossRef CAS.
- G. Wei, L. Wang, Z. Liu, Y. Song, L. Sun, T. Yang and Z. Li, J. Phys. Chem. B, 2005, 109, 23941 CrossRef CAS.
- G. Wei, H. Zhou, Z. Liu, Y. Song, L. Wang, L. Sun and Z. Li, J. Phys. Chem. B, 2005, 109, 8738 CrossRef CAS.
- (a) K. Keren, M. Krueger, R. Gilad, G. B. Yoseph, U. Sivan and E. Braun, Science, 2002, 297, 72 CrossRef CAS; (b) K. Keren, R. S. Berman and E. Braun, Nano Lett., 2004, 4, 323 CrossRef CAS; (c) G. A. Burley, J. Gierlich, M. R. Mofid, H. Nir, S. Tal, Y. Eichen and T. Carell, J. Am. Chem. Soc., 2006, 128, 1398 CrossRef CAS.
- (a) M. B. Renfrow, N. Naryshkin, L. M. Lewis, H. T. Chen, R. H. Ebright and R. A. Scott, J. Biol. Chem., 2003, 279, 2825 CrossRef; (b) Y. Lu, Inorg. Chem., 2006, 45, 9930 CrossRef CAS; (c) J. J. Welch, F. J. Rauscher III and T. A. Beerman, J. Biol. Chem., 1994, 269, 31051 CAS.
- (a) K. F. S. Luk, A. H. Maki and R. J. Hoover, J. Am. Chem. Soc., 1975, 97, 1241 CrossRef CAS; (b) R. H. Jensen and N. Davidson, Biopolymers, 1966, 4, 17 CrossRef CAS; (c) T. Yamane and N. Davidson, Biochim. Biophys. Acta, 1962, 55, 609 CAS; (d) Z. Hossain and F. Huq, J. Inorg. Biochem., 2002, 91, 398 CrossRef CAS; (e) R. M. Izatt, J. J. Christensen and J. H. Rytting, Chem. Rev., 1971, 71, 439 CrossRef CAS.
- (a) E. S. Kryachko and F. Remacle, J. Phys. Chem. B, 2005, 109, 22746 CrossRef CAS; (b) E. S. Kryachko and F. Remacle, Nano Lett., 2005, 5, 735 CrossRef.
- (a) H. M. Joshi, P. S. Shirude, V. Bansal, K. N. Ganesh and M. Sastry, J. Phys. Chem. B, 2004, 108, 11535 CrossRef CAS; (b) A. Gourishankar, S. Shukla, K. N. Ganesh and M. Sastry, J. Am. Chem. Soc., 2004, 126, 13186 CrossRef CAS.
- (a) M. L. Bianconi, J. Biol. Chem., 2003, 278, 18709 CrossRef CAS; (b) A. Künne, M. Sieber, D. Meierhans and R. K. Allemann, Biochemistry, 1998, 37, 4217 CrossRef CAS.
- P. R. Selvakannan, A. Swami, D. Srisathiyanarayanan, P. S. Shirude, R. Pasricha, A. B. Mandale and M. Sastry, Langmuir, 2004, 20, 7825 CrossRef CAS.
- M. Moskovits and B. Vlckova, J. Phys. Chem. B, 2005, 109, 14755 CrossRef CAS.
- (a) C. L. Schofield, A. H. Haines, R. A. Field and D. A. Russell, Langmuir, 2006, 22, 6707 CrossRef CAS; (b) J. Zhang, D. Roll, C. D. Geddes and J. R. Lakowicz, J. Phys. Chem. B, 2004, 108, 12210 CrossRef CAS.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207 CrossRef CAS.
- C. M. Ritchie, K. R. Johnsen, J. R. Kiser, Y. Antoku, R. M. Dickson and J. T. Petty, J. Phys. Chem. C, 2007, 111, 175 CrossRef CAS.
- C. J. Loweth, W. B. Caldwell, X. Peng, A. P. Alivisatos and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 1808 CrossRef CAS.
Footnote |
| † Present address: Tata Chemicals Innovation Centre, Baner Road, Pune 411 045, India. |
| This journal is © The Royal Society of Chemistry 2009 |