The role of nucleophilic catalysis in chemistry and stereochemistry of ribonucleosideH-phosphonate condensation†
Michal
Sobkowski
*a,
Jacek
Stawinski
b and
Adam
Kraszewski
a
aInstitute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704, Poznan, Poland. E-mail: msob@ibch.poznan.pl; Fax: +48 61 8520 532; Tel: +48 61 852 8503
bDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, S-106 91, Stockholm, Sweden
First published on 29th October 2008
Abstract
The efficiency and stereoselectivity of condensation of ribonucleoside 3′-H-phosphonates with alcohols were investigated as a function of amines used for the reaction. It was found that irrespective of the presence or absence of nucleophilic catalysts, the Dynamic Kinetic Asymmetric Transformation (DYKAT) was the major factor responsible for the stereoselective formation of the DP(SP) isomers of the H-phosphonate diesters, and a mechanistic rationalization of this observation was proposed. In addition, studies on the reactions carried out in the presence of various bases led to the conclusion that certain sterically hindered pyridines, e.g.2,6-lutidine, may act as nucleophilic catalysts in the condensation of ribonucleoside 3′-H-phosphonates with alcohols.
Introduction
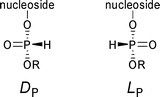
P-Chiral oligonucleotide analogues (e.g. phosphorothioates,2 phosphoramidates,3 methylphosphonates,4 or boranophosphates5) having defined configuration at the phosphorus atom find diverse applications in investigations of nucleic acid interactions with other biologically important molecules, for example proteins, RNA, and DNA.6 Such P-chiral oligonucleotides may also be considered as potential drugs for nucleic acid-based therapies,7 that could permit a more precise tuning of oligonucleotide interactions with the biological targets than is possible with the currently used pools of P-diastereomers. This should also relieve problems of potential variation of therapeutic and toxic effects resulting from different ratios of P-diastereomers produced in various batches of oligonucleotide drugs.There are several strategies to stereocontrolled synthesis of P-chiral oligonucleotides.8 One of them, stereoselective (or more precisely, diastereoselective) condensation of ribonucleosideH-phosphonates,9 attracted our attention due to its simplicity and high efficiency. It makes use of commercially available H-phosphonate synthons which are condensed with nucleosides under standard reaction conditions commonly used for the synthesis of H-phosphonate diesters to provide DP diastereomers‡ as major products.
Recently, we have proposed a Dynamic Kinetic Asymmetric Transformation (DYKAT) as a possible mechanism for the stereoselectivity observed in these reactions.1 According to this model, diastereomers of nucleosideH-phosphonic–pivalic mixed anhydrides 2 exist in a rapid equilibrium, and one of them, namely the LP(SP) diastereomer, is significantly more reactive towards nucleosides (or alcohols) than the other one (Fig. 1 and Chart 1). To simplify mechanistic considerations, in our earlier studies the role of nucleophilic and base catalysis by the amines was consciously neglected. However, since the participation of nucleophilic catalysis in condensation of H-phosphonates is a well-established phenomenon,11–15 it was important to examine and to assess its impact on the asymmetric induction in the reactions investigated. In this paper we present studies on the role of nucleophilic catalysis in the chemistry and stereochemistry of condensation of ribonucleosideH-phosphonates with alcohols.
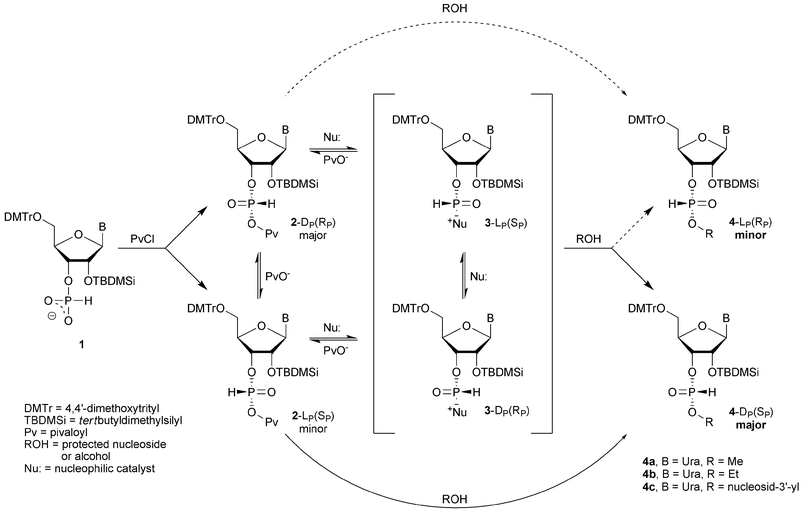 | ||
| Fig. 1 Putative routes of the reaction during stereoselective condensation of ribonucleosideH-phosphonate monoester 1 with alcohols and nucleosides according to the DYKAT mechanism in the absence (curved arrows) and in the presence (central pathways) of a nucleophilic catalyst. | ||
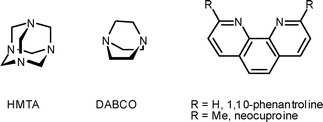 | ||
| Chart 1 | ||
Results and discussion
In routine condensations of nucleosideH-phosphonates pyridine or quinoline (either neat or diluted with non-basic solvent) is used as a basic component of the reaction mixture. Both of these weakly basic heterocyclic amines (pKa 5.2 and 4.9, respectively) secure fast and quantitative formation of H-phosphonate diesters due to their ability to act as nucleophilic catalysts.11,12 In contrast to this, in the presence of more powerful nucleophilic catalysts, e.g.NMI or DMAP,§nucleosideH-phosphonates are prone to P-acylation that compromises the diester formation.12 Also strongly basic tertiary amines (e.g.TEA, pKa 11.0) are usually avoided since these can promote undesired base-catalysed bis-acylation of H-phosphonate monoesters,13,15 while in the presence of less basic tertiary amines (e.g. DMA, pKa 5.1) the condensations are effective but sluggish (at least 10 times slower than those for pyridine), presumably due to lack of nucleophilic catalysis.12 Thus, it was somewhat surprising that 2,6-lutidine (pKa 6.7), which is usually considered as poorly nucleophilic base,16,17 promoted condensations of ribonucleosideH-phosphonates with similar efficiency as pyridine or quinoline.1 Moreover, the stereochemistry of the reactions performed in the presence of 2,6-lutidine was the same as that with pyridine. The above called into question the commonly accepted non-nucleophilic character of 2,6-lutidine and prompted us to consider the involvement of nucleophilic catalysis as an additional process in the DYKAT mechanism (Fig. 1).Since the involvement of P–N+ adducts of type 3 (Fig. 1) in ribonucleosideH-phosphonate diester formation has to be crucial for the rate of condensation as well as for stereochemical outcome of the reaction (Fig. 2), we undertook investigations to pinpoint the cases in which amines acted solely as base catalysts or as base and nucleophilic catalysts during H-phosphonate condensations. To this end, the reactions of H-phosphonate 1 with ethanol were carried out in the presence of selected tertiary amines, various pyridine derivatives, and strong nucleophilic catalysts. The obtained data (Table 1) showed that irrespective of significant differences in the yields and stereoselectivity observed for different amines, the same DP(SP) diastereomer of diester 4 was always formed as the main product. In the light of our earlier studies,1,18 these results might suggest that in the absence of nucleophilic catalysts, the previously described DYKAT mechanism operated at the level of the mixed anhydride 2 (Fig. 2, Path B), while in the nucleophile-catalyzed reactions, an analogous DYKAT took place at the level of adducts of type 3 (Fig. 2, Path A2).¶
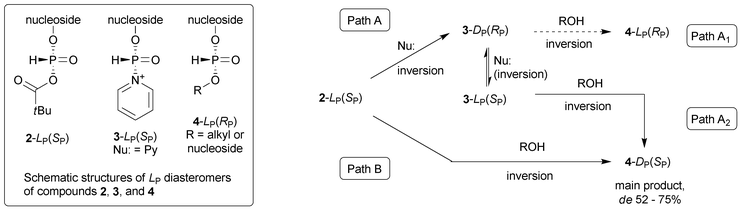 | ||
| Fig. 2 Possible stereochemistry of esterification of the more reactive LP(SP) diastereomer of ribonucleosideH-phosphonic—pivalic mixed anhydride 2 in the presence (Path A) and absence (Path B) of nucleophilic catalysts. | ||
| Entry | Amine | pKa (H2O)a | pKa (DMSO) | pKa (ACN) | pKHBb | dec,d (DP) | Yield of diester (%)d |
|---|---|---|---|---|---|---|---|
| a Aqueous pKa data, unless otherwise indicated, are taken from ref. 37. b Hydrogen bonding basicity. pKHB = logK(formation of HB complex); larger values correspond to greater basicity.38 c One should note that the difference between de values, for instance de 52% and de 75%, corresponds to over two-fold increase of the stereoselectivity measured as a ratio of diastereomers (i.e.∼3 : 1 vs.∼7 : 1, respectively). d Determined via integration of the corresponding 31P NMR signals. e For structure, see Chart 1. f Estimated, assuming an additive and similar methyl and ethyl groups effect on the pKa39 and a linear correlation between α,α′-steric hindrance and pKa40 of substituted pyridines. | |||||||
| Strong nucleophilic catalysts | |||||||
| 1 | MPO | 2.119 | 3.520 | 12.421 | 62% | 27 | |
| 2 | HMTA e | 5.2 | 1.922 | 57% | 66 | ||
| 3 | NMI | 7.0 | 14.323 | 2.724 | 60% | 84 | |
| 4 | DABCO e | 8.7 | 8.925 | 18.326 | 2.622 | 53% | 42 |
| 5 | DMAP | 9.7 | 7.927 | 17.728 | 2.829 | 53% | 85 |
| Heteroaromatic amines | |||||||
| 6 | Pyrazine | 0.7 | 1.229 | 39% | 55 | ||
| 7 | Pyrimidine | 1.2 | 1.429 | 39% | 70 | ||
| 8 | Tetramethylpyrazine | 3.6 | 59% | 100 | |||
| 9 | Quinoline | 4.9 | 12.028 | 1.929 | 63% | 100 | |
| 10 | 1,10-Phenanthroline e | 4.9 | 66% | 100 | |||
| 11 | 2,6-Di-tert-butyl-pyridine | 5.030 | 1.031 | 47% | 70 | ||
| 12 | Pyridine | 5.2 | 3.227 | 12.528 | 1.929 | 62% | 100 |
| 13 | 2-Picoline | 5.9 | 4.027 | 13.932 | 2.029 | 69% | 100 |
| 14 | 4-Picoline | 6.0 | 3.827 | 14.532 | 2.129 | 68% | 100 |
| 15 | Neocuproine e | 6.2 | 64% | 100 | |||
| 16 | 2,5-Lutidine | 6.4 | 68% | 100 | |||
| 17 | 3,4-Lutidine | 6.5 | 4.327 | 14.732 | 2.229 | 63% | 100 |
| 18 | 2,6-Lutidine | 6.7 | 4.427 | 14.432 | 2.129 | 70% | 100 |
| 19 | 2,4-Lutidine | 6.7 | 4.527 | 15.032 | 70% | 100 | |
| 20 | 2,4,6-Collidine | 7.5 | 15.028 | 2.329 | 68% | 100 | |
| 21 | EDIPP | (7.6)f | 52% | 92 | |||
| 22 | (−)-Nicotine | 8.0 | 65% | 100 | |||
| 23 | (±)-Nicotine | 8.0 | 64% | 100 | |||
| Tertiary amines | |||||||
| 24 | DMA | 5.1 | 2.533 | 11.428 | 0.534 | 56% | 100 |
| 25 | N-Methylmorpholine | 7.4 | 15.635 | 1.722 | 70% | 91 | |
| 26 | TEA | 11.0 | 9.036 | 18.828 | 2.022 | 75% | 74 |
| 27 | DIPEA | 11.4 | 1.122 | 71% | 89 | ||
Additionally, these experiments confirmed the earlier findings11–15 that neither powerful nucleophilic catalysts nor strongly basic tertiary amines could promote quantitative condensations of H-phosphonates with alcohols. However, in contrast to the literature data,11–15 there was no (or very little) side product formation, and the 31P NMR spectra of the reaction mixtures revealed only presence of the expected diester 4 and unreacted monoester 1 (Fig. 3). This lack of by-product formation was tentatively attributed to low concentration of the amines in the reaction mixtures (0.3 M or ca. 2.5%).
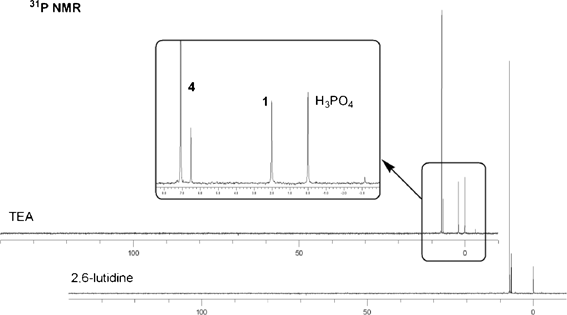 | ||
| Fig. 3 31P NMR spectra of the reaction of H-phosphonate 1 with ethanol (3 equiv.) promoted by PvCl (1.5 equiv.) in DCM containing 3 equiv. of 2,6-lutidine or TEA. The minor signal (ca. 1.5%) at −3.2 ppm in the upper spectrum is in the region of P-acylated compounds. | ||
In order to find sources for the incomplete condensations that have been carried out in the presence of the amines examined herein, the reactivity of pivaloyl chloride towards nucleosides was investigated in separate experiments. It was found that TEA and pyridine derivatives when used alone did not promote significant acylation of nucleosides, however, in the presence of strong nucleophilic amines (e.g.DMAP) or TEA–pyridine mixtures, pivaloyl chloride was rapidly consumed in the acylation of 5′-OH or N3-H functions of uridine.41 These side reactions could compete with the formation of the mixed anhydride 2 and, at least partly, could be responsible for incomplete condensations. However, H-phosphonate condensations were also not quantitative in the presence of tertiary aliphatic amines alone (Table 1, entries 25–27), i.e. under the conditions in which the acylation of nucleoside components was negligible.41 This issue was addressed in additional experiments, which indicated that the mixed anhydride 2 might undergo deacylation by pivalic acid with regeneration of the starting H-phosphonate monoester 1 and formation of pivalic anhydride (a poor activator of H-phosphonates42). The rate of deacylation was found to correlate well with the ability of an amine conjugated acid to form hydrogen bonds (quantified as a pKHB value||) rather than with the amine basicity expressed by pKa. A plausible rationale, which could account for the obtained results involved an increased contribution of general acid catalysis during decomposition of the mixed anhydride 2 by amines having high pKHB (Fig. 4).43
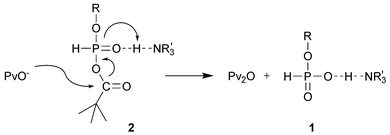 | ||
| Fig. 4 A possible participation of general acid catalysis in deacylation of the mixed anhydride 2 by pivalic acid. | ||
Thus, it can be tentatively concluded that pivaloyl chloride promoted coupling of H-phosphonates with alcohols in the presence of strongly nucleophilic amines, and/or those of high H-bonding basicity, did not go to completion due to consumption of the condensing agent (PvCl) in the acylation of nucleosides or due to formation of unreactive pivalic anhydridevia a partial deacylation of the mixed anhydride 2.
In contrast, pyridine and most of its derivatives investigated secured quantitative condensations (with an exception of pyridine derivatives bearing branched substituents in both α positions)** despite significant differences in their pKa (3.6–8.0) and considerably high pKHB values (1.9–2.3). Although it might be argued that the high pKHB of pyridines should be associated with high catalytic activity of their conjugate acids which should lead to deacylation of the mixed anhydride 2 (Fig. 4), apparently it was not the case in the reactions discussed. A plausible explanation of the excellent yields obtained for the most of the pyridines examined could be the participation of nucleophilic catalysis, i.e. the involvement of intermediate phosphonopyridinium adducts of type 3 (Fig. 2) which, as monofunctional entities, should undergo a nucleophilic attack at the phosphorus centre only. While this is readily understandable in the case of pyridine derivatives with unhindered endocyclic nitrogen atoms (i.e. having at least one α position unsubstituted), the question might arise, whether this could hold also for α,α′-dimethylpyridines?
Although 2,6-lutidine and its derivatives are usually considered as poor nucleophiles,16,17 they can act as nucleophiles under mild conditions undergoing, for instance, N-alkylation with alkyl halides,44 alkyl iodonium triflate45 or radical cations,46 or N-sulfonation with triflic anhydride.47 Notably, in phosphorus chemistry the lack of nucleophilic properties of 2,6-lutidine was observed for P(V) compounds, e.g. for phosphoroiodidates,16 while whether nucleophilic catalysis by this base may operate for H-phosphonates, remains to be determined. Since P(V) and P(III) compounds differ significantly in electrophilicity,48 and the steric hindrance around the phosphorus atom in H-phosphonates is clearly lower than that in P(V) compounds, significant differences in their reactivity towards hindered pyridine derivatives cannot be excluded.
To get a better insight into this problem, condensations of H-phosphonate 1 were performed in the presence of EDIPP (a peralkylated 2,6-diisopropylpyridine derivative, pKa∼7.6)49 and DTBP (2,6-di-tert-butylpyridine, pKa 5.0) for which the nucleophilicity might be safely excluded on steric grounds. The yields of H-phosphonate diester 4 obtained in these reactions (92 and 70%) were similar to those found for trialkyl amines, while low stereoselectivity (de ca. 50%) was similar to that observed for tertiary aniline derivatives (e.g. DMA, de 56%). In contrast, all the other pyridine derivatives, including α,α′-dimethylpyridines, differed only slightly in stereoselectivity and invariably gave quantitative condensations of H-phosphonate 1. Thus, it seems reasonable to assume that the main route for H-phosphonate diester formation in the presence of 2,6-lutidine derivatives could still involve the nucleophilic catalysis (preventing in this way deacylation of the mixed anhydride 2, and in consequence, the yield deterioration), and that only bulky alkyl substituents in α,α′ positions were able to suppress the nucleophilic properties of pyridine.
In additional experiments the condensations of uridineH-phosphonate 1 with ethanol performed in the presence of mixtures of 2,6-lutidine with more nucleophilic amines (pyridine, NMI, DMAP, MPO) were investigated (Fig. 5). In neither case were any specific effects due to the nucleophilic amine noted, and the yields and stereoselectivity of the condensations were proportional to the weighted average of the values obtained for each amine used separately. This lent support to the aforementioned assumption that the same mechanism (i.e. nucleophilic catalysis) was operating for 2,6-lutidine and for other amines of known nucleophilic character.
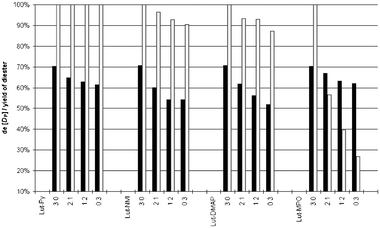 | ||
| Fig. 5 Diastereomeric excess (solid bars) of the DP(SP) diastereomer of H-phosphonate diester 4b formed in the presence of mixtures of amines, and the total yield of diester 4b (a sum of diastereomers, open bars). Reaction conditions: 0.05 mmol of 1 (B = Ura) + EtOH (3 equiv.) + amines (the number of molar equivalents specified on the x axis) + PvCl (1.5 equiv.) in DCM (0.5 mL). | ||
Kinetic quenching experiments
To probe the involvement of nucleophilic catalysis in the DYKAT mechanism, kinetic quenching experiments for various amines were carried out using large excess of methanol. Under such reaction conditions we expected to observe significant changes in the ratio of diastereomers (with a possible reversal of stereoselectivity1) of the produced H-phosphonate diester 4a as a result of substantial increase in the rate of esterification of the reactive intermediates (mixed anhydride 2 and amine adduct 3). Indeed, a remarkable decrease in stereoselectivity was observed for the reactions involving pyridine or 2,6-lutidine as bases (Table 2). Such results can be interpreted as a partial change of the DYKAT into the Dynamic Thermodynamic Resolution (DYTR) mechanism of the asymmetric induction due to acceleration of the esterification at high concentration of MeOH.1 In contrast, in the presence of the poorly nucleophilic amines, the stereoselectivity under the kinetic quenching conditions decreased only slightly.| Entry | Amine | “Standard” 3 equiv. of MeOH 3 equiv. of amine [1] = 100 mM | “Kinetic quenching” 2500 equiv. of MeOH, 30 equiv. of amine [1] = 10 mM | ||
|---|---|---|---|---|---|
| de (DP)a | Yield of diester (%)a | de (DP)a | Yield of diester (%)a | ||
| a Determined via integration of the corresponding 31P NMR signals. b Advantage of the LP diastereomer. c 3 + 3 equiv. or 15 + 15 equiv. | |||||
| 1 | Pyridine | 63 | 100 | −10b | 100 |
| 2 | 2,6-Lutidine | 68 | 100 | 12 | 100 |
| 3 | EDIPP | 47 | 100 | 66 | 100 |
| 4 | DMA | 56 | 96 | 42 | 100 |
| 5 | TEA | 69 | 73 | 51 | 93 |
| 6 | Proton sponge | 49 | 95 | 45 | 95 |
| 7 | Pyridine + TEA 1:1c | 62 | 89 | 60 | 88 |
| 8 | 2,6-Lutidine + TEA 1:1c | 68 | 80 | 61 | 91 |
| 9 | DMA + TEA 1:1c | 60 | 84 | 58 | 94 |
Thus, it seems that the nucleophilic catalysis (Table 2, entries 1 & 2) speeded up the esterification of intermediates 3 more efficiently than their epimerization, while for the base catalysed reactions (entries 3–6 & 9) or in the presence of highly basic amines (e.g.TEA, pKa 11.0; entries 7 & 8), the rate of epimerization was always significantly higher than that of esterification, even in the presence of large excess of an alcohol. Interestingly, it seems that the behaviour of a given amine in a kinetic quenching experiment might be exploited as a marker of its nucleophilic properties towards H-phosphonates, according to the following rule of thumb: the higher the stereoselectivity of ribonucleosideH-phosphonate condensation in neat methanol, the lower the nucleophilicity of the amine used for the reaction.
Conclusions
In the previous paper in this series we reported that stereoselectivity in condensations of ribonucleosideH-phosphonates 1 with alcohols originated from the Dynamic Kinetic Asymmetric Transformation (DYKAT).1 The data presented in this paper confirmed this conclusion and suggested that the equilibrium between the diastereomers of nucleosideH-phosphonic—pivalic mixed anhydride (2-DP⇌2-LP) was significant for the stereochemical outcome of the reaction only in the absence of nucleophilic catalysis. In the presence of nucleophilic amines, however, the DYKAT mechanism was governed most likely by the 3-DP⇌3-LP equilibrium between the putative P–N+ intermediates. In most instances this path was also essential for quantitative yield of the condensation.Pyridine derivatives (excluding those with a large steric hindrance around the nitrogen atom) secured practically quantitative yields of the condensations along with reasonable high stereoselectivity (de 60–70%). Noteworthy, pyridine derivatives with methyl groups in the α positions (e.g.2,6-lutidine) also provided fast, clean and highly stereoselective condensations, and thus indicated that the esterification of H-phosphonate monoesters in the presence of these bases might proceed with the intermediacy of the P–N+ adducts of type 3 (i.e. involving nucleophilic catalysis; Fig. 1 and 2). To the best of our knowledge this would be the first documented example of manifestation of nucleophilic properties of 2,6-dimethylpyridines in SN2(P) reactions.
For practical purposes, among investigated bases, 2,6-lutidine was found to be the amine of choice (quantitative yield of condensations, high stereoselectivity, and easy availability) and is advised to be used in stereoselective ribonucleoside 3′-H-phosphonate diesters formation.
Experimental section
Methods and materials
31P NMR spectra were recorded at 121 MHz on a Varian Unity BB VT spectrometer. 31P NMR experiments were carried out in 5 mm tubes using 0.5 mL of the reaction mixture and the spectra were referenced to 2% H3PO4 in D2O (external standard). The quantities of phosphorus-containing compounds were determined via integration of the corresponding 31P NMR signals. Diastereomeric excess was calculated with accuracy of ±1.5 percentage points (an average of 3 measurements).Commercial (Sigma-Aldrich, Alfa Aesar, Merck, POCh-Poland) reagents and were used as purchased unless otherwise noted. Ethanol was distilled over magnesium. Dichloromethane and pyridine were refluxed over P2O5, distilled, and stored over 4 Å molecular sieves until they contained below 20 ppm of water (Karl Fischer coulometric titration, Metrohm 684 KF coulometer). Anhydrous triethylamine (TEA) was distilled and kept over CaH2. Commercial p-methoxypyridine N-oxide (MPO) hydrate was rendered anhydrous by co-evaporation with dry acetonitrile (1×) and dry toluene (2×). Other liquid amines were refluxed for 1 hour with 2,4,6-triisopropylbenzenesulfonyl chloride (TPS-Cl) and distilled under reduced pressure. 4-Ethyl-2,6-diisopropyl-3,5-dimethylpyridine (EDIPP)49 was a gift from Prof. A. T. Balaban, Texas A&M University. UridineH-phosphonate 150 was obtained according to the published method. Racemization of S-(−)-nicotine was done according to ref. 51. Immediately prior to all reactions, H-phosphonate 1 was rendered anhydrous by dissolving in toluene (3 mL/0.05 mmol) and evaporation of this solvent under reduced pressure. After drying under vacuum (15 min, 0.5 Torr), the flask was filled with air, dried up by passing through Sicapent (Merck).
General procedure for condensation of H-phosphonates of type 1 with alcohols
Nucleoside H-phosphonate 1 (0.05 mmol) was dissolved in 0.5 mL of DCM and amine (3 equiv.) and EtOH or MeOH (3 equiv.) were added, followed by PvCl (1.5 equiv.). The reaction mixture was transferred to an NMR tube and the 31P NMR spectra were recorded within 1 hour.Kinetic quenching experiments
Nucleoside H-phosphonate 1 (0.05 mmol) was dissolved in DCM (0.3–0.5 mL), and TEA (0.2 equiv.) and PvCl (1.2 equiv.) were added successively. The formation of the mixed anhydride 2 was confirmed by 31P NMR spectroscopy (δP 1.47 & 1.56) and the reaction mixture was utilized within one hour (no degradation products were found within that period).The solution (0.3 mL) of the intermediate 2 (0.5 mmol; generated as described above) was added dropwise by a syringe to a septum-sealed flask containing vigorously stirred methanol (5 mL) and an amine (2.4% v/v or w/v). After ca. 30 s toluene (15 mL) was added and the mixture was evaporated almost to dryness under vacuum at temperature <40 °C (such procedure was obligatory for strongly basic tertiary amines in order to avoid transesterification52 of the product). The oily residue was dissolved in DCM (0.5 mL) and analyzed by 31P NMR spectroscopy.
Acknowledgements
The financial support from the Polish Ministry of Science and Higher Education is gratefully acknowledged.References
- M. Sobkowski, A. Kraszewski and J. Stawinski, Tetrahedron: Asymmetry, 2007, 18, 2336–2348 CrossRef CAS.
- W. J. Stec and G. Zon, Tetrahedron Lett., 1984, 25, 5275–5278 CrossRef CAS; W. B. Cruse, S. A. Salisbury, T. Brown, R. Cosstick, F. Eckstein and O. Kennard, J. Mol. Biol., 1986, 192, 891–905 CrossRef CAS.
- J. Nilsson and J. Stawinski, Chem. Commun., 2004, 2566–2567 RSC.
- L. A. Wozniak, M. Janicka and M. Bukowiecka-Matusiak, Eur. J. Org. Chem., 2005, 5189–5197 CrossRef CAS.
- J. Tomasz, B. R. Shaw, K. Porter, B. F. Spielvogel and A. Sood, Angew. Chem., Int. Ed., 1992, 31, 1373–1375 CrossRef; Y. Jin and G. Just, Tetrahedron Lett., 1998, 39, 6433–6436 CrossRef CAS.
- P. Guga, Curr. Top. Med. Chem., 2007, 7, 695–713 CrossRef CAS.
- J. B. Opalinska and A. M. Gewirtz, Nat. Rev. Drug Discovery, 2002, 1, 503–514 CrossRef CAS.
- Y. X. Lu, Mini-Rev. Med. Chem., 2006, 6, 319–330 CrossRef CAS.
- H. Almer, J. Stawinski and R. Stromberg, Nucleic Acids Res., 1996, 24, 3811–3820 CrossRef CAS.
- M. Sobkowski, J. Stawinski and A. Kraszewski, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 1301–1307 CrossRef CAS; M. Sobkowski, J. Stawinski and A. Kraszewski, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 1363–1375 CrossRef CAS; M. Sobkowski, J. Stawinski and A. Kraszewski, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 1377–1389 CrossRef CAS; M. Sobkowski, J. Stawinski and A. Kraszewski, ‘A Convenient Stereochemical Notation for P-Chiral Nucleotide Analogs’, in Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley & Sons, New York, 2007, A.1E.1–A.1E.16 Search PubMed.
- V. A. Efimov and I. Y. Dubey, Bioorg. Khim., 1990, 16, 211–218 Search PubMed; P. J. Garegg, J. Stawinski and R. Stromberg, J. Chem. Soc., Perkin Trans. 2, 1987, 1209–1214 RSC.
- I. Y. Dubey, V. A. Efimov, T. V. Lyapina and D. M. Fedoryak, Bioorg. Khim., 1992, 18, 911–919 Search PubMed; V. A. Efimov, I. Y. Dubey and O. G. Chakhmakhcheva, Nucleosides Nucleotides, 1990, 9, 473–477.
- S. Sigurdsson and R. Stromberg, J. Chem. Soc., Perkin Trans. 2, 2002, 1682–1688 RSC; S. Sigurdsson and R. Stromberg, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1–12 CrossRef CAS.
- A. Kraszewski and J. Stawinski, ‘Aryl Nucleoside H-Phosphonates. Versatile Intermediates in the Synthesis of Nucleotides and Their Analogues’, in Trends in Organic Chemistry, Research Trends, Trivandrum India, 2003, vol. 10, pp. 1–19 Search PubMed.
- P. J. Garegg, T. Regberg, J. Stawinski and R. Stromberg, Nucleosides Nucleotides, 1987, 6, 655–662 CAS.
- J. Stawinski, R. Stromberg and R. Zain, Tetrahedron Lett., 1992, 33, 3185–3188 CrossRef CAS.
- C. W. Downey and M. W. Johnson, Tetrahedron Lett., 2007, 48, 3559–3562 CrossRef CAS.
- M. Sobkowski, J. Jankowska, J. Stawinski and A. Kraszewski, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 1469–1484 CrossRef CAS; M. Sobkowski, J. Jankowska, J. Stawinski and A. Kraszewski, in Collection Symposium Series, ed. M. Hocek, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Prague, 2005, vol. 7, pp. 183–187 Search PubMed.
- V. I. Rybachenko, K. Yu Chotii, V. V. Kovalenko and G. Shreder, Russ. J. Phys. Chem. (Engl. Transl.), 2003, 77, 1695–1698.
- L. Chmurzynski and Z. Warnke, Aust. J. Chem., 1993, 46, 185–194 CAS.
- L. Chmurzynski, G. Wawrzyniak, Z. Warnke and Z. Pawlak, J. Heterocycl. Chem., 1997, 34, 215–220 CrossRef CAS.
- J. Graton, F. Besseau, M. Berthelot, E. Raczynska and C. Laurence, Can. J. Chem., 2002, 80, 1375–1385 CrossRef CAS.
- Z. Pawlak, J. Mol. Struct., 1986, 143, 369–374 CrossRef CAS.
- L. Cecchi, F. De Sarlo and F. Machetti, Eur. J. Org. Chem., 2006, 4852–4860 CrossRef CAS.
- R. L. Benoit, D. Lefebvre and M. Frechette, Can. J. Chem., 1987, 65, 996–1001 CAS.
- J. F. Coetzee and G. R. Padmanabhan, J. Am. Chem. Soc., 1965, 87, 5005–5010 CrossRef CAS.
- D. Augustin-Nowacka, M. Makowski and L. Chmurzynski, J. Chem. Thermodyn., 2002, 34, 391–400 CrossRef CAS.
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Maemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS.
- M. Berthelot, C. Laurence, M. Safar and F. Besseau, J. Chem. Soc., Perkin Trans. 2, 1998, 283–290 RSC.
- H. P. Hopkins, D. V. Jahagirdar, P. S. Moulik, D. H. Aue, H. M. Webb, W. R. Davidson and M. D. Pedley, J. Am. Chem. Soc., 1984, 106, 4341–4348 CrossRef CAS.
- R. L. Benoit, M. Frechette and D. Lefebvre, Can. J. Chem., 1988, 66, 1159–1162 CAS.
- D. Augustin-Nowacka and L. Chmurzynski, Anal. Chim. Acta, 1999, 381, 215–220 CrossRef CAS.
- R. L. Benoit, M. J. MacKinnon and L. Bergeron, Can. J. Chem., 1981, 59, 1501–1504 CAS.
- E. Marquis, J. Graton, M. Berthelot, A. Planchat and C. Laurence, Can. J. Chem., 2004, 82, 1413–1422 CrossRef CAS.
- P. Beltrame, G. Gelli and A. Loi, Gazz. Chim. Ital., 1980, 110, 491–494 CAS.
- I. M. Kolthoff, M. K. Chantoon and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23–28 CrossRef CAS.
- D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solution, Butterworth, London, 1965 Search PubMed; CRC Handbook of Chemistry and Physics, CRC Press LLC, Boca Raton, 87th edn, 2007 Search PubMed.
- R. W. Taft, D. Gurka, L. Joris, P. von Schleyer and J. W. Rakshys, J. Am. Chem. Soc., 1969, 91, 4801–4808 CrossRef CAS.
- A. S. Chia and R. F. Trimble, J. Phys. Chem., 1961, 65, 863–866 CAS; J. Epsztajn and R. M. K. Marcinkowski, Polish J. Chem., 1979, 53, 601–609 Search PubMed; A. Gero and J. J. Markham, J. Org. Chem., 1951, 16, 1835–1838 CrossRef CAS.
- H. C. Brown and B. Kanner, J. Am. Chem. Soc., 1966, 88, 986–992 CrossRef CAS; B. Kanner, Heterocycles, 1982, 18, 411–419 CrossRef CAS.
- M. Sobkowski, in Collection Symposium Series, ed. M. Hocek, Institute of Organic Chemistry and Biochemistry Academy of Sciences of the Czech Republic, Prague, 2008, vol. 10, pp. 277–281 Search PubMed.
- R. Stromberg and J. Stawinski, Nucleic Acids Res. Symp. Ser., 1987, 18, 185–188 Search PubMed.
- M. Sobkowski, J. Stawinski and A. Kraszewski, Part 5 of this series, New J. Chem., submitted.
- F. Yang, X. L. Xu, Y. H. Gong, W. W. Qiu, Z. R. Sun, J. W. Zhou, P. Audebert and J. Tang, Tetrahedron, 2007, 63, 9188–9194 CrossRef CAS; M. Szafran, Z. Dega-Szafran, B. Nowak-Wydra and M. Pietrzak, J. Mol. Struct., 2001, 563–564, 555–564 CrossRef CAS.
- T. Umemoto and Y. Gotoh, Bull. Chem. Soc. Jpn., 1991, 64, 2008–2010 CrossRef CAS.
- V. D. Parker, E. T. Chao and G. Zheng, J. Am. Chem. Soc., 1997, 119, 11390–11394 CrossRef CAS.
- R. W. Binkley and M. G. Ambrose, J. Org. Chem., 1983, 48, 1776–1777 CrossRef CAS.
- J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS.
- A. T. Balaban, I. Ghiviriga, E. W. Czerwinski, P. De and R. Faust, J. Org. Chem., 2004, 69, 536–542 CrossRef CAS.
- J. Jankowska, M. Sobkowski, J. Stawinski and A. Kraszewski, Tetrahedron Lett., 1994, 35, 3355–3358 CrossRef CAS.
- Y. Tsujino, S. Shibata, A. Katsuyama, T. Kisaki and H. Kaneko, Heterocycles, 1982, 19, 2151–2154 CrossRef CAS.
- M. Sobkowski, J. Stawinski, A. Sobkowska and A. Kraszewski, J. Chem. Soc., Perkin Trans. 2, 1994, 1803–1808 RSC.
Footnotes |
| † Stereochemistry of internucleotide bond formation by the H-phosphonate method. Part 4.1 |
| ‡ For the compounds presented in this paper the DP descriptor refers to a structure in which the P–H bond is directed to the right in the Fischer projection, and in the LP one, to the left. The full DP/LP notation is described in ref. 10. |
| § Abbreviations: DABCO, 1,4-diazabicyclo[2.2.2]octane; DIPEA, diisopropylethylamine; DMAP, 4-(N,N-dimethylamino)pyridine; DMA, N,N-dimethylaniline; DTBP, 2,6-di-tert-butylpyridine; EDIPP, 4-ethyl-2,6-diisopropyl-3,5-dimethylpyridine; HMTA, hexamethylenetetramine; Lut, 2,6-lutidine; MPO, 4-methoxypyridine N-oxide; NMI, N-methylimidazole; PvCl, pivaloyl chloride; Py, pyridine; TEA, triethylamine. |
| ¶ Involving a rapid 3-DP⇌3-LP equilibrium in which the more reactive diastereomer 3-DP was esterified preferentially. |
| || The pKHB measures the relative strength of the acceptor in hydrogen-bonded complex formation with a reference acid (H-bonding basicity). pKa and pKHB may be unrelated.38 |
| ** Two heteroaromatic amines, pyrazine and pyrimidine, were apparently too weakly basic (pKa 0.7 and 1.2, respectively) to be efficient promoters of the condensations investigated since a significant detritylation was observed during the course of reactions, even in the presence of 6 equiv. of an amine (c≈ 5%). These amines were thus excluded from further investigations. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |