Two polyaminophenolic fluorescent chemosensors for H+ and Zn(II). Spectroscopic behaviour of free ligands and of their dinuclear Zn(II) complexes†
Gianluca
Ambrosi
a,
Cristina
Battelli
a,
Mauro
Formica
a,
Vieri
Fusi
*a,
Luca
Giorgi
a,
Eleonora
Macedi
a,
Mauro
Micheloni
*a,
Roberto
Pontellini
a and
Luca
Prodi
b
aInstitute of Chemical Sciences, University of Urbino, P.za Rinascimento 6, I-61029, Urbino, Italy
bDepartment of Chemistry, University of Bologna, Via Selmi 2, Bologna, Italy. E-mail: vieri@uniurb.it
First published on 31st October 2008
Abstract
The UV-Vis and fluorescence optical properties of the two polyamino-phenolic ligands 3,3′-bis[N,N-bis(2-aminoethyl)aminomethyl]-2,2′-dihydroxybiphenyl (L1) and 2,6-bis{[bis(2-aminoethyl)amino]methyl}phenol (L2) were investigated in aqueous solution at different pH values as well as in the presence of Zn(II) metal ion. Both ligands show two diethylenetriamine units separated by the 1,1′-bis(2-phenol) (BPH) or the phenol (PH) for L1 and L2, respectively. Both ligands are fluorescence-emitting systems in all fields of pH examined, with L1 showing a higher fluorescence emission than L2. In particular, the emission of fluorescence mainly depends on the protonation state of the phenolic functions and thus on pH. The highest emitting species is H3L3+ for both systems, where the BPH is monodeprotonated (in L1) and the PH is in the phenolate form (in L2). On the contrary, when BPH and PH are in their neutral form both ligands show the lowest fluorescence, since H-bonds occurring between the phenol and the closest tertiary amine functions decrease fluorescence. The Zn(II)-dinuclear species are also fluorescent in the pH range where they exist; the highest emitting species being [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ which are present in a wide range of pH including the physiological one. Fluorescence experiments carried out at physiological pH highlighted that, in the case of L1, the presence of Zn(II) ion in solution produces a simultaneous change in λem with a drop in fluorescence due to the formation of the [Zn2(H−2L1)]2+ species, while, in the case of L2, it gives rise to a strong CHEF effect (a twenty-fold enhancement was observed) due to the formation of the [Zn2(H−1L2)]3+ species. These results, supported by potentiometric, 1H and 13C NMR experiments, are of value for the design of new efficient fluorescent chemosensors for both H+ and Zn(II) ions.
Introduction
The development of chemosensors is in continuous expansion due to their usefulness in many fields; they have a wide range of applications, such as environmental monitoring, process control, food and beverage analysis, medical diagnosis and others.1–8 Due to their use in many disciplines, they are very attractive for chemists, biologists, physicists and material scientists. For example, in biochemistry, clinical and medical sciences, and cell biology, freely mobile sensor molecules are employed extensively in microscopy, offering the possibility of performing real-space measurements.9,10Among the different chemosensors, the fluorescence-based ones present many advantages: fluorescence measurements are usually very sensitive, low-cost, easily performed and versatile, offering submicrometer spatial resolution and submillisecond temporal resolution.11–19 The versatility of fluorescence-based sensors originates also from the wide number of parameters that can be tuned in order to optimize the convenient signal. In most cases, changes in luminescence intensity represent the most directly detectable response to target recognition; more recently, however, other properties such as excited-state lifetime and fluorescence anisotropy have also been preferred as diagnostic parameters, since they are less affected by environmental and experimental conditions.
Phenol and poly-phenols show well known optical properties which mainly depend on their protonation degree;20,21 in our lab, several polyamino-phenolic ligands of different topologies have been synthesized. In this study, we wanted to extend our knowledge to the spectroscopic properties of two of them to identify their possible applications as chemosensors for suitable guests. In this case, we focused our attention on the two previously synthesized amino-phenolic ligands L1 and L2 (Chart 1). They were chosen for several reasons: they have similar topology; they both show two diethylenetriamine (dien) units separated by a phenolic aromatic spacer, the 1,1′-bis(2-phenol) group (BPH) and the phenol for L1 and L2, respectively; in addition, they easily form dinuclear species with transition metal ions. The molecular skeleton of both ligands affords the formation of preorganized dinuclear Zn(II) species where the two Zn(II) ions can cooperate in binding guests; in particular, it has been demonstrated that in some dinuclear species such as the [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ ones, the two zinc ions show, in both systems, an equal coordination environment, are displaced at fixed different distances and are able to add guests to saturate the coordination requirement of the two zinc ions (see Scheme 1).

![Coordination scheme for Zn(ii) in the [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ complexes.](/image/article/2009/NJ/b810228g/b810228g-s1.gif) | ||
| Scheme 1 Coordination scheme for Zn(II) in the [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ complexes. | ||
Although zinc is an essential metal ion in human life and plays a fundamental role in many biological functions, for example in the alkaline phosphatase or carbonic anhydrase enzymes,22 excess zinc can be very harmful, as it can lead to many health problems.23 For this reason, easy recognition of the zinc ion is key mainly and as a result many fluorescent molecular sensors have been developed in recent years, also to allow its in vivo mapping.24
In this work, we have studied the NMR, UV-Vis and fluorescence properties of the free ligands as well as of their zinc complexes in aqueous solution. The aim has been to detect if the optical properties of these systems are affected by pH as well as by the presence of Zn(II) in solution.
Experimental
Synthesis
Ligand 3,3′-bis[N,N-bis(2-aminoethyl)aminomethyl]-2,2′-dihydroxybiphenyl (L1) and 2,6-bis{[bis-(2-aminoethyl)amino]methyl}phenol (L2) were prepared as previously described.25EMF measurements
Equilibrium constants for protonation and complexation reactions with L2 were determined by pH-metric measurements (pH = −log[H+]) in 0.15 M NaCl at 298.1 ± 0.1 K, using the fully automatic equipment that has already been described; the EMF data were acquired with the PASAT computer program.26 The combined glass electrode was calibrated as a hydrogen concentration probe by titrating known amounts of HCl with CO2-free NaOH solutions and determining the equivalent point by Gran’s method,27 which gives the standard potential E° and the ionic product of water (pKw = 13.73(1) at 298.1 K in 0.15 M NaCl, Kw = [H+][OH−]). At least three potentiometric titrations were performed for each system in the pH range 2–11, using different molar ratios of Zn(II)/L2 ranging from 1:1 to 2:1. All titrations were treated either as single sets or as separate entities, for each system; no significant variations were found in the values of the determined constants. The HYPERQUAD computer program was used to process the potentiometric data.28Spectroscopic experiments
1H and 13C NMR spectra were recorded on a Bruker Avance 200 instrument, operating at 200.13 and 50.33 MHz, respectively, and equipped with a variable temperature controller. The temperature of the NMR probe was calibrated using 1,2-ethanediol as calibration sample. For the spectra recorded in D2O, the peak positions are reported with respect to HOD (4.75 ppm) for 1H NMR spectra, while dioxane was used as reference standard in 13C NMR spectra (δ = 67.4 ppm). Fluorescence spectra were recorded at 298 K with a Varian Cary Eclipse spectrofluorimeter. UV absorption spectra were recorded at 298 K with a Varian Cary-100 spectrophotometer equipped with a temperature control unit.The fluorescence quantum yields (Φf) of the highest fluorescent species were calculated as reported in ref. 29 using 2-aminopyridine as standard reference.
Results and discussion
Solution studies
Ligands L1 and L2 as well as the Zn(II)/L systems were studied by fluorescence spectroscopy in aqueous solution at different pH values to investigate the fluorescence properties of both ligands and how these are affected by protonation and the presence of Zn(II) ion. 1H and 13C NMR experiments on the free L1 as well as those reported for the Zn(II)/L1 system25a aided in understanding the role played by both protonation and Zn(II). The fluorescence quantum yields (Φf) of the highest fluorescent species are reported in Table 1.| Φ f | |
|---|---|
| H3L13+ | 0.34 |
| H3L23+ | 0.01 |
| [Zn2H−2L1]2+ | 0.24 |
| [Zn2H−1L2]3+ | 0.08 |
Similar 1H NMR studies carried out on L2 and Zn(II)/L2 system are reported in refs. 30 and 31, respectively. Moreover, further studies on the UV-Vis absorption properties of both L and Zn(II)/L systems were performed in aqueous solution in addition to those already reported.25,30,31
L1 and L2 in aqueous solution at different pH values
| Reaction | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K K |
|---|---|
| a Values in parentheses are the standard deviations on the last significant figure. | |
| L + H+ = HL+ | 10.04(1)a |
| HL+ + H+ = H2L2+ | 9.87(1) |
| H2L2+ + H+ = H3L3+ | 9.12(1) |
| H3L3+ + H+ = H4L4+ | 7.59(1) |
| H4L4+ + H+ = H5L5+ | 2.50(3) |
| Zn2+ + L + 2H+ = ZnH2L4+ | 28.42(1) |
| Zn2+ + L + H+ = ZnHL3+ | 23.82(2) |
| Zn2+ + L = ZnL2+ | 14.67(2) |
| Zn2+ + L = Zn(H−1L)+ + H+ | 5.05(2) |
| 2Zn2+ + L = Zn2(H−1L)3+ + H+ | 17.17(1) |
| 2Zn2+ + L + H2O = Zn2(H−1L)OH2+ + 2H+ | 8.34(3) |
| 2Zn2+ + L + 2H2O = Zn2(H−1L)(OH)2+ + 3H+ | −1.63(3) |
| Zn2(H−1L)3+ + OH− = Zn2(H−1L)OH2+ | 4.90 |
| Zn2(H−1L)OH2+ + OH− = Zn2(H−1L)(OH)2+ | 3.76 |
As reported in the literature, BPH shows emission of fluorescence depending on the degree to which it is deprotonated;20 in particular, it shows the most intense fluorescence in its monodeprotonated form and the least intense in its neutral one (more than six times lower), while the dianionic form, although fluorescent, is obtainable only at very high pH values (pH > 15).20,32
The fluorescence spectra of L1 (λexc = 287 nm) recorded in aqueous solution in the pH range 2–12 are reported in Fig. 1; the trend of the fluorescence emission intensity (◆) vs. pH (λexc = 287 nm) is reported in Fig. 2(a) together with the maximum absorption (⋯) and the emission (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) wavelength trend. Fig. 2(b) reports the trend of the absorption titration at λ = 308 nm (●) together with the distribution curves for the species of L1 (—) as a function of pH.
) wavelength trend. Fig. 2(b) reports the trend of the absorption titration at λ = 308 nm (●) together with the distribution curves for the species of L1 (—) as a function of pH.
 | ||
| Fig. 1 Fluorescence spectra of L1 at different pH values. | ||
![Fluorescent emission titration (λexc = 287 nm, λem = 403 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 308 nm (●) and distribution curves of the species (—) (b) as function of pH in aqueous solution: [L1] = 5.0 × 10−5 M, I = 0.15 M NaCl, T = 298.1 K.](/image/article/2009/NJ/b810228g/b810228g-f2.gif) | ||
| Fig. 2 Fluorescent emission titration (λexc = 287 nm, λem = 403 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 308 nm (●) and distribution curves of the species (—) (b) as function of pH in aqueous solution: [L1] = 5.0 × 10−5 M, I = 0.15 M NaCl, T = 298.1 K. | ||
Excitation of L1 acid solution at pH 2 (λexc = 287 nm) gives rise to a fluorescence emission band of very low intensity (λem = 403 nm) attributed to the BPH fluorophore. The intensity of the fluorescence emission of the compound is highly dependent on the protonation state of the ligand (see Fig. 1 and 2(a)); however the shape and the λem of the spectra are substantially pH-independent. In this pH range, the free BPH group shows a similar fluorescent behavior produced by the monoanionic excited state of BPH.20,32 Taking into account that the fluorescence of L1 is due to the BPH fluorophore, this suggests that also in L1 the changes in fluorescence emission reflect only the ground states acid–base equilibrium.33 For this reason, no indication of the excited state proton transfer reaction was found and, as reported for free BPH, the fluorescence is due to the monoanionic excited state of BPH in L1.
In the fluorescence spectra, the emission remains substantially very low and constant (Fig. 2(a)) at acidic pH values (2 ≤ pH ≤ 5) while it starts increasing at pH 5 in concomitance with the appearance of the H3L13+ species in solution, reaching a maximum intensity at pH 7.4–8.4 with the complete formation of the H3L13+ species. A small decrease can be observed in the alkaline range up to pH 10.5 at which the less protonated species appear in solution; on the contrary, when the pH is increased to 11 emission rises again, reaching maximum intensity at pH 12 with the presence in solution of the monoanionic H−1L1− species.
Bearing in mind the previous studies on BPH,20,32 the trend of the emission intensity in the range of pH 2–8 can be easily explained by the deprotonation of the neutral BPH unit to form its monoanionic species that occurs with the formation of the H3L13+ species. In other words, in the protonated species H5L15+ and H4L14+, BPH is present in its neutral form while in the H3L13+ species it has lost one of the acidic protons forming the highest emitting species (Φ = 0.34, Table 1). These results are in agreement with those already obtained by UV-Vis absorption studies which revealed that the deprotonation of one of the hydroxyl functions of BPH occurred in the pH range involving the passage from H4L14+ to H3L13+ species.25a This was highlighted, as reported in Fig. 2(b), by the change in absorption at 308 nm which increases when the monoanionic form of BPH is present in solution and is further underlined by the variation in the trend of the maximum of the absorption wavelength (Fig. 2(a)) as a function of pH; both figures highlight that the changes take place in the field of pH where the H3L13+ species forms. Although the absorption and emission wavelength maxima as well as the absorption at 308 nm remain constant, increasing the pH to form lesser protonated species than H3L13+, there is a small decrease (about 25% at pH 10) in fluorescence intensity occurring with the formation of the H2L12+, HL1+ and neutral L1 species (Fig. 2(a)). This trend could be explained by the formation of a H-bond network involving BPH and the closer nitrogen atoms. In fact, as reported for free BPH,20,32 the formation of an intramolecular H-bond interaction occurring between the two oxygen atoms of BPH in stabilizing the hydrogen atom in the monoanionic species gives the greatest fluorescence intensity, while, on the contrary, the formation of intermolecular H-bonds with H-accepting molecules, such as water, gives rise to a very fast nonradiating process throughout the H-bond, thus leading to a decrease in the fluorescence (this occurs for example in the neutral form of excited BPH). In addition, it has been demonstrated that in the presence of strong proton-accepting molecules such as triethylamine (TEA), the formation of H-bonding between TEA and a hydrogen atom of BPH once again leads to a decrease in fluorescence.20b In our case, it is presumable that similar H-bonding between the BPH oxygen and the closer nitrogen atoms of the dien units is formed, thus decreasing fluorescence; however, the formation of this type of H-bond cannot be the favourite situation since only a slight drop in fluorescence was observed. Moreover, two different H-bonds could be suggested in the case of L1: viaOH⋯N as well as viaO−⋯HN+; in other words, in the H2L12+, HL1+ and L1 species, a partial stabilization of the acidic hydrogen atom of the monoanionic BPH unit could also take place with the closer N atom (c in Scheme 2), but also a partial ammonium character of the closer N atom could give rise to the same quenching H-bond effect with the BPH unit (b in Scheme 2). In any case, the form (a) shown in Scheme 2 is the favoured form and it is the only one present in the H3L13+ as well as in the H−1L1− species where the highest fluorescence is reached. In addition, the absence of fluorescence changes even at highly alkaline pH values once again demonstrates that the full deprotonation of BPH in L1 is not reachable under our experimental conditions.
 | ||
| Scheme 2 Possible H-bond interactions for the neutral L1 species. | ||
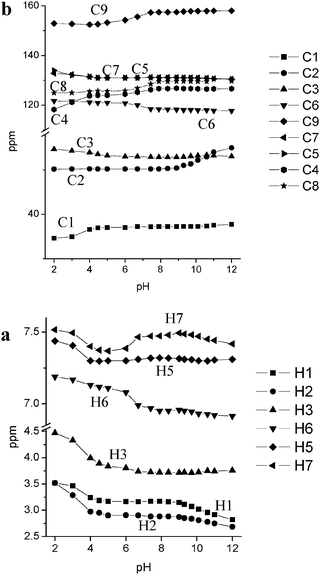 | ||
| Fig. 3 Experimental NMR chemical shifts in aqueous solution of L1 as a function of pH: 1H NMR (a); 13C NMR (b). | ||
In the pH range 4–6 the H4L4+ species is prevalent and, as demonstrated both by UV and fluorescence experiments, the fifth protonation step occurs at the BPH group. This was also confirmed in the NMR experiments by the downfield shift of the H6 signal in the para position to the phenolic oxygens and by the upfield shift of the H7 protons in the 1H NMR spectra, as well as by the accompanying upfield shift of C4, C8 and C9 and downfield shift of C6 in the 13C NMR spectra. The strong upfield shift exhibited by the signal of H7 could be related not only to a protonation process of the BPH unit but also to a change in the angle between the two aromatic rings that probably is affected by the protonation degree of L1 leading the formation of a new H-bond network involving the neutral BPH and the unprotonated tertiary amine functions, as depicted in Fig. 4 for the H4L14+ species; this almost entirely quenches the fluorescence (see above). The protonation step giving the H5L5+ species, occurring below pH 4, basically causes a downfield shift of protons H2 and H3 together with an upfield shift in the signals of the carbon atoms C1 and C4, suggesting that it takes place on the tertiary amine groups. Once again the H7 and H5 resonances, both of which shift downfield, are perturbed by this protonation step, highlighting the formation of a H-bond network with the closer amine functions on the BPH unit different from the previous one; this affects the chemical shift, modifying the electron density of the neutral BPH unit as well as the angle between the two aromatic rings.
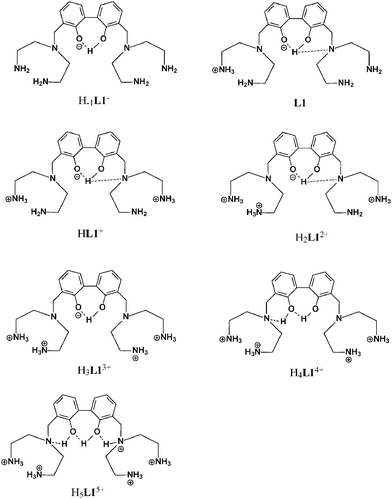 | ||
| Fig. 4 Location of acidic hydrogen atoms in the protonated species of L1. | ||
A protonation scheme arising from NMR experiments is summarized in Fig. 4.
) wavelength trends. Fig. 5(b) reports the trend for the absorption titration at λ = 290 nm (●) together with the distribution curves of the species of L2 (—) as a function of pH obtained by potentiometry.
![Fluorescence emission titration (λex = 280 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 290 nm (●) and distribution curves of the species (—) (b); of L2 as function of pH in aqueous solution: [L2] = 5.0 × 10−5 M, I = 0.15 M NaCl, T = 298.1 K.](/image/article/2009/NJ/b810228g/b810228g-f5.gif) | ||
| Fig. 5 Fluorescence emission titration (λex = 280 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 290 nm (●) and distribution curves of the species (—) (b); of L2 as function of pH in aqueous solution: [L2] = 5.0 × 10−5 M, I = 0.15 M NaCl, T = 298.1 K. | ||
The acidic solutions of L2 up to pH 6 are barely fluorescent, as also reported for the free neutral phenol (PH), while fluorescence increases with the formation of the H3L23+ species, reaching maximum emission at pH 8 together with the maximum presence in solution of the H3L23+ species; at higher pH values, the emission drops, reaching a plateau at pH higher than 11 with the formation of the neutral L2 species. The fluorescence of ligand L2, which is lower compared to that of L1 (see Table 1), is highly dependent on the protonation state of the ligand as seen before for L1. The acidic proton distribution in the several protonated species of L2 obtained by UV-Vis, potentiometry and NMR studies was previously reported and the scheme is reported in Fig. S1 of the ESI;† the most fluorescent H3L23+ species is the one in which the phenol is deprotonated (i.e.phenolate) and the four acidic protons are located on the primary amine functions; this is the same situation found for the H3L13+ species where there are no H-bond interactions with the closest amine functions, thus affording the highest emission quantum yield also in the H3L23+ species. It should be noted that in the free PH, the anion presents a much lower fluorescence intensity than the neutral species.21 In this case the opposite behaviour was observed; this could be explained (see also below) by a decrease in the solvation viaH-bond network of the phenolate oxygen atom by the water molecules in the H3L23+ species in comparison with the free PH anion.21a In other words, the presence of the two protonated dien units linked to the PH group modifies the accessibility of the solvent molecules to the phenolate oxygen atom decreasing its quenching effect and thus increasing the emissive relaxation decay of the PH anion. As reported, an acidic proton redistribution was observed in the less protonated species involving at least a tertiary amine function that becomes protonated. This ammonium group, found mainly in the neutral L2 species, is stabilized viaH-bond with the close phenolate oxygen atom (see Fig. S1, ESI†). For this reason, as for ligand L1, the formation of H-bonding with the amine function leads to a decrease in its fluorescence.
This H-bond interaction, which is also monitorable through the UV-Vis spectra (see Fig. 5(b)), is also highlighted by the change in the maximum of the absorption and emission wavelengths (Fig. 5(a)) as a function of pH. λmax and λem shifted in different directions, increasing the Stokes shift when the phenol becomes phenolate (pH ≥ 5, λmax and λem shift towards lower and higher energies, respectively), while an opposite trend was observed at higher pH values (λmax and λem shift towards higher and lower energies, respectively) with the formation of the neutral zwitterionic L2 species in which a strong H-bond between the closest tertiary ammonium and phenolate groups was suggested. Taking into account the trend and shift in both λmax and λem, it can be suggested that the fluorescence is yielded by light emission decay from the phenolate excited state of all L2 species to different ground states, characterized by the formation of strong intramolecular H-bonds.
In conclusion, although L1 is a much more efficient fluorescent system than L2 (Table 1), both ligands show fluorescence emission depending on the protonation state of the aromatic functions. In particular, the highest emitting species are due to the monodeprotonated form of BPH of L1 as well as to the phenolate species of PH of L2, both of which are achieved in the H3L3+ species; on the contrary, the neutral BPH and PH species are very low fluorescence emitters. The presence of the closer tertiary amine function affects the emission quantum yield in some species by forming intramolecular H-bonding with the close phenol oxygen atom of both systems. The H-bonding induces a nonradiative relaxation process of the excited species, yielding a decrease in the fluorescence in both ligands. This H-bonding is weaker in L1viaOH⋯N as well as O−⋯HN+, and for this reason only a relatively low efficiency of fluorescence quenching could be observed, while it takes place strongly viaO−⋯HN+ in L2 giving an almost total quenching of the fluorescence of L2. Taking into account these results, both ligands behave as chemosensors of H+ in that they are able to change their optical absorption and fluorescence properties as a function of pH.
Coordination of Zn(II)
The coordination behaviour of both systems towards Zn(II) was potentiometrically studied and the results obtained are reported in ref. 25 and 31; as for basicity, the Zn(II)/L2 system had been studied in NMe4Cl ionic medium,31 thus we performed new potentiometric measurements to obtain the stability constants for the Zn(II)/L2 system under the same experimental conditions as the Zn(II)/L1 system (0.15 mol dm−3NaCl aqueous solution at 298.1 K). The potentiometrically determined stability constants for the equilibrium reactions of L2 with Zn(II) are reported in Table 2. The species formed as well as the values of the stability constants evaluated are similar to those previously reported and thus the discussion can be outlined in the same way. The main difference found was the formation of the [Zn2(H−1L2)(OH)2]+ species in this ionic medium which was not previously detected. The addition of the second OH− anion to [Zn2(H−1L2)OH]2+ is quite high (logK = 3.76) suggesting that it is probably bound in a bridge disposition between the Zn(II) ions. The distribution diagrams for the Zn(II)-complexed species for both 2Zn(II)/L systems are reported in Fig. 6 for L1 and in Fig. 8 for L2 as a function of pH. However, the results previously discussed can be summarized in this way: (i) the dinuclear species are prevalent in solution and the only species existing at pH higher than 7 is a L/Zn(II) with a 1:2 molar ratio; (ii) the most prevalent species are [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ for L1 and L2, respectively; (iii) these dinuclear species have similar molecular skeletons indicating a preorganized dinuclear Zn(II) species in which the two Zn(II), similarly coordinated, can cooperate in binding suitable guests (see Scheme 1).
) wavelength trends. Fig. 6(b) reports the trend for the absorption titration at λ = 295 nm (●) together with the distribution curves for the species of the 2Zn(II)/L1 system (—) as a function of pH. Moreover, fluorescence titration was carried out by adding increasing amounts of Zn(II) to a HEPES buffer (pH = 7.4) solution of L1 and the spectra are reported in Fig. 7. The fluorescence quantum yield of the highly emitting species is reported in Table 1.
![Fluorescence emission titration (λex = 283 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 295 nm (●) and distribution curves of the species (–) (b); as a function of pH in aqueous solution: [L1] = 5.0 × 10−5 M, [Zn(ii)] = 10−4 M, I = 0.15 M NaCl, T = 298.1 K.](/image/article/2009/NJ/b810228g/b810228g-f6.gif) | ||
| Fig. 6 Fluorescence emission titration (λex = 283 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 295 nm (●) and distribution curves of the species (–) (b); as a function of pH in aqueous solution: [L1] = 5.0 × 10−5 M, [Zn(II)] = 10−4 M, I = 0.15 M NaCl, T = 298.1 K. | ||
![Fluorescence spectra of the Zn(ii)/L1 system in aqueous buffer (HEPES, 5 × 10−2 M) solution at pH = 7.4, obtained by adding several amounts of Zn(ii) up to 2 equivalents with respect to [L1] = 5.0 × 10−5 M.](/image/article/2009/NJ/b810228g/b810228g-f7.gif) | ||
| Fig. 7 Fluorescence spectra of the Zn(II)/L1 system in aqueous buffer (HEPES, 5 × 10−2 M) solution at pH = 7.4, obtained by adding several amounts of Zn(II) up to 2 equivalents with respect to [L1] = 5.0 × 10−5 M. | ||
The UV-Vis absorption spectra of solutions containing 2Zn(II)/L1 recorded at different pH values were discussed previously;25a they showed spectral profiles indicating the deprotonation of BPH and simultaneous coordination of the Zn(II) ions; in these new experiments, some further aspects can be discussed. The absorption λmax shifts toward lower energy when monitored from acidic (free ligand) to basic pH values (Zn(II)-complexes); up to the presence in solution of the Zn(II)-mononuclear species it moves from 280 to 305 nm, while the appearance in solution of the dinuclear species, at approximately pH 6, gives rise to a change in the λmax which shifts from 305 nm in the presence of the mononuclear [Zn(HL1)]3+ species to 298 nm with the complete formation of the more stable [Zn2(H−2L1)]2+ species at pH = 7.4. This λmax is preserved also at higher pH values where only dinuclear Zn(II)-complexed species are present in solution. The shift in λmax observed from the mono- to the di-nuclear species can be ascribed to the full deprotonation of BPH which loses both acidic hydrogen atoms in the Zn(II)-dinuclear species, affording the bi-negative form of BPH; this result is in agreement with the studies previously reported for the Zn(II)-dinuclear species of L1. The changes in absorption from the mono-negative BPH to the bi-negative species are also visible in Fig. 6(b), where a change in absorptivity can also be observed when the dinuclear species appear in solution. These changes are in agreement with a change in the protonation degree of BPH and thus to its full deprotonation and simultaneous coordination of each Zn(II) ion by one phenolate oxygen atom of the BPH unit as already reported. The fluorescence experiments gave rise to analogous results, with fluorescence increasing at values starting from acidic pH and reaching maximum intensity in the field of pH 7.4–8.4 with the maximum presence in solution of the [Zn2(H−2L1)]2+ species, then decreasing at higher pH values and reaching a plateau at pH > 11 with the presence in solution of the di-hydroxylated [Zn2(H−2L1)(OH)2] species (Fig. 6). It is interesting to note that, unlike the free L1, the λem changes (λexc = 283 nm) by changing the pH, and as in the absorption experiments the change occurs at the pH values where there is the formation of the Zn(II)-dinuclear species. Specifically, λem shifts from 403 nm (free ligand) to 379 nm with the formation of the [Zn2(H−2L1)]2+ species, while remaining constant in the other dinuclear species. Once again, this trend can be related to the full deprotonation of BPH, as retrieved in the crystal structure of the [Zn2(H−2L1)(H2O)2]2+ previously reported, which produces changes in the ground as well as in the excited state of BPH. Moreover, the formation of the hydroxylated [Zn2(H−2L1)OH]+ and [Zn2(H−2L1)(OH)2] species produces a drop in fluorescence emission without changing the λem (Fig. 6(a)); this is due to an increase in electron density of the BPH unit by coordinating the OH− species which, as reported in similar cases, increases a thermal relaxation negatively affecting emission decay mechanisms.20b The change in λem occurring with the formation of the dinuclear [Zn2(H−2L1)]2+ is well highlighted by titrating a buffer (pH = 7.4) solution of L1, adding increasing amounts of Zn(II) up to 2 equivalents (Fig. 7); as shown in the figure, the λem shifts toward higher energy by adding Zn(II) but, at the same time, the fluorescence of the new species formed decreases by about 30% and thus none chelation-enhanced fluorescence (CHEF) effects were observed for this system.
![Fluorescence emission titration (λex = 279 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 287 nm (●) and distribution curves of the species (—) (b); as a function of pH in aqueous solution: [L2] = 5.0 × 10−5 M, [Zn(ii)] = 1.0 × 10−4 M, I = 0.15 M NaCl, T = 298.1 K.](/image/article/2009/NJ/b810228g/b810228g-f8.gif) | ||
| Fig. 8 Fluorescence emission titration (λex = 279 nm) (◆), absorption wavelength trend (⋯), and emission wavelength trend () (a); absorption titration at λ = 287 nm (●) and distribution curves of the species (—) (b); as a function of pH in aqueous solution: [L2] = 5.0 × 10−5 M, [Zn(II)] = 1.0 × 10−4 M, I = 0.15 M NaCl, T = 298.1 K. | ||
UV-Vis absorption spectra of solutions containing 2Zn(II)/L2 at different pH values show, as previously reported, spectral profiles due to the deprotonated form of PH. However, also in this case, some further aspects can be discussed. Observing the λmax of the spectra from acidic to alkaline pH values (Fig. 8(a)), a shift in the λmax from 273 (free ligand) to 286 nm (complexed ligand) occurs at pH > 5 with the appearance in solution of the [Zn2(H−1L2)]3+ species, in agreement with the deprotonation of PH as previously demonstrated. The value of λmax 286 nm is enough preserved also at higher pH values where only a little decrease is shown with the appearance in solution of the dihydroxylated species (λmax = 282 at pH = 12). On the contrary, in the 6–8 pH range, where the [Zn2(H−1L2)]3+ species is prevalent in solution, a slight increase in absorption with respect to the free ligand can be observed, while a marked increase is visible at higher pH values with the formation of the hydroxylated [Zn2(H−1L2)OH]2+ species (see Fig. 8). This finding could be explained by a different disposition of L2 in forming the Zn–O–Zn cluster system (O is the phenolate oxygen atom) in the [Zn2(H−1L2)]3+ and [Zn2(H−1L2)OH]2+ species. In fact, while it was demonstrated that the hydroxylated [Zn2(H−1L2)OH]2+ species shows the OH− displaced in a bridged disposition between the two Zn(II) ions,30 on the contrary, a coordination environment without secondary bridging ligands could be hypothesized in the [Zn2(H−1L2)]3+ species. In the latter, the fifth coordination site of each Zn(II) ion could be saturated by a water molecule or by a chloride anion of the ionic medium. This may be the reason for the increase in absorption of the [Zn2(H−1L2)OH]2+ with respect to the [Zn2(H−1L2)]3+ species.
Analysis of the fluorescence experiments gives additional information; examining the maximum of λem (λexc = 275 nm) from acidic to alkaline field of pH, a shift of the λem is observable (see Fig. 8) at pH > 5; λem moves from 354 nm, typical of the free ligand, reaching a constant value (308 nm) at pH 6, with the full formation of the [Zn2(H−1L2)]3+ species. This change in λem is coupled with an increase in fluorescence, which shows its highest emission in the range of the [Zn2(H−1L2)]3+ species. These changes are in agreement with the simultaneous deprotonation of the phenolic oxygen atom due to the Zn(II) complex formation and its bridging coordination between the two Zn(II) ions, as phenolate. At pH > 9, a further change in the λem can be highlighted, since it shifts from 308 to 325 nm in concomitance with the appearance of the [Zn2(H−1L2)OH]2+ species in solution; this occurs without observing any significant change in fluorescence intensity. This result may be related, as above, to a different disposition of the secondary ligands in the two complexed [Zn2(H−1L2)]3+ and [Zn2(H−1L2)OH]2+ species that could be responsible of the different λem in the dinuclear [Zn2(H−1L2)]3+ and [Zn2(H−1L2)OH]2+ species. As previously discussed for the [Zn2(H−2L1)(OH)2] species, the increase in the total electron density of the complex in the dihydroxylated [Zn2(H−1L2)(OH)2]+ species affects fluorescence at higher pH values.
The Zn(II)-L2 dinuclear complexes showed very interesting fluorescent properties; in fact, although free L2 exhibits emitting species in the same range of pH of the Zn(II)-dinuclear one, the fluorescence intensity of the latter is higher, giving a strong CHEF effect. This effect, occurring to L2 in the presence of Zn(II), is highlighted in Fig. 9, which reports the fluorescence spectra of L2 obtained by adding several amounts of Zn(II) in aqueous buffer pH = 7.4 solution. At this pH value, the species formed in the presence of Zn(II) is the [Zn2(H−1L2)]3+ species. As can be observed in Fig. 9, the free ligand shows low fluorescence emission with a λem centered at 347 nm; by adding Zn(II), the emission increases and λem shifts toward higher energy. The spectra preserve the same profile when adding up to 2 equivalents of Zn(II), reaching a constant emission and λem of 308 nm, in concomitance with the complete formation of the [Zn2(H−1L2)]3+ species. (see Fig. 8(b)). At this pH the emission quantum yield is more than twenty-fold higher for the [Zn2(H−1L2)]3+ species than for the free ligand; furthermore, a similar CHEF effect was also found in other fields of pH such as 8 and 10, at which the dinuclear species are formed, thus highlighting the sensing role of L2 towards Zn(II) in aqueous solution in a biologically important range of pH. For this reason, L2 can be considered a potential chemosensor for Zn(II).
![Fluorescence spectra of the Zn(ii)/L2 system in aqueous buffer (HEPES, 5 × 10−2 M) solution at pH = 7.4, obtained by adding several amounts of Zn(ii) up to 2 equivalents with respect to [L2] = 5.0 × 10−5 M.](/image/article/2009/NJ/b810228g/b810228g-f9.gif) | ||
| Fig. 9 Fluorescence spectra of the Zn(II)/L2 system in aqueous buffer (HEPES, 5 × 10−2 M) solution at pH = 7.4, obtained by adding several amounts of Zn(II) up to 2 equivalents with respect to [L2] = 5.0 × 10−5 M. | ||
Conclusions
The studies highlighted that the intensity of the fluorescence of both ligands depends on the protonation state of the phenolic functions, and in the case of L2, the λem is also affected by protonation while this does not occur for L1. For this behavior, both ligands are suitable chemosensors of H+ in that they are able to change their optical absorption and fluorescence properties as function of pH.For both systems the most fluorescent species is the same: H3L3+ in which the BPH unit of L1 is in its mono-deprotonated form, while PH is present as phenolate form in L2; on the contrary, when BPH and PH are in their neutral form both ligands show the lowest fluorescence. While these results are in agreement with those found for free BPH (the more fluorescent species is the monoanionic species of BPH), this finding is opposite to that for free PH where the neutral species is the most fluorescent. This can be explained by a lower solvation of the phenolate oxygen atom in the H3L23+ species which limits the quenching effect occurring viaH-bond with the water molecules. The presence of the tertiary amine function close to the phenol oxygen affects the emission quantum yield of those species in which the formation of intramolecular H-bonds is possible, highlighting that the formation of H-bonds has a quenching effect to the fluorescence in these systems.
The Zn(II)-dinuclear species are fluorescent in the field of pH where they exist; the highest emitting species are the [Zn2(H−2L1)]2+ and the [Zn2(H−1L2)]3+ species, respectively; they are prevalent in a wide range of pH including the physiological one. In the [Zn2H−2L1]2+, the presence of the dianionic form of BPH produces a blue shift of λem in the fluorescence experiments, in comparison with the free ligand. The interaction with guests such as OH− perturbs the emission but not the absorption of the dinuclear species.
In the [Zn2(H−1L2)]3+ species a slight blue shift in λem can also be observed, as well as, a decrease in fluorescence brought about by the addition of an anionic guest such as OH−.
The main result retrieved is that both L1 and L2 sense the Zn(II) in aqueous solution at physiological pH 7.4 by fluorescence; at this pH the [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ species are prevalent in solution. The [Zn2(H−2L1)]2+ species shows a simultaneous change in the λem with a drop in fluorescence, but real and efficient sensing was obtained by using ligand L2 which, in the presence of two equivalents of Zn(II), gives rise to a strong CHEF effect (a twenty-fold increase) with the formation of the [Zn2(H−1L2)]3+ species; in this case, a similar CHEF effect was also found in other fields of pH such as 8 and 10, highlighting the sensing role of L2 towards Zn(II) in aqueous solution in a biologically important range of pH.
Concluding, both systems behave as chemosensors for both H+ and Zn(II) and their investigation has given much useful information for the design of more efficient systems. Moreover, taking into account that both [Zn2(H−2L1)]2+ and [Zn2(H−1L2)]3+ dinuclear species show the highest fluorescence intensity and that they are the most suitable hosting species for guests, they are a very interesting platform for the sensing of guest species.
Acknowledgements
The authors thank the Italian Ministero dell’Istruzione dell’Università e della Ricerca (MIUR), PRIN2007 for financial support.References
- (a) Fluorescent Chemosensors of Ion and Molecule Recognition, eds. J.-P. Desvergne and A. W. Czarnik, NATO-ASI Series, Kluwer Academic Publishers, 1996 Search PubMed; (b) U. E. Spichiger-Keller, Chemical Sensors and Biosensors for Medical and Biological Applications, Wiley-VCH, 1998 Search PubMed; (c) Fluorescent Chemosensors for Ion and Molecule Recognition, ed. A. W. Czarnik, ACS, Washington, DC, 1992 Search PubMed.
- (a) Chem. Rev., 2000, 100, 2477–2738, special issue on Chemical Sensors, eds. A. B. Ellis and D. R. Walt Search PubMed; (b) Coord. Chem. Rev., 2000, 205, 1–232, special issue on luminescent sensors, ed. L. Fabbrizzi Search PubMed; (c) special issue on synthetic receptors as sensors, ed. A. V. Aslin, Tetrahedron, 2004, 60, 11055–11316 CrossRef CAS.
- (a) Special issue on luminescent sensors, eds. A. P. deSilva and P. Tecilla, J. Mater. Chem., 2005, 15, 2617–2976 RSC; (b) A. P. de Silva, G. D. McClean, T. S. Moody and S. M. Weir, in Handbook of Photochemistry and Photobiology, ed. H. S. Nalwa, American Institute of Physics, 2003, vol. 3, p. 217 Search PubMed.
- L. Fabbrizzi, M. Licchelli and A. Taglietti, Dalton Trans., 2003, 3471 RSC.
- K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 2161 CrossRef CAS.
- R. Martinez-Manez and F. Sancenon, Chem. Rev., 2003, 103, 4419 CrossRef CAS.
- (a) K. J. Albert, N. S. Lewis, C. L. Schauer, G. A. Sotzing, S. E. Stitzel, T. M. Vaid and D. R. Walt, Chem. Rev., 2000, 100, 2595 CrossRef CAS; (b) W. Göpel, Sens. Actuators B, 1998, 52, 125 CrossRef; (c) J. J. Lavigne and E. V. Anslyn, Angew. Chem., Int. Ed., 2001, 40, 3118 CrossRef; (d) A. D’Amico, C. Di Natale and R. Paolesse, Sens. Actuators B, 2000, 68, 324 CrossRef.
- (a) L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59 CrossRef CAS; (b) C. Bargossi, M. C. Fiorini, M. Montalti, L. Prodi and N. Zaccheroni, Coord. Chem. Rev., 2000, 208, 17–32 CrossRef CAS; (c) L. Prodi, New J. Chem., 2005, 29, 20 RSC.
- (a) “Advanced Concepts in Fluorescent sensing, Part A: Small Molecule Sensing”, in Topics in Fluorescence Spectroscopy, vol. 9 Search PubMed; (b) “Advanced Concepts in Fluorescent sensing, Part B: Macromolecular Sensing”, eds. C. D. Geddes and J. R. Lakowicz, Springer, 2005, vol. 10 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, 1999 Search PubMed.
- R. Y. Tsien, Annu. Rev. Neurosci., 1989, 12, 227 CrossRef CAS.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef.
- Special Issue on Chemical sensors, guest editors A. B. Ellis and D. R. Walt, Chem. Rev., 2000, 100, 2477–2738 CrossRef CAS.
- K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 2161 CrossRef CAS.
- L. Fabbrizzi, M. Licchelli and A. Taglietti, Dalton Trans., 2003, 3471 RSC.
- W. T. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589 RSC.
- (a) L. Prodi, New J. Chem., 2005, 29, 20 RSC; (b) G. Farrugia, S. Iotti, L. Prodi, M. Montalti, N. Zaccheroni, P. B. Savage, V. Trapani, P. Sale and F. I. Wolf, J. Am. Chem. Soc., 2006, 128, 344–350 CrossRef CAS; (c) G. Farrugia, S. Iotti, L. Prodi, N. Zaccheroni, M. Montalti, P. Savage, G. Andreani, V. Trapani and F. I. Wolf, J. Fluorescence DOI:10.1007/s10895-008-0374-6; (d) M. Montalti, L. Prodi and N. Zaccheroni, J. Mater. Chem., 2005, 15, 2810–2814 RSC.
- Theme Issue: Fluorescent Sensors, J. Mater. Chem., 2005, 15, 2617–2976 RSC.
- L. Basade-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993 RSC.
- (a) J. W. Bridges, P. J. Creaven and R. T. Wiliams, Biochem. J., 1965, 96, 872 CAS; (b) J. Mohanty, H. Pal and A. V. Sapre, Bull. Chem. Soc. Jpn., 1999, 72, 2193 CrossRef CAS.
- (a) J. Feitelson, J. Phys. Chem., 1964, 68, 691; (b) M. Krauss, J. O. Jensen and H. F. Hameka, J. Phys. Chem., 1994, 9955 CrossRef CAS.
- (a) J. E. Coleman, in Zinc Enzymes, Birkauser, Boston, MA, 1986, ch. 4, pp. 49–58 Search PubMed; (b) Lindskog, in Zinc Enzymes, Birkauser, Boston, MA, 1986, ch. 22, pp. 307–316 Search PubMed; (c) E. Erksson, T. A. Jones and A. Liljas, in Zinc Enzymes, Birkauser, Boston, MA, 1986, ch. 23, pp. 317–328 Search PubMed; (d) A. C. Sen, C. K. Tu, H. Thomas, G. C. Wynns and Silvermann, in Zinc Enzymes, Birkauser, Boston, MA, 1986, ch. 24, pp. 329–340 Search PubMed; (e) Y. Pocker, N. Janjic and C. H. Miao, in Zinc Enzymes, Birkauser, Boston, MA, 1986, ch. 25, pp. 341–356 Search PubMed; (f) R. G. Khalifah, J. I. Rogers and J. Mukherjee, in Zinc Enzymes, Birkauser, Boston, MA, 1986, ch. 26, pp. 357–370 Search PubMed.
- J. M. Berg and Y. Shi, Science, 1996, 271, 1081 CrossRef CAS.
- (a) P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205 CrossRef CAS; (b) S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 7831 CrossRef CAS; (c) E. M. Nolan and S. J. Lippard, Inorg. Chem., 2004, 43, 8310 CrossRef CAS; (d) L. Rodriquez, J. C. Lima, J. Parola, R. Meitz, R. Aucejo, E. Garcia-España, J. M. Linares, C. Soriano and J. Alarcón, Inorg. Chem., 2008, 47, 6173 CrossRef.
- (a) G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, A. Guerri, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2007, 46, 309 CrossRef CAS; (b) P. Dapporto, M. Formica, V. Fusi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2000, 39, 4663 CrossRef CAS.
- M. Fontanelli and M. Micheloni, I Spanish–Italian Congress Thermodynamics of Metal Complexes, Univ. of Valencia, Spain, Peñiscola, June 3–6, 1990, p. 41.
- (a) G. Gran, Analyst, 1952, 77, 661 RSC; (b) F. J. Rossotti and H. Rossotti, J. Chem. Educ., 1965, 42, 375 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- (a) J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef; (b) M. T. Gandolfi, A. Credi, M. Montalti and L. Prodi, Handbook of Photochemistry, 3rd edn, CRC Press, Boca Raton, FL Search PubMed.
- N. Ceccanti, M. Formica, V. Fusi, L. Giorgi, M. Micheloni, R. Pardini, R. Pontellini and M. R. Tiné, Inorg. Chim. Acta, 2001, 321, 153 CrossRef CAS.
- P. Dapporto, M. Formica, V. Fusi, L. Giorgi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2001, 40, 6186 CrossRef CAS.
- (a) H. Pal and T. N. Das, J. Phys. Chem., 2003, 107, 5876 Search PubMed; (b) M. Jonsson, J. Lind and G. Merényi, J. Phys. Chem., 2002, 106, 4758 Search PubMed; (c) M. Jonsson, J. Lind and G. Merényi, J. Phys. Chem., 2003, 107, 5878 Search PubMed.
- (a) A. Testa, J. Photochem. Photobiol. A: Chem., 1992, 64, 73 CrossRef CAS; (b) G. Wenska, B. Skalski, Z. Gdaniec, R. W. Adamiak, J. Matulic-Adamic and L. Beigelman, J. Photochem. Photobiol. A: Chem., 2000, 133, 169–176 CrossRef CAS.
- (a) J. C. Batchelor, J. H. Prestegard, R. J. Cushley and S. R. Lipsy, J. Am. Chem. Soc., 1973, 95, 6558; (b) A. R. Quirt, J. R. Lyerla, I. R. Peat, J. S. Cohen, W. R. Reynold and M. F. Freedman, J. Am. Chem. Soc., 1974, 96, 570 CrossRef CAS; (c) J. C. Batchelor, J. Am. Chem. Soc., 1975, 97, 3410 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: Location of acidic hydrogen atoms in the protonated species of L2. See DOI: 10.1039/b810228g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |