DOI:
10.1039/B809654F
(Paper)
New J. Chem., 2009,
33, 157-163
The formation of silver nanofibres by liquid/liquid interfacial reactions: mechanistic aspects
Received
(in Montpellier, France)
9th June 2008
, Accepted 22nd August 2008
First published on 23rd October 2008
Abstract
The liquid/liquid interfacial reaction (LLIR) between silver nitrate in aqueous solution and ferrocene in organic solution has been investigated: the resultant silver deposit is found to contain long, well-defined nanometre scale fibres, together with thin silver nanowire networks. In situoptical microscopy and ex situscanning electron microscopy indicate that the 1D growth of the interfacial deposits is due to recrystallisation of the structure formed initially. Geometric factors are found to exert a larger effect on the 1D growth of silver by LLIRs compared to the electrochemical mechanism previously suggested by Scholz et al.
1. Introduction
Nanostructures (i.e. structures with at least one dimension in the range of 1 to 100 nm) have attracted increasing attention because of their unusual chemical and physical properties. There has been particular interest in methods of forming one dimensional (1D) nanostructures, including nanowires, because such structures provide a better model system for investigating the dependence of electronic transport, optical and mechanical properties on size confinement and dimensionality.1 Strategies for achieving 1D growth have been summarized by Xia et al.,2 these include: (i) use of the intrinsically anisotropic crystallographic structure of a solid;3 (ii) introduction of a liquid/solid interface to reduce the symmetry of a seed;4 or use of supersaturation control to modify the growth habit of a seed;5 (iii) use of various templates with 1D morphologies to direct the formation of nanostructures6–9 (iv) assembly of zero dimensional nanostructures (i.e.nanoparticles);10 (v) use of appropriate capping reagent(s) to kinetically control the growth rates of various facets of a seed.11,12 Another interface, the liquid/liquid (L/L) interface, can also be used to limit the growth of materials, as in (ii) above, or to assemble the symmetry of nanoparticles (NPs), as in (iv) above, where liquid/liquid interfacial reactions (LLIRs) are involved.
Metal NPs can be grown at the L/L interface either electrochemically, by applying a voltage across the L/L interface when sufficient electrolytes are present in each phase,13 or by spontaneous chemical reaction where the electron exchange between redox couples present in the oil and water phases is normally accompanied with biphasic ion exchange. Using the former approach, gold NPs,14 platinum NPs15 and pyrrole oligomers16 have been prepared electrochemically at the interface between immiscible electrolyte solutions. The second approach, using spontaneous deposition, can be traced back to Faraday’s formation of colloidal gold at the water/carbon disulfide boundary. In this case, particles can either be formed, or pre-formed particles can be assembled,17 at the L/L interface. A surprising variation in particle morphology has been reported at the L/L interface. Most interfacial deposits appear to consist of spherical NPs, which assemble to form films, or aggregate into larger structures if no stabilising ligands are present.18,19 The intrinsic difficulty in studying the larger-scale structure is in finding an appropriate microscopic technique to probe particle morphology in situ. Recent studies have described the preparation of well-defined metal and metal oxide NPs at the toluene/water interface, with X-ray scattering being used to study the nanometre-scale assembly of Au NPs into an ordered interfacial film.20,21Silver deposition by LLIR, the focus of this manuscript, has been described in a number of previous reports. Silver assembly (as opposed to formation) in the presence of surfactants at the water/dichloromethane interface produces a “metal liquid-like film”,22 whose structure has been described as micron-scale flocs of silver NPs.23,24Assembly of silver NPs at an aqueous/chloroform interface in the presence of thiol species has also been described.25 Other reports have suggested that more unusual structures are observed for Ag assembly and/or deposition at the L/L interface. Agitation of aqueous silver hydrosols, during their assembly at the water/toluene interface, has been reported to form “2D networks of uniform diameter nanowires”.26 The formation of silver deposits, by interfacial reduction with an organic phase electron donor, gives rise to intergrown “whisker” structures, although in this case the geometry is not uniform.27 The latter article noted that a transition between 1D and 2D growth could be tuned according to the experimental conditions of the spontaneous LLIR. By choosing appropriate organic solvents and concentrations of the reagents in the two phases, either silver whiskers (with radii from about 50 nm to 50 microns) or ultrathin Ag films were observed. Herein, we present further investigations into the spontaneous LLIR between Fc in various organic solvents and aqueous AgNO3 solutions, where long and well-defined Ag nanofibres were found under appropriate reaction conditions. The morphological evolution and reaction mechanism are also discussed, based on the micrographic observations.
2. Experimental
Silver nitrate (AgNO3, BDH Chemicals, GPR), ferrocene (Fc, 99%, Alfa Aesar), 1,2-dichoroethane (DCE, Rathburn, HPLC), nitrobenzene (NB, 99%, Sigma) and toluene (99%, Fisher Scientific) were used directly without further treatment. Typically, 3.3 mM of silver nitrate solution was prepared with deionised water from an Elga “Purelab Ultra” (Elga, Marlow, UK) system. Ferrocene was dissolved in DCE, or other organic solvents, as the organic phase for the interfacial reactions. The aqueous solution of AgNO3 and one of the organic Fc solutions were placed together in a glass tube with dimensions 75 mm (height) × 25 mm (diameter), following the sequence that the higher density phase was added prior to the light one. The mixture was then kept still at ambient temperature. The interfacial deposit was collected after 48 h of reaction, and transferred onto glass slides and dried in air. It was then washed with acetone and deionised water separately, and dried at ambient temperature before further analysis. Copper grids with holey carbon films (S147-4, Agar Scientific) were employed to collect samples for analysis viatransmission electron microscopy (TEM) and high resolution transmission microscopy (HRTEM). The samples were rinsed with both acetone and deionised water, and were dried in air in order to remove the remained contaminants.
In situ
optical microscopy was performed by a Leica DMIL optical microscope fitted with a Sony CCD-IRIS camera on an anti-vibration system (Active vibration isolation system TS-200, HWL Scientific Instruments GmbH). The X-ray diffraction (XRD) analysis was carried out using an Oxford Diffraction System (Xcalibur 2, Mo-Kα = 0.7093 Å), and XL 30 FEG Philips and ESEM XL30 Philips electron microscopes were employed at 15 kV for scanning electron microscopy (SEM). TEM and HRTEM were performed with a Tecnai F30 FEG-TEM system operating at 300 kV.
3. Results
The deposition of Ag resulting from the LLIR between AgNO3 in water and Fc in organic solvent can be written as:27| | | Fc(o) + Ag+(w) + X−(w)→ Fc+(o) + X−(o) + Ag(σ) | (1) |
where the subscripts “σ”, “w”, and “o” in the reactions represent interfacial, aqueous and organic phases, respectively, and the anion X− is added to balance the charge since no Fc+ transfer to the aqueous phase was believed to occur (in the experiments reported herein, X− is nitrate). The reaction was monitored in situ by an optical microscope placed on the active anti-vibration system. As shown in Fig. 1(a), at the beginning of the reaction (ca. 1 min), only separate particles were observed at the L/L interface. Many particles were rapidly generated, and started aggregating, after about five minutes of contact between the two phases (Fig. 1(b)). After 10 min, fractal-like aggregates appeared at the interface, as illustrated by Fig. 1(c), and the fluidity at the interface became obstructed after 25 min of reaction, owing to the appearance of large Ag agglomerates (see Fig. 1(d)). After 24 h of reaction (Fig. 1(e)), the L/L interface became somewhat solidified, and a grey coloured deposit was seen. The interfacial deposits became a bit denser compared to Fig. 1(e) when the reaction time was extended to 48 h (shown in Fig. 1(f)), but no visible fibre-like deposit was found under the optical microscopy. The process was also investigated ex situ by SEM, and the morphological evolution of the Ag deposit is illustrated by Fig. 2, although an important point to note here is that the extraction and drying of the sample could induce changes in morphology. The interfacial deposit seen after twenty five minutes of reaction appeared as micron-scale “flakes” with a few nuclei on the surface as displayed in Fig. 2(a), indicative of a possible destruction of an originally compact 2D interfacial layer during the sample collection. After 1 h of the reaction, some 1D Ag deposits can be differentiated from others (see Fig. 2(b)). After 4 h of reaction (Fig. 2(c)), some of the “microflakes” are found with holes and irregular edges in the background and co-exist with the 1D Ag nanostructures, which are not seen at shorter or longer reaction times. After 48 h, long Ag nano-scale fibres are observed, as illustrated in Fig. 2(d), together with some short 1D nanostructures and “microflakes”. The inset shows that the nanofibre is rather smooth and well-defined at a larger magnification. The Ag nanofibres shown in Fig. 2(d) are measured and give an average diameter of 171 ± 4 nm (N = 13) with a mean aspect ratio of ca. 174, where the largest aspect ratio from the other micrographs is observed to be ca. 450. Fig. 2(e) further reveals that the growth of the nanofibres originates from defects, such as independent nuclei or the edges of the “microflakes”etc. TEM and HRTEM micrographs offer the means of observation under higher magnification. Fig. 3(a) suggests that some of the “microflakes” observed under SEM are actually composed of networks of thinner 1D nanostructures, termed “nanowires” in the following text, with an average diameter of 14.8 ± 3.7 nm (N = 163), where the distribution of the diameter values visible in Fig. 3(a) is shown in Fig. 3(b). Fig. 3(a) also illustrates the evolution from the 2D film to 1D nanostructures, where a few branches of nanowires are observed to extend from a piece of film higlighted by the circle in the figure. The triangular highlights in Fig. 3(c) suggest that the growth of the nanowires originates from the triangular nuclei, and the 1D extension from the nuclei is “welded” on meeting other wires as shown by the appearance of lattice fringes under TEM (circular highlight). The HRTEM image in Fig. 3(d) indicates that the wires are nanocrystalline. From the micrographs presented in Fig. 2 and 3, one can summarize that the microflakes (the 2D growth owing to the presence of the L/L interface) are formed in the early stages of the LLIR, a process followed by 1D growth at the active sites of the interfacial layer after a long-term LLIR, such as the edge of the layer or the tips of triangular nuclei. The intermediate stage (holey microflakes with irregular edges displayed in Fig. 2(c)) at 4 h of the LLIR, suggests the appearance of a parallel process which might be either the dissolution of the as-formed microflakes; alternatively the secondary aggregation or growth of the nuclei may occur, to form thin nanowire networks shown in Fig. 3(a). Considering that complete microflakes and nanofibres are seen in the SEM micrographs at 48 h of the LLIR, and the nanowire networks can only be seen under TEM, the visible networks in Fig. 3(c) would appear to arise from the dissolution of the as-formed 2D layers. The LLIR process is therefore viewed as involving: (i) formation of Ag nuclei at the L/L interface; (ii) agglomeration of the as-prepared Ag nuclei to form 2D flakes due to the constraint of the L/L interface; (iii) dissolution of some of the initial Ag nuclei while secondary nucleation or growth occurs elsewhere. Some of the larger 2D structures can even be dissolved if the nuclei are depleted; (iv) the structural defects, including the tips of the triangular nuclei, independent nuclei and the edges of the microflakes, offer active sites for new nucleation, leading to 1D growth. The diameter of the 1D nanostructures is normally dependant on the size of the protuberance of the defects, for example larger nuclei for the nanofibres, compared to the corner of the smaller triangular nuclei for the nanowires.
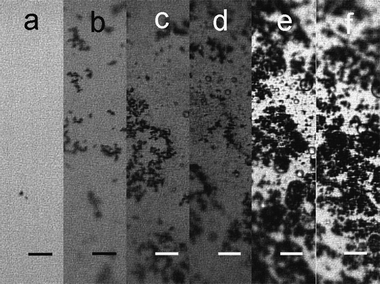 |
| | Fig. 1
Optical micrographs recorded during the formation of Ag interfacial deposits by LLIR: (a) at ca. 1 min, (b) at 5 min, (c) at 25 min, (d) at 1 h, (e) at 24 h, (f) at 48 h. The length of the scale bars in the figure is 50 microns. | |
 |
| | Fig. 2
SEM micrographs of the Ag interfacial deposits collected at different times during the LLIR process: (a) at 25 min, (b) at 1 h, (c) at 4 h, (d) at 48 h, the inset shows the diameter of the nanowire is around 100–200 nm; (e) at 48 h, 1D growth stems from the defects of the interfacial deposit. | |
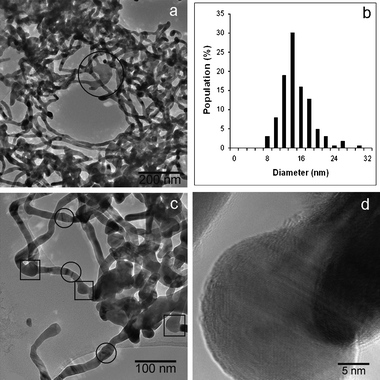 |
| | Fig. 3
TEM and HRTEM micrographs of the Ag interfacial deposit of the LLIR reaction between 3.3 mM AgNO3 aqueous solution and 5 mM Fc in DCE at 48 h: (a) nanowire networks in the deposit, the highlighted part shows the 1D growth from a piece of a 2D thin film, (b) distribution of the diameter of the nanowires, (c) the connection of the nanowires, the parts highlighted with “□” indicate that nanowires protrude from portions that were originally nanoparticulate. The other parts highlighted with “○” denote the “welding” positions of the nanowires; (d) HRTEM image of the nanowires, which indicates the nanowires are composed of crystalline silver. | |
Different organic solvents, such as NB and toluene, were used instead of DCE in the LLIR process. A thin and continuous film was observed at the NB/water interface in a Teflon container after 24 h of LLIR between 3.3 mM AgNO3 solution and 1 mM Fc in NB (see Fig. 4(a)), in accordance with the results described by Scholz and Hasse.27 In contrast, a radial pattern gradually appeared after a period of time in a glass container (see Fig. 4(b)), in possible association with random vibrations. The pattern was quite stable over time, and exhibited a “self-recovery” capability from external disruption of the interface. If the LLIR was forced to stop “half way” by the depletion of the silver salt, a ring-like thin film extending from the surface of the glass tube with some irregular deposit in the centre was seen, indicative of the adhesion of Ag nuclei on the hydrophilic surface of glass (as shown in Fig. 4(c)). Fig. 4(d) shows that no pattern appeared if the LLIR was carried out on an active anti-vibration table for 48 h. Hence, the pattern shown in Fig. 4(b) can be interpreted in terms of: (i) the adsorption of the initial Ag nuclei on the surface of glass tube; (ii) growth of the Ag deposit to form a ring-like interfacial deposit layer as shown in Fig. 4(c); (iii) this process continues and consequently the interfacial layer is able to cover the whole L/L interface; (iv) vibrations cause standing waves to form at the L/L interface, and the interfacial layer is then easily folded since its outer edge is “pinned” to the surface of the glass container. However, when the Ag nuclei are adjacent to the Teflon surface, no adsorption occurs, and the Ag layer formed by the LLIR “floats” on the L/L interface, hence no such standing waves are set up. Consequently no radial pattern appeared after 48 h of reaction.
The microstructure of the interfacial deposits in water/NB and water/toluene was observed under SEM. Fig. 5(a) shows that the interfacial deposit collected from the NB/water interface displays an analogous microstructure to that in Fig. 2(d). Long and well-defined nanofibres and other short 1D nanostructures are observed with the presence of “microflakes”. In contrast, no 1D growth is seen after the LLIR between 3.3 mM AgNO3 solution and 5 mM Fc in toluene, but only smoother “microflakes” are shown in Fig. 5(b) under SEM. The X-ray diffraction patterns of the Ag interfacial deposits formed by the LLIRs between 3.3 mM AgNO3 solution and 5 mM Fc in DCE, NB and toluene are presented in Fig. 6. Reflections assigned to Ag (111), (200), (220), (311), (222) planes are marked in the plot (Fm3m, a = 4.08 Å, JCPDF No. 02-1167). The crystallite size is estimated from the broadening of X-ray diffraction peaks by Scherrer’s equation:28
| | Bcrystallite = kλ/(L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) cosθ) | (2) |
where
λ is the wavelength of the X-ray,
θ is the Bragg angle,
L is the average crystallite size measured in a direction perpendicular to the surface of the specimen, and
k is a constant taken to be 0.9. The crystallite size calculated from the (111) reflection of the interfacial deposit of DCE/water system is 2.2 nm, which is the same as that in the
toluene/water system. The crystallite size for the interfacial deposit from the NB/water system is 3.1 nm. Other polar and nonpolar organic
solvents, such as
1,2-dichlorobenzene and
silicone oil (data not shown), were also employed to perform the LLIR with the same concentrations of reactants, and the morphology of the interfacial deposits also follows the same trend, in that polar organic
solvents favour the formation of 1D Ag deposits at L/L interfaces.
 |
| | Fig. 4 The Ag film obtained by interfacial reaction between 3.3 mM AgNO3 aqueous solution and 1 mM Fc in NB (after reaction for 24 h): (a) in Teflon container, (b) in glass container, (c) stopped half-way owing to the depletion of AgNO3 in glass container, (d) the LLIR in a glass container on an active anti-vibration table. | |
 |
| | Fig. 5
SEM micrographs of the Ag interfacial deposits at different LLIR systems: (a) 3.3 mM AgNO3 aqueous solution with 5 mM Fc in NB, (b) 3.3 mM AgNO3 aqueous solution with 5 mM Fc in toluene. | |
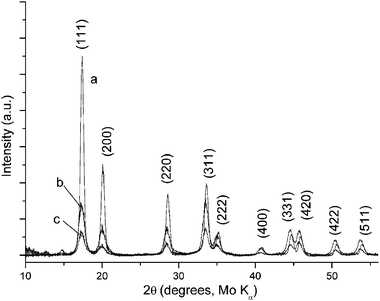 |
| | Fig. 6
X-Ray diffraction patterns of the Ag interfacial deposits by the LLIR between 3.3 mM AgNO3 aqueous solution and 5 mM Fc in (a) DCE, (b) NB and (c) toluene. | |
The effect of varying the concentrations of the reagents was investigated. As shown in Fig. 7(a) and (b), a lower concentration of AgNO3 solutions was employed to react with 5 mM Fc in DCE. The 0.33 mM AgNO3 (c+Ag/cFc = 0.066) exhibited a tendency to 1D growth, while the 0.07 mM AgNO3 solution (c+Ag/cFc = 0.014) basically formed Ag aggregates. If the concentration of Fc was varied, as illustrated in Fig. 7(c) and (d), 1D growth could be seen but was not fully developed in the case of 3.3 mM AgNO3 solution reacted with 0.5 mM (c+Ag/cFc = 6.6) and with 0.1 mM Fc in DCE (c+Ag/cFc = 33). The influence of the organic solvent on the resultant composition of the aqueous phase was also investigated via the visible absorbance of the aqueous phase (Fig. 8). The toluene/water system shows the highest transfer of Fc+ to water, whereas the NB/water displays the weakest spectral response. The transfer is also found to be proportional to the concentration of Fc employed in the DCE solutions (data not shown).
 |
| | Fig. 7
SEM micrographs of the Ag interfacial deposits as a function of reagent concentration: (a) 0.33 mM AgNO3 aqueous solution with 5 mM Fc in DCE, (b) 0.07 mM AgNO3 aqueous solution with 5 mM Fc in DCE, (c) 3.3 mM AgNO3 aqueous solution with 0.5 mM Fc in DCE, (d) 3.3 mM AgNO3 aqueous solution with 0.1 mM Fc in DCE. | |
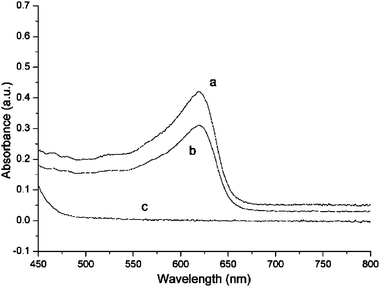 |
| | Fig. 8
UV-Vis
absorbance spectra of the aqueous solutions recorded after 48 h of LLIR between 3.3 mM AgNO3 aqueous solution and 5 mM Fc in (a) toluene, (b) DCE and (c) NB, respectively (deionised water as reference). The absorbance maximum of Fc+ appears at about 620 nm. | |
4. Discussion
Scholz and Hasse27 have proposed an electrochemical mechanism for the deposition of silverviaLLIR, where the reaction occurs at the L/L interface (see eqn (1), above). In the treatment of Scholz et al., the nuclei at the L/L interface were viewed as disc-shaped microelectrodes, where the current (i) to the equivalent disc-shaped silver/electrolyte interface was described by:| | 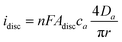 | (3) |
where n is the number of electrons transferred, F is the Faraday constant, Adisc is the surface area of the disc, ca and Da the bulk concentration and diffusion coefficient of species a, respectively, and r is the radius of the disc. When the same crystal is growing in a 1D mode, a cylinder electrode was used by Scholz and Hasse to approximate the current flow across the silver/liquid interface:
| |
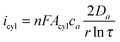
| (4) |
where
τ =
Dat/
r2, and
t is the time.
Acyl and
r are the surface area and radius of the cylinder, respectively. Since the oxidative process (oxidation of Fc) should balance the reductive one (reduction of Ag
+) at all times, Scholz and Hasse assumed the 1D Ag structure must protrude into the organic phase for the above-named fluxes to balance under conditions of excess
silver ion, since equating
(3) and (4) leads to:
| |
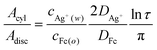
| (5) |
where
DAg+ and
DFc are the diffusion coefficients of Ag
+ in the aqueous phase and Fc in the organic phase, respectively.
Eqn (5) indicates that the higher concentration ratio of Ag
+ to Fc increases the ratio of
Acyl to
Adisc and favours 1D growth
, whereas the converse case should lead to 2D films. Note that the Ag
+ reduction can occur at a site distant from Fc oxidation, if the electrical conductivity of the deposit is sufficient.
The data presented in Fig. 7 act as experimental tests of eqn (5): from inspection of the deposits, it is clear that the initial concentration ratio of the LLIR is not the sole factor controlling deposit morphology (cf.eqn (5)), and the concentration of AgNO3 itself actually exerts a large effect on the final deposit. Combined with the observation (in Fig. 3) that no 1D Ag nanostructures were found in the first 25 min of the LLIR, which also suggests that a secondary crystallisation step is involved in the 1D growth, this leads to the conclusion that the actual mechanism is more complicated than the simple electrochemical process suggests. However, the experiments on the effect of organic solvents demonstrate that the use of non-polar solvents suppressed the formation of 1D structures. The morphological evolution seen here suggests that 2D thin films are formed initially at the L/L interface, followed with a transformation from 2D to 1D growth, in the case of more polar organic solvents.
The spontaneous LLIR between Fc in the organic phase and Ag+ in the aqueous solution initially generates Ag nuclei. An associated transfer of Fc+ from the organic phases (i.e.DCE, NB or toluene) to water, or of nitrate in the reverse direction, must occur to preserve electroneutrality. The visible absorbance band (see Fig. 8) centred on 620 nm is attributed to Fc+, on the basis of a previous report,29 but the extent of transfer is a function of the polarity of the solvent: the least polar solvent (toluene) is least able to solvate the Fc+ nitrate ion pair, hence in the toluene case, transfer of Fc+ to the aqueous phase predominates.
This observation, combined with the change to 2D morphology on using the less polar solvent, leads to the following mechanism being postulated for the 1D growth mode. Ag nuclei are initially generated by the spontaneous LLIR, and form a 2D interfacial layer because of the constraint of the L/L interface. After that, a transformation from 2D layers to 1D nanostructures occurs as a higher flux of reactants to the deposit can be sustained by radial diffusion.30 Consequently, parts of the 2D structures appear to dissolve, accompanied with the emergence of 1D nanostructures. The 1D growth is observed to occur at active sites with high surface energy, such as independent nuclei, the edges of 2D structures or the corner of the small triangular crystals, which show surprisingly well-defined long nanofibres without any branches. Nanowires from the Ag triangular crystals can even connect to form nanowire networks. The evolution of the 1D process is, however, suppressed in less polar solvents since it requires the (unfavourable) formation of a ferrocenium nitrate ion pair in the organic phase, by transfer of the nitrate, or the transfer of the ferrocenium to the aqueous phase. The latter process is more favourable, but the extent of transfer depends on the distance from the interface where the ferrocenium ion is formed. In the case of a 2D Ag deposit, the ferrocenium ion is formed adjacent to the interface and is readily transferred. By contrast, if 1D growth occurs, the ferrocenium ion may be formed some distance from the aqueous phase (a distance determined by the length of the structure). We therefore suggest that the driving force behind the morphological change observed in the deposit is the solvation of the ions formed by the LLIR.
5. Conclusions
The liquid/liquid interfacial reaction (LLIR) between silver nitrate in aqueous solution and ferrocene in various organic solvents has been investigated: long and well-defined silver nanofibres and thin nanowire networks were obtained in more polar media. In situoptical microscopy and ex situscanning electron microscopy indicate that the 1D growth of the interfacial deposits is due to directed recrystallization, where geometric factors associated with the flux to the growing deposit, and energetic factors, associated with the solvation of the ions generated, play an important role.
Acknowledgements
The authors thank the financial support from the UK Engineering & Physical Science Research Council (EPSRC, grant EP/C509773/1).
References
- Y. N. Xia and P. D. Yang, Adv. Mater., 2003, 15, 351 CrossRef CAS.
- Y. N. Xia, P. D. Yang, Y. G. Sun, Y. Y. Wu, B. Mayers, B. Gates, Y. D. Yin, F. Kim and Y. Q. Yan, Adv. Mater., 2003, 15, 353 CrossRef CAS.
- B. Gates, B. Mayers, B. Cattle and Y. N. Xia, Adv. Funct. Mater., 2002, 12, 219 CrossRef CAS.
- T. J. Trentler, K. M. Hickman, S. C. Goel, A. M. Viano, P. C. Gibbons and W. E. Buhro, Science, 1995, 270, 1791 CrossRef CAS.
- Y. Y. Wu and P. D. Yang, J. Am. Chem. Soc., 2001, 123, 3165 CrossRef CAS.
- K. Luo, C. T. Walker and K. J. Edler, Adv. Mater., 2007, 19, 1506 CrossRef CAS.
- J. H. Song, Y. Y. Wu, B. Messer, H. Kind and P. D. Yang, J. Am. Chem. Soc., 2001, 123, 10397 CrossRef CAS.
- M. Lai and D. J. Riley, Chem. Mater., 2006, 18, 2233 CrossRef CAS.
- R. A. W. Dryfe, E. C. Walter and R. M. Penner, ChemPhysChem, 2004, 5, 1879 CrossRef CAS.
- D. Wyrwa, N. Beyer and G. Schmid, Nano Lett., 2002, 2, 419 CrossRef CAS.
- Y. G. Sun, B. Mayers, T. Herricks and Y. N. Xia, Nano Lett., 2003, 3, 955 CrossRef CAS.
- A. Tao, F. Kim, C. Hess, J. Goldberger, R. R. He, Y. G. Sun, Y. N. Xia and P. D. Yang, Nano Lett., 2003, 3, 1229 CrossRef CAS.
- M. Guainazzi, G. Silvestri and G. Serravalle, J. Chem. Soc., Chem. Commun., 1975, 200 RSC.
- Y. F. Cheng and D. J. Schiffrin, J. Chem. Soc., Faraday Trans., 1996, 92, 3865 RSC.
- A. Trojanek, J. Langmaier and Z. Samec, J. Electroanal. Chem., 2007, 599, 160 CrossRef CAS.
- V. J. Cunnane and U. Evans, Chem. Commun., 1998, 2163 RSC.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458 CrossRef CAS.
- R. A. W. Dryfe, Phys. Chem. Chem. Phys., 2006, 8, 1869 RSC.
- C. Johans, K. Kontturi and D. J. Schiffrin, J. Electroanal. Chem., 2002, 526, 29 CrossRef CAS.
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas, V. V. Agrawal and P. Saravanan, J. Phys. Chem. B, 2003, 107, 7391 CrossRef CAS.
- C. N. R. Rao and K. P. Kalyanikutty, Acc. Chem. Res., 2008, 41, 489 CrossRef CAS.
- D. Yogev and S. Efrima, J. Phys. Chem., 1988, 92, 5754 CrossRef CAS.
- D. Yogev, M. Deutsch and S. Efrima, J. Phys. Chem., 1989, 93, 4174 CrossRef CAS.
- H. Schwartz, Y. Harel and S. Efrima, Langmuir, 2001, 17, 3884 CrossRef CAS.
- J. K. Sakata, A. D. Dwoskin, J. L. Vigorita and E. M. Spain, J. Phys. Chem. B, 2005, 109, 138 CrossRef CAS.
- T. Maddanimath, A. Kumar, J. D’Arcy-Gall, P. G. Ganesan, K. Vijayamohanan and G. Ramanath, Chem. Commun., 2005, 1435 RSC.
- F. Scholz and U. Hasse, Electrochem. Commun., 2005, 7, 541 CrossRef CAS.
-
C. Suryanarayana and M. G. Norton, X-Ray Diffraction A Practical Approach, Plenum Press, New York and London, 1998, vol. 6 Search PubMed.
- H. Hotta, S. Ichikawa, T. Sugihara and T. Osakai, J. Phys. Chem. B, 2003, 107, 9717 CrossRef CAS.
-
C. M. A. Brett and A. M. O. Brett, Electrochemistry, Principles, Methods and Applications, Oxford University Press, Oxford, 1993, ch. 5 Search PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.