Gold imidazolium-based ionic liquids, efficient catalysts for cycloisomerization of γ-acetylenic carboxylic acids
Florentina Neaţua, Vasile I. Pârvulescua, Véronique Micheletb, Jean-Pierre Gênetb, Alexandre Goguetc and Christopher Hardacrecd
aDepartment of Chemical Technology and Catalysis, University of Bucharest, B-dul Regina Elisabeta 4-12, 030016, Bucharest, Romania. E-mail: v_parvulescu@chem.unibuc.ro; Fax: +40 21 4010241
bLaboratoire de Synthèse Sélective Organique et Produits Naturels, ENSCP, UMR 7573, 11 rue P. et M. Curie, F-725231, Paris Cedex 05, France. E-mail: veronique-michelet@enscp.fr; Fax: +33 1 44071062
cCenTACat School of Chemistry and Chemical Engineering, Queen’s University, Stranmillis Road, Belfast, Northern Ireland, UK BT9 5AG
dQUILL School of Chemistry and Chemical Engineering, Queen’s University, Stranmillis Road, Belfast, Northern Ireland, UK BT9 5AG. E-mail: c.hardacre@qub.ac.uk; Fax: +44 28 90974687
First published on 20th October 2008
Abstract
Ionic liquid stabilized gold(III) chloride is shown to be a very active catalyst in the cyclization of sterically hindered and unhindered acetylenic carboxylic acid substrates even in the absence of a base.
Introduction
Ionic liquids (ILs) have received significant attention recently because they exhibit several advantages over molecular solvents with respect to their environmental impact.1 Catalytic reactions in ILs have been examined for at least 20 years and have been used for a wide variety of carbon–carbon bond forming reactions, which were the first to be undertaken in this media, and a number of good reviews cover the area of ILs.2–6 The extensive interest stems from the fact that the properties of ionic liquids may be tuned in order to suit a particular application by varying the cation–anion combination systematically and thereby are useful engineering solvents. In addition, for chemical reactions, the ionic liquid provides an ionic environment which can significantly alter the reactivity and selectivity of processes compared with molecular solvents.7The transition metal-catalyzed cyclization of 4-alkynoic acids constitutes a major route8 for the construction of 5-membered exocyclic lactones and has been the subject of a large number of investigations.9–14 Recently, a very attractive route to perform this reaction under mild conditions in the presence of gold was reported.15,16 In our laboratory, the catalytic properties of AuCl and AuCl3 for gem-substituted substrates, in the absence of base, was described15 as well as the use of two heterogeneous systems Au2O317 and Au/beta.18 These systems were found to be active for both substituted and unsubstituted substrates. In addition, the optimized heterogeneously catalyzed system was found to be recyclable.22 Whilst gold has been shown to be highly active, it commonly undergoes significant deactivation due to the ease by which it may change its oxidation state and, in the case of nanoparticles, the catalyst particle size leading to instability in the form of the active catalyst.19 Therefore, modalities which can enhance the stability are extremely important for practical applications.20 These can be in the form of the solvent used or the nature of the support.21 For example, stabilization of gold in the form of nanoparticles in ionic liquids by imidazolium derivatives has been reported.22–25
In this report the behavior of a 4 wt% Au/beta catalyst and a series of 1,3-dialkylimidazolium tetrachloroaurate salts in ionic liquids as catalysts for the cyclization of acetylenic substrates has been studied. Gold has recently been reported as a catalyst in ionic liquids in the hydration of phenylacetylene26,27 and the syntheses of substituted 3(2H)-furanones,27 2,5-dihydrofurans28 and substituted indoles.29 However, with the exception of the Co2(CO)8-catalyzed intramolecular and intermolecular Pauson–Khand annelation using 1-butyl-3-methylimidazolium hexafluorophosphate ([C4mim][PF6]) and tetrafluoroborate ([C4mim][BF4]) ionic liquids as solvents, no other related reactions to the cyclization of acetylenic substrates have been reported in ILs, to date.30
Experimental
Catalysts preparation
 | ||
| Fig. 1 Schematic of the cation of the basic ionic liquid (BIL) used as solvent for the cyclization of the functionalized acetylenic substrates 1a and 1b. | ||
Catalyst characterization
The solid catalysts were characterized by nitrogen adsorption–desorption isotherms at 77 K (Micromeritics ASAP 2000) after out-gassing the samples at 393 K for 12 h.The XPS spectra were recorded using a Kratos Axis UltraDLD spectrometer with monochromatic Al-Kα radiation. The data were analyzed using Casa-XPS (v2.3.13) employing a Shirley-background subtraction prior to fitting. EXAFS data were collected at the Synchrotron Radiation Source in Daresbury, UK, using station 9.3. The spectra were recorded at the Au LIII edge using a double crystal Si(111) monochromator. Scans were collected and averaged. Data were processed using EXCALIB which was also used to convert raw data into energy vs. absorption data. EXBROOK was used to remove the background. The analysis of the EXAFS was performed using EXCURV98.34 The gold concentration was determined by ICP-AES.
Catalytic tests
Typically 0.26 mmol of acetylenic acid was stirred with 2.5 mol% [Cnmim][AuCl4] dissolved in 0.5 g [Cnmim]Cl in air at room temperature, until completion of the reaction. After the completion of the reaction, the reaction products were extracted three times with diethyl ether and the solvent completely removed under vacuum to give the corresponding lactone. 1H and 13C NMR were recorded on a Bruker AV 300 instrument operating at 300 Hz to identify the products. The measured NMR spectra for 3-phenyl-5-methylene-γ-butyrolactone (2a), 3-n-butyl-3-ethoxycarbonyl-5-methylene-γ-butyrolactone (2b), 3-methoxycarbonyl-5-methylene-3-(3′-phenylprop-2′-enyl)-γ-butyrolactone (2c), 3-allyl-3-methoxycarbonyl-5-methylene-γ-butyrolactone (2d), 3-benzyl-3-ethoxycarbonyl-5-methylene-γ-butyrolactone (2e) and 3-methoxycarbonyl-5-methylene-γ-butyrolactone (2f) were in good agreement were those reported previously.19–21 In all cases, no cyclization is observed in the absence of any of the gold catalysts irrespective of the solvent used.Results and discussion
Catalysts characterization
The beta zeolite used in these experiments had a surface area of 464 m2 g−1 and a pore volume of 0.96 cm3 g−1. After the deposition of gold (4 wt%) the surface area decreased to 383 m2 g−1 and the pore volume to 0.80 cm3 g−1. TEM analysis of this material shown an uniform size distribution with an average of 3 nm. The characterization of IL-stabilized gold(III) chloride was examined using TEM, XPS and EXAFS. Fig. 2 shows the XPS spectra of the Au 4f photoelectron emission corresponding to the mixture of the catalyst ([C6mim][AuCl4]) with the ionic liquid [C6mim]Cl after the reaction. The binding energies of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4f7/2 and Au4f5/2 levels were located at 90.0 eV and 86.3 eV, respectively. EXAFS of [C6mim][AuCl4] dissolved in [C6mim]Cl showed a single peak at 0.22 nm associated with 4 chlorine atoms in the first coordination shell. During reaction, this peak decreases slightly and a small decrease in the white line of the XANES is observed. This variation may indicate that chlorine is being replaced by a lighter element such as coordination by the substrate during reaction, as would be expected. TEM analysis is in agreement with the XPS and EXAFS measurements and showed no nanoparticle formation. These observations are consistent with the presence of a dissolved Au(III) species with ∼4 chlorines in the first coordination sphere.35
4f7/2 and Au4f5/2 levels were located at 90.0 eV and 86.3 eV, respectively. EXAFS of [C6mim][AuCl4] dissolved in [C6mim]Cl showed a single peak at 0.22 nm associated with 4 chlorine atoms in the first coordination shell. During reaction, this peak decreases slightly and a small decrease in the white line of the XANES is observed. This variation may indicate that chlorine is being replaced by a lighter element such as coordination by the substrate during reaction, as would be expected. TEM analysis is in agreement with the XPS and EXAFS measurements and showed no nanoparticle formation. These observations are consistent with the presence of a dissolved Au(III) species with ∼4 chlorines in the first coordination sphere.35![The XPS spectra in the Au 4f region of the [C6mim][AuCl4] after reaction.](/image/article/2009/NJ/b812580e/b812580e-f2.gif) | ||
| Fig. 2 The XPS spectra in the Au 4f region of the [C6mim][AuCl4] after reaction. | ||
Catalytic tests
Table 1 shows the activity of Au/beta in a range of ionic liquids for the cyclization of the functionalized acetylenic substrates 1a and 1b.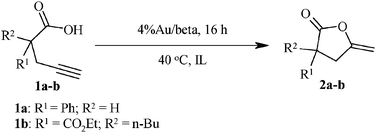
Although cycloisomerization of substrate 1a did occur in the BIL and [P66614][HSO4] with conversions of 75 and 70%, respectively, (Table 1, entries 1, 2), the ILs could not be separated from the reaction products due to the high solubility of the ionic liquid–substrate mixture in diethyl ether. In the case of the reaction performed in [C4mim][NTf2] and [P66614][NTf2], the ionic liquid could only be partially separated from the reactants and products and showed conversion of the substrate of 80% and 70%, respectively (Table 1, entries 3, 4). In each case, the conversions were obtained from NMR determination in the ionic liquid. [C6mim]Cl was found to separate efficiently from diethyl ether and the cycloisomerization of 1a (Table 1, entry 5) resulted in the desired compound 2a in 79% isolated yield.
Due to the ease of workup [C6mim]Cl was also examined as a medium for the cyclization of 1b over Au/beta. From an analysis of the 1H NMR, a conversion of 91% was obtained with an isolated yield of the lactone of 58% (Table 1, entry 6). Similar activities and selectivities were also found in conventional molecular solvents, such as acetonitrile (Table 1, entry 7).
The heterogeneously catalyzed reaction results were compared with the use of the ionic liquid as a catalyst in the form of the tetrachloroaurate based ionic liquids. Since [C6mim][AuCl4] is a solid at room temperature, [C6mim]Cl was used as solvent. The results are summarised in Table 2.

To date, it has only been possible to exclude a base from the homogeneous reaction conditions if the two substituents on a tetrahedral centre were of a significant size, as understood by the Thorpe–Ingold effect.36 In the case of the ionic liquid catalyst system, all the γ-acetylenic carboxylic acids examined were cleanly transformed to the corresponding γ-alkylidene γ-butyrolactones. Moreover, similar activity and selectivity was found for the ionic liquid catalyst compared with the homogeneous AuCl catalysts system in acetonitrile.15 Irrespective of the alkenyl side chain length the lactones were isolated in 85–96% yields (Table 2, entries 1–5) even at room temperature. In all cases, the catalytic amount of gold ionic liquid used in these reactions was equivalent to the amount used under heterogeneous conditions. No side reactions were observed on the alkenyl side chains during the course of the reaction.
Using the gold based ionic liquid system, complete transformations of 2-prop-2-ynylmalonic acid monomethyl ester 1f with an 84% isolated yield (Table 2, entry 6) and of 2-phenylpent-4-ynoic acid 1a with an 96% isolated yield (Table 2, entry 7) after 1 h at room temperature were obtained. Similar results were found for the reaction of 1a in the presence of [C2mim][AuCl4], [C4mim][AuCl4] and [C18mim][AuCl4] at room temperature. These results for unsubstituted substrates are comparable with those obtained under heterogeneous Au2O3 conditions (3 h),21 or homogeneous AuCl/K2CO3 conditions (2 h)20 and show significant advantages over other homogeneous gold chloride based catalysts.19 In the case of the homogeneous catalysts, the formation of degradation products or the corresponding methylketone was observed for the sterically unhindered substrates and the reaction only occurred in the presence of a base. In the ionic liquid catalyzed reactions, excellent reactivity was found without the need for additives. Furthermore, the IL-catalysts were recycled three times without any loss in conversion or yields.
A comparison of the homogeneous reaction (Table 2, entries 7 and 1) with that of the heterogeneous reaction (Table 1, entries 5 and 6) indicates that the former is more active requiring a lower temperature and resulting in a higher conversion/yield after 1 h. This is reflected in the turnover frequencies (TOF) for the homogeneous catalysts compared with the supported catalysts which are ∼19.8 h−1 (at RT) and ∼1.3 h−1 (at 40 °C), respectively, for the formation of 2b, for example. The lower rate is unlikely to be due to mass transfer limitations in the heterogenous case which has been observed in other solid catalyzed reactions in ionic liquids37 as the acetonitrile reaction (Table 1, entries 6 and 7) also shows reduced activity. The higher TOF may be expected due to the inaccessibility of the bulk gold atoms; however, even taking into account the lower dispersion of the heterogeneous catalyst, which contains 3 nm gold particles, the homogeneous catalyst has a higher intrinsic activity compared with the heterogeneous system.
Scheme 1 shows a proposed mechanism for the gold based ionic liquid catalyzed reaction. From the XPS and EXAFS, the active catalyst is thought to be Au3+ which activates the acetylenic group.20 The ionic liquid provides a medium which eliminates the use of a base. This role of the ionic liquid may be to shift the acid equilibrium of the starting material to favour the carboxylate anion, either by hydrogen bonding with the chloride, for example, or via stabilizing the anion in the ionic environment. This increase in concentration of the active intermediate allows reaction and activation of the carbon–carbon triple bond by the Au3+ centre and nucleophilic addition to the alkyne. After the cyclization a hydrogen mediated demetalation ends the catalytic cycle. It should be noted that the scheme does not preclude the possibility that the cation of the ionic liquid may play an important role in creating the active catalyst. For example, imidazolium based ionic liquids have been shown to form carbenes with homogeneous catalysts in C–C bond forming reactions.10
![The mechanism of cyclization of acetylenic carboxylic acid using Au3+ derived catalysts. It should be noted that representation of the gold as [Au] is for illustrative purposes only and will exist in the form of a [AuClx]y+ complex.](/image/article/2009/NJ/b812580e/b812580e-s1.gif) | ||
| Scheme 1 The mechanism of cyclization of acetylenic carboxylic acid using Au3+ derived catalysts. It should be noted that representation of the gold as [Au] is for illustrative purposes only and will exist in the form of a [AuClx]y+ complex. | ||
Conclusions
Gold chloride based ionic liquids have been shown to be very active catalysts in the cyclization of acetylenic substrates. The ionic liquid medium prevents nanoparticle formation via stabilization of the gold in the form of isolated species by chlorine coordination. The ionic liquid also allows activation of the carboxylic acid in the starting material and provides a medium which eliminates the need for added base in order to cyclize unhindered acetylenic carboxylic acid.Acknowledgements
This work was financially supported by the Consiliul National al Cercetarii Stiintifice din Invatamantul Support (CNCSIS) and the Centre National de la Recherche Scientifique (CNRS), a Portfolio partnership from the EPSRC and an EU transnational grant. CCLRC are thanked for providing EXAFS beamtime.References
- J. A. Boon, J. A. Levinsky, J. I Pflug and J. S. Wilkes, J. Org. Chem., 1986, 51, 480 CrossRef CAS.
- Ionic Liquids IIIA and B: Fundamentals, Progress Challenges and Opportunities–Properties and Structure, eds. R. D. Rogers and K. R. Seddon, American Chemical Society, Washington DC, 2005 Search PubMed.
- J. S. Wilkes, J. Mol. Catal., A: Chem., 2004, 214, 11 CrossRef CAS.
- Ionic Liquids in Synthesis, eds. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2007 Search PubMed.
- C. Hardacre, Annu. Rev. Mater. Res., 2005, 35, 29 CrossRef CAS.
- V. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 106, 2615 CrossRef.
- C. Hardacre, J. D. Holbrey, M. Nieuwenhuyzen and T. G. A. Youngs, Acc. Chem. Res., 2007, 40, 1146 CrossRef CAS.
- (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS; (b) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS; (c) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS; (d) for Ru-catalyzed intermolecular reactions, see C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311 Search PubMed.
- H. Imagawa, Y. Fujikawa, A. Tsuchihiro, A. Kinoshita, T. Yoshinaga, H. Takao and N. Nishizawa, Synlett, 2006, 639 CrossRef CAS.
- C. H. Oh, H. J. Yi and J. H. Lee, New J. Chem., 2007, 31, 835 RSC.
- R. W. Spencer, T. F. Tam, E. Thomas, V. J. Robinson and A. Krantz, J. Am. Chem. Soc., 1986, 108, 5589 CrossRef CAS.
- Z. Huo, N. T. Patil, T. Jin, N. K. Pahadi and Y. Yamamoto, Adv. Synth. Catal., 2007, 349, 680 CrossRef CAS; Z. Ahmed, U. Albrecht and P. Langer, Eur. J. Org. Chem., 2005, 3469 CrossRef CAS; A. Duchêne, J. Thibonnet, J.-L. Parrain, E. Anselmi and M. Abarbri, Synthesis, 2007, 597 CAS.
- M. Jimenez-Tenotio, M. C. Puerta, P. Valerga, F. J. Moreno-Dorado, F. M. Guerra and G. M. Massanet, Chem. Commun., 2001, 2324 RSC.
- S. Elgafi, L. D. Field and B. A. Messerle, J. Organomet. Chem., 2000, 607, 97 CrossRef CAS.
- (a) E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J.-P. Genêt and V. Michelet, J. Am. Chem. Soc., 2006, 128, 3112 CrossRef CAS; (b) E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J.-P. Genêt and V. Michelet, ARKIVOC, 2007, v, 67.
- H. Harkat, J.-M. Weibel and P. Pale, Tetrahedron Lett., 2006, 47, 6273 CrossRef CAS ; for a recent Au-catalyzed cyclization of γ- and δ-acetylenic acids, see: E. Marchal, P. Uriac, B. Legoin, L. Toupet and P. van de Weghe, Tetrahedron, 2007, 63, 9979 Search PubMed.
- P. Y. Toullec, E. Genin, S. Antoniotti, J.-P. Genêt and V. Michelet, Synlett, 2008, 707 CAS.
- F. Neaţu, Z. Li, R. Richards, P. Y. Toullec, J.-P. Genêt, K. Dumbuya, J. M. Gottfried, H.-P. Steinrück, V. I. Pârvulescu and V. Michelet, Chem.–Eur. J., 2008, 14, 9412 CrossRef.
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212 CrossRef CAS and references therein.
- R. J. David, Science, 2003, 301, 926 CrossRef CAS , and references therein.
- (a) Z.-R. Tang, J. K. Edwards, J. K. Bartley, S. H. Taylor, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 249, 208 CrossRef CAS; (b) S. Carrettin, A. Corma, M. Iglesias and F. Sanchez, Appl. Catal., A, 2005, 291, 247 CrossRef CAS.
- K.-S. Kim, D. Demberelnyamba and H. Lee, Langmuir, 2004, 20, 556 CrossRef CAS.
- H. Itoh, K. Naka and Y. Chujo, J. Am. Chem. Soc., 2004, 126, 3026 CrossRef CAS.
- R. Tatumi and H. Fujihara, Chem. Commun., 2005, 83 RSC.
- M. Hasan, I. V. Kozhevikov, M. R. H. Siddiqui, A. Steiner and N. Winterton, Inorg. Chem., 1999, 38, 5637 CrossRef CAS.
- M. Deetlefs, H. G. Raubenheimer and M. W. Esterhuysen, Catal. Today, 2002, 72, 29 CrossRef CAS.
- Y. Liu, M. Liu, S. Guo, H. Tu, Y. Zhou and H. Gao, Org. Lett., 2006, 8, 3445 CrossRef CAS.
- O. Aksin and N. Krause, Adv. Synth. Catal., 2008, 350, 1106 CrossRef CAS.
- I. Ambrogio, A. Arcadi, S. Cacchi, G. Fabrizi and F. Marinelli, Synlett, 2007, 1775 CAS.
- P. Mastrorilli, C. F. Nobile, R. Paolillo and G. P. Suranna, J. Mol. Catal., A: Chem., 2004, 214, 103 CrossRef CAS.
- C. M. Gordon, J. D. Holbrey, A. R. Kennedy and K. R. Seddon, J. Mater. Chem., 1998, 8, 2627 RSC; J. D. Holbrey and K. R. Seddon, J. Chem. Soc., Dalton Trans., 1999, 2133 RSC; A. Downard, M. J. Earle, C. Hardacre, S. E. J. McMath, M. Nieuwenhuyzen and S. J. Teat, Chem. Mater., 2004, 16, 43 CrossRef CAS.
- P. Bonhôte, A. P. Dias, N. Papageorgiou, K. Kalyanasundram and M. Grätzel, Inorg. Chem., 1996, 5, 1168 CrossRef.
- U. Fröhlich, PhD Thesis, Queens University, Belfast, 2005; U. Fröhlich, P. Goodrich, H. Q. N. Gunaratne, C. Hardacre, A. McKeown and K. R. Seddon, to be published; C. Paun, J. Barklie, P. Goodrich, H. Q. N. Gunaratne, A. McKeown, V. I. Pârvulescu and C. Hardacre, J. Mol. Catal., A: Chem., 2007, 269, 64 Search PubMed.
- N. Binstead, EXCURV98: CCLRC Daresbury Laboratory computer program, 1998.
- H. Kitagawa, N. Kojima and T. Nakajima, J. Chem. Soc., Dalton Trans., 1991, 3121 RSC.
- C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080 RSC.
- P. N. Davey, S. A. Forsyth, H. Q. N. Gunaratne, C. Hardacre, A. McKeown, S. E. J. McMath, D. W. Rooney and K. R. Seddon, Green Chem., 2005, 7, 224 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |