Novel thiophene-conjugated indoline dyes for zinc oxide solar cells
Takuya Dentania, Yasuhiro Kubotaa, Kazumasa Funabikia, Jiye Jinb, Tsukasa Yoshidac, Hideki Minourac, Hidetoshi Miurad and Masaki Matsui*a
aDepartment of Materials Science and Technology, Faculty of Engineering, Gifu University, Yanagido, Gifu 501-1193, Japan. E-mail: matsui@apchem.gifu-u.ac.jp; Fax: +81 58 293 2794; Tel: +81 58 293 2601
bDepartment of Chemistry, Faculty of Science, Shinshu University, 3-1-1 Asahi, Matsumoto, Nagano 390-8621, Japan
cEnvironmental and Renewable Energy System Division, Graduate School of Engineering, Gifu University, Yanagido, Gifu 501-1193, Japan
dChemicrea Co. Ltd., 2-1-6 Sengen, Tsukuba, Ibaragi 305-0047, Japan
First published on 16th October 2008
Abstract
The application of a series of thiophene-conjugated indoline dyes for zinc oxide solar cells, prepared by the one-step cathode deposition template method, was examined. The introduction of thiophene ring(s) into D131-type indoline dye improved the cell performance due to their appropriate energy levels and bathochromic shift in the UV-vis absorption band on zinc oxide. It is important for the oxidation potential (Eox) of dyes to have a more positive value than ca. 0.25 V vs. Fc/Fc+ in acetonitrile in order to show a high (>70%) incident photon-to-current efficiency.
Introduction
Organic dyes, such as coumarins,1 styryls,2 polyenes,3 dimethylfluorenyl-containing derivatives4 and indoline derivatives,5 have been reported to act as good sensitizers for titanium oxide. Bathochromic organic dyes, such as squaryliums,6 phthalocyanines7 and heptamethinecyanines,8 have also been reported to sensitize semiconductors. In particular, D149 has been reported to show the highest solar-light-to-electricity conversion efficiency (η) of 9.0% among organic dyes.5a One promising approach to improve the performance of sensitizers is the expansion of the π-conjugation system to absorb more photons. The introduction of ethylene and thiophene units into chromophores is a good methodology to expand π-conjugation.9 On the other hand, a convenient preparation process for zinc oxide thin films has been reported.10 The key point of this method is the formation of porous zinc oxide films at low temperature (<70 °C). Indoline dyes D131, D102 and D149, in which cyanoacrylic, monorhodanic and double rhodanic acids are used as anchor moieties, respectively, are known to show good performances.5f We report herein the application of novel thiophene-conjugated indoline dyes having a series of anchor moieties to zinc oxide dye-sensitized solar cells.Results and discussion
Synthesis of indoline dyes
Thiophene-conjugated indoline dyes 20–28 were synthesized, as shown in Scheme 1. Compound 1 was allowed to react with NBS (2) to give 3, followed by a reaction with thiophene boronic acids esters 4–7 to provide 8–11, which were formylated to give 12–15. These compounds were allowed to react with cyanoacetic, mono- and double-rhodanic acids 16–19 to provide 20–28. D131, D102 and D149 were prepared in a similar way.
UV-vis absorption and fluorescence spectra
The UV-vis absorption and fluorescence spectra of 20–28, D131, D102 and D149 are shown in Fig. 1, Fig. 2 and Fig. 3. The results are also listed in Table 1. All the indoline dyes showed first and second absorption bands at around 500 and 400 nm, respectively. The first absorption maximum (λmaxfirst) of monothiophene derivatives 20, 23, 24, 25 and 26 were more bathochromic than thiophene-free derivatives D131, D102 and D149. Interestingly, no further bathochromic shift was observed for di- and trithiophene derivatives 21, 22, 27 and 28 compared to monothiophene derivatives 20, 23, 24, 25 and 26, respectively. The molar absorption coefficients at the first absorption band (εfirst) of 20–28 were less than those of thiophene-free derivatives D131, D102 and D149. The half-widths of 20–28 (99–146 nm) were larger than those of D131, D102 and D149 (65–79 nm). No marked difference in εfirst among mono-, di- and trithiophene derivatives was observed, being in the range 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 to 47900. The second absorption maximum (λmaxsecond) of thiophene derivatives 20–28 showed a bathochromic shift, and at the same time, their molar absorption coefficients (εsecond) were larger with increasing numbers of thiophene units. No remarkable differences in the UV-vis absorption spectra between 20 and 23, and between 25 and 26, were observed. The fluorescence maximum (Fmax) showed a bathochromic shift by the introduction of a thiophene unit.
700 to 47900. The second absorption maximum (λmaxsecond) of thiophene derivatives 20–28 showed a bathochromic shift, and at the same time, their molar absorption coefficients (εsecond) were larger with increasing numbers of thiophene units. No remarkable differences in the UV-vis absorption spectra between 20 and 23, and between 25 and 26, were observed. The fluorescence maximum (Fmax) showed a bathochromic shift by the introduction of a thiophene unit.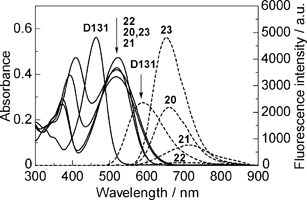 | ||
| Fig. 1 UV-vis absorption and fluorescence spectra of indoline dyes D131, 20, 21, 22 and 23 at a concentration of 1.0 × 10−5 mol dm−3 in chloroform. Solid and dotted lines represent UV-vis absorption and fluorescence spectra, respectively. | ||
 | ||
| Fig. 2 UV-vis absorption and fluorescence spectra of indoline dyes D102 and 24 at a concentration of 1.0 × 10−5 mol dm−3 in chloroform. Solid and dotted lines represent UV-vis absorption and fluorescence spectra, respectively. | ||
 | ||
| Fig. 3 UV-vis absorption and fluorescence spectra of indoline dyes D149, 25, 26, 27 and 28 at a concentration of 1.0 × 10−5 mol dm−3 in chloroform. Solid and dotted lines represent UV-vis absorption and fluorescence spectra, respectively. | ||
| Compound | λmax/nm (ε)a | Fmax/nma | RFIb | Eoxvs. Fc/Fc+ in MeCN/V | Eox−E0–0vs. Fc/Fc+ in MeCN/V |
|---|---|---|---|---|---|
| a Measured on 1.0 × 10−5 mol dm−3 of substrate in chloroform at 25 °C.b Relative fluorescence intensity.c Not measured due to low solubility. | |||||
| D131 | 463 (55400) | 591 | 83 | +0.41 | −2.00 |
| 325 (15600) | |||||
| 20 | 519 (43300) | 659 | 77 | +0.25 | −1.86 |
| 373 (27300) | |||||
| 21 | 517 (37700) | 712 | 27 | +0.23 | −1.81 |
| 393 (38300) | |||||
| 22 | 519 (41700) | 701 | 4 | +0.22 | −1.83 |
| 409 (47100) | |||||
| 23 | 523 (47300) | 653 | 203 | +0.25 | −1.85 |
| 373 (29100) | |||||
| D102 | 514 (54700) | 621 | 68 | +0.37 | −1.83 |
| 368 (25200) | |||||
| 24 | 548 (41400) | 702 | 80 | +0.25 | −1.75 |
| 388 (37100) | |||||
| D149 | 550 (68000) | 636 | 100 | +0.30 | −1.79 |
| 395 (32000) | |||||
| 25 | 571 (43500) | 717 | 78 | +0.24 | −1.67 |
| 410 (34800) | |||||
| 26 | 568 (45600) | 713 | 49 | —c | —c |
| 408 (40500) | |||||
| 27 | 564 (42000) | 743 | 10 | —c | —c |
| 405 (41900) | |||||
| 28 | 550 (47900) | 727 | 3 | —c | —c |
| 412 (48900) |
Electrochemical properties
The oxidation potentials (Eox) of D131, D102, D149 and 20–25 were measured by using an Ag/Ag+ electrode in acetonitrile to compare the energy levels of Eox and Eox−E0–0 of the dyes, the I−/I3− potential, and the conduction band of zinc oxide. Fc/Fc+ was used as a standard. The Eox of ferrocene was observed at +0.13 V vs. Ag/Ag+ in acetonitrile. Fig. 4 shows that the Eox of 20 was observed at +0.38 V vs. Ag/Ag+ in acetonitrile, corresponding to +0.25 V vs. Fc/Fc+ in acetonitrile. The I−/I3− potential level was observed at +0.09 V vs. Ag/Ag+ in acetonitrile, corresponding to −0.04 V vs. Fc/Fc+ in acetonitrile. | ||
| Fig. 4 The electrochemical measurement of 20 in acetonitrile (2 ml) containing tetrabutylammonium perchlorate (0.1 mol dm−3). Ag/Ag+ in acetonitrile was used as a reference electrode. Platinum wire was used as the working and counterelectrode. The scan rate was 100 mV s−1. | ||
The potential level of Eox−E0–0, where E0–0 represents the intersection of the normalized absorption and fluorescence spectra in solution, is considered to correspond to the LUMO energy level.9 The E0–0 of 20 was observed at 589 nm, corresponding to 2.11 eV. Therefore, the Eox−E0–0 value of 20 was calculated to be −1.86 V vs. Fc/Fc+ in acetonitrile. The energy levels of free indoline dyes measured in solution differed from those of adsorbed ones. Unfortunately, ferrocene and indoline dyes on a zinc oxide-coated ITO electrode did not give distinct redox responses due to a slow charge transfer process. Hence, the Eox of ferrocene and indoline dyes could not be determined. The Eox and Eox−E0–0 of all the indoline dyes are listed in Table 1.
UV-vis absorption and IR spectra of 20
The UV-vis absorption spectra of 20 are shown in Fig. 5. The λmaxfirst of 20 in chloroform and on zinc oxide were observed at 519 and 449 nm, respectively. Thus, large hypsochromic shift of λmaxfirst was observed on zinc oxide. The λmaxfirst of 20 in the presence of an equimolar amount of triethylamine (TEA) in chloroform was observed at 470 nm, there being slightly more bathochromic than that on zinc oxide. | ||
| Fig. 5 The UV-vis absorption spectra of 20 in chloroform and on zinc oxide. | ||
FTIR spectra of 20 are shown in Fig. 6. The IR spectrum of 20 in a potassium bromide disk showed an absorption band at around 1680 cm−1, which was assigned to a carbonyl stretching absorption. When indoline dye 20, adsorbed onto a zinc oxide film, was scraped off and its IR spectrum was measured in a potassium bromide disk, the absorption band at around 1680 cm−1 disappeared and new absorption was observed at around 1600 cm−1. This spectrum is similar to that of the triethylammonium salt of 20, in which the absorption band at around 1600 cm−1 is assigned to the asymmetric stretch absorption of the carboxylate anion. It was also observed that indoline dye 20 showed negative solvatochromism in solution (λmaxfirst = 516 (toluene), 519 (chloroform), 529 (dichloromethane), 465 (DMSO), 453 (acetonitrile) and 464 nm (methanol)). These results indicate that the hypsochromic shift of 20 on zinc oxide is mainly attributed to the formation of a bidentate complex between the carboxylate and zinc. The polar zinc oxide surface could also be attributed to the hypsochromic shift.
 | ||
| Fig. 6 FTIR spectra of 20: (a) 20, (b) 20 in the presence of zinc oxide and (c) the triethylammonium salt of 20. | ||
Photoelectrochemical properties
The cell performance of D131-type indoline dyes was examined. The normalized UV-vis absorption spectra on zinc oxide and action spectra are shown in Fig. 7. The results are also listed in Table 2. The cell performance was improved in the presence of cholic acid (CA), a co-adsorbate that can inhibit the aggregation of dyes on zinc oxide due to carboxylic acid and hydrophobic moieties. The UV-vis absorption spectra of 20 and 23 in the absence and presence of CA are depicted in Fig. 7(a). In the case of 20, a broad absorption at around 530 nm decreased in the presence of CA, indicating the prevention of aggregation on zinc oxide. Meanwhile, only slight differences in the absorption bands between the absence and presence of CA were observed for 23. The η values of 20 and 23 in the absence of CA were 3.13 and 3.30%, respectively (Table 2, runs 3 and 7). Those in the presence of CA were 3.78 and 3.36%, respectively (Table 2, runs 2 and 6). Thus, the η values of 20 and 23 were improved by 21 and 2% in the presence of CA, respectively. These results suggest that the hexyl group in 23 is very effective in inhibiting aggregation on zinc oxide. In the cases of 21 and 22, aggregation formation decreased in the presence of CA, resulting in an improved cell performance (Table 2, runs 4 and 5). The absorption bands of 20, 21, 22 and 23 on zinc oxide were more bathochromic than that of D131, as shown in Fig. 7(b). The action spectra show the sensitization of zinc oxide by 20, 21, 22 and 23 at around 550 nm, whereas no sensitization was observed for D131 at around 550 nm, as depicted in Fig. 7(c). The incident photon-to-current efficiency (IPCE) in the presence of CA was in the following dye order: 20 (83.1%) > 23 (78.2%), D131 (77.8%) > 21 (69.1%) > 22 (55.5%) (Table 2, runs 1, 2, 4–6). The short-circuit photocurrent densities (Jsc) of 21 (8.15 mA cm−2), 20 (8.09 mA cm−2), 23 (7.42 mA cm−2) and 22 (6.69 mA cm−2) were higher than that of D131 (5.55 mA cm−2). The fill factor (ff) was lowered by introducing a thiophene unit. Consequently, the η value was in the following order of dyes: 20 (3.78%) > 23 (3.36%), 21 (3.19%) > D131 (2.60%) > 22 (2.08%). Thus, an improvement in cell performance was successfully observed for a series of D131-type thiophene-conjugated indoline dyes. The improved cell performance of 20, 21 and 23, compared with D131, mainly came from the bathochromic shift in the absorption band and a high IPCE (>70%) to increase Jsc. | ||
| Fig. 7 (a) Normalized UV-vis absorption spectra of 20 and 23 on zinc oxide in the absence and presence of CA, (b) normalized UV-vis absorption spectra of D131, 20, 21, 22 and 23 on zinc oxide in the presence of CA and (c) action spectra of D131, 20, 21, 22 and 23 in the presence of CA. | ||
| Run | Compound | CAa | λmax/nm | Abs.b | IPCE (%) | Jsc/mA cm−2 | Voc/V | ff | ηc (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Equivalents of cholic acid with respect to dye.b Absorbance at absorption maximum on zinc oxide.c Action spectra and I–V characteristics under AM 1.5 irradiation (100 mW cm−2). | |||||||||
| 1 | D131 | 2 | 405 | 3.36 | 77.8 | 5.55 | 0.66 | 0.71 | 2.60 |
| 2 | 20 | 2 | 449 | 2.48 | 83.1 | 8.09 | 0.69 | 0.68 | 3.78 |
| 3 | 20 | 0 | 459 | 1.96 | 71.4 | 7.09 | 0.65 | 0.68 | 3.13 |
| 4 | 21 | 2 | 461 | 2.16 | 69.1 | 8.15 | 0.63 | 0.62 | 3.19 |
| 5 | 22 | 2 | 457 | 2.44 | 55.5 | 6.69 | 0.59 | 0.53 | 2.08 |
| 6 | 23 | 2 | 450 | 2.07 | 78.2 | 7.42 | 0.66 | 0.68 | 3.36 |
| 7 | 23 | 0 | 452 | 2.07 | 76.2 | 7.58 | 0.65 | 0.67 | 3.30 |
| 8 | D102 | 2 | 476 | 3.20 | 77.1 | 9.00 | 0.65 | 0.66 | 3.88 |
| 9 | 24 | 2 | 514 | 1.94 | 48.6 | 7.33 | 0.62 | 0.63 | 2.83 |
| 10 | 24 | 0 | 539 | 1.54 | 42.4 | 6.44 | 0.59 | 0.58 | 2.20 |
| 11 | D149 | 2 | 521 | 3.08 | 81.2 | 11.08 | 0.68 | 0.57 | 4.23 |
| 12 | 25 | 2 | 547 | 1.76 | 43.4 | 6.85 | 0.54 | 0.64 | 2.35 |
| 13 | 25 | 0 | 555 | 1.65 | 35.9 | 5.38 | 0.50 | 0.62 | 1.68 |
| 14 | 26 | 2 | 546 | 1.25 | 37.5 | 5.85 | 0.62 | 0.68 | 2.45 |
| 15 | 26 | 0 | 561 | 1.21 | 37.7 | 5.55 | 0.57 | 0.65 | 2.07 |
| 16 | 27 | 2 | 546 | 1.26 | 29.7 | 4.40 | 0.59 | 0.66 | 1.71 |
| 17 | 28 | 2 | 542 | 1.45 | 26.1 | 3.67 | 0.56 | 0.67 | 1.36 |
Next, the cell performance of D102 and 24 was examined (Table 2, runs 8–10). The UV-vis absorption and action spectra are shown in Fig. 8. The absorption band of 24 was more bathochromic than that of D102. The absorption spectrum of 24 in the absence of CA clearly showed a broad absorption at around 600 nm, suggesting the formation of aggregates. Fig. 8(b) shows the sensitization of zinc oxide by 24 at around 630 nm. However, the IPCE value of 24 was lower than that of D102 so as not to increase Jsc. The open-circuit voltage (Voc) and ff of 24 were lower than those of D102 (Table 2, runs 8 and 9). Thus, no improvement in cell performance was observed for D102-type thiophene-conjugated indoline dyes.
 | ||
| Fig. 8 (a) Normalized UV-vis absorption spectra of D102 and 24 on zinc oxide in the absence and presence of CA, and (b) action spectra of D102 and 24 in the presence of CA. | ||
Finally, the cell performance of D149-type indoline dyes was examined. The UV-vis absorption and action spectra of D149, 25, 26, 27 and 28 are shown in Fig. 9. In this case, the difference in the UV-vis absorption bands of 25 and 26 in the absence and presence of CA was small compared with those in the cases of 20 and 23, as shown in Fig. 9(a). The η values of 25 and 26 in the presence of CA were higher than those in the absence of CA (Table 2, runs 12–15). Fig. 9(b) shows that 25, 26, 27 and 28 are more bathochromic than D149. Fig. 9(c) indicates that though the sensitization of zinc oxide was observed for 25, 26, 27 and 28 at around 670 nm, their IPCE values were lower than that of D149. Thus, no improvement in cell performance was observed for the series of D149-type thiophene-conjugated indoline dyes (Table 2, runs 11, 12, 14, 16 and 17).
 | ||
| Fig. 9 (a) Normalized UV-vis absorption spectra of 25 and 26 on zinc oxide in the absence and presence of CA, (b) normalized UV-vis absorption spectra of D149, 25, 26, 27 and 28 on zinc oxide in the presence of CA, and (c) action spectra of D149, 25, 26, 27 and 28 in the presence of CA. | ||
Relationship between IPCE and Eox, Eox−E0–0
To examine why only D131-type thiophene-conjugated indoline dyes showed improved cell performances, the relationship between IPCE and energy levels was examined. Fig. 10(a) shows the relationship between IPCE and Eox. Indoline dyes D131, 20, 21, 23, D102 and D149, of which the Eox levels were more positive than the ca. +0.25 V vs. Fc/Fc+ in acetonitrile, showed high (>70%) IPCE values. The potential level of I−/I3− was observed at −0.04 V vs. Fc/Fc+ in acetonitrile. Fig. 10(b) shows the relationship between IPCE and Eox−E0–0. It was also found that indoline dyes D131, 20, 21, 23, D102 and D149 showed high IPCE values. It is reported that the potential levels of the conduction band of titanium oxide and the I−/I3− redox are −0.5 and +0.4 V vs. NHE, respectively, there being an energy gap of 0.9 V.9,11 The conduction band level of zinc oxide is similar to that of titanium oxide. Therefore, the level of zinc oxide is considered to be −0.94 V vs. Fc/Fc+ in acetonitrile, which is much more positive than the Eox−E0–0 levels of all the indoline dyes, the energy gap between Eox−E0–0 and the conduction band levels being larger than 0.7 V. It is suggested that an energy gap larger than 0.2 V between Eox and I−/I3−, and Eox−E0–0 and the conduction band levels, respectively, are required.9 Thus, though the Eox−E0–0 level of all the indoline dyes are sufficiently negative, their Eox levels are critical for the sensitization cycle to proceed. No marked difference in the Eox levels among the thiophene-conjugated derivatives 20 (+0.25 V), 21 (+0.23 V), 22 (+0.22 V), 23 (+0.25 V), 24 (+0.25 V) and 25 (+0.24 V) was observed in solution. However, their Eox level on zinc oxide could differ from that in solution. As the Eox level of D131 (+0.41 V vs. Fc/Fc+ in MeCN) was more positive than those of D102 (+0.37 V) and D149 (+0.30 V) in solution, those of D131-type derivatives 20, 21, 22 and 23 might be more positive than those of D102- and D149-type derivatives 24, 25, 26, 27 and 28 on zinc oxide. The Eox levels of D102, 20, 21 and 22 were observed at +0.37, +0.25, +0.23 and +0.22 V vs. Fc/Fc+ in acetonitrile, respectively. This suggests that the Eox level can negatively shift with increasing numbers of thiophene units on zinc oxide. Therefore, the Eox levels of D131-type mono- and dithiophene derivatives 20, 21 and 23 could be more positive than those of D102- and D149-type derivatives, and the redox potential of I−/I3− on zinc oxide could show an improved cell performance. As a result, indoline dyes 20, 21 and 23 could show better performances than D131 due to larger Jsc values. The Eox levels of D102- and D149-type thiophene-conjugated indoline dyes 24, 25, 26, 27 and 28 might be too negative on zinc oxide, despite their bathochromic shift in the UV-vis absorption spectrum on zinc oxide. In order to improve the performance of indoline dyes, it is important to design derivatives of them having more positive Eox level.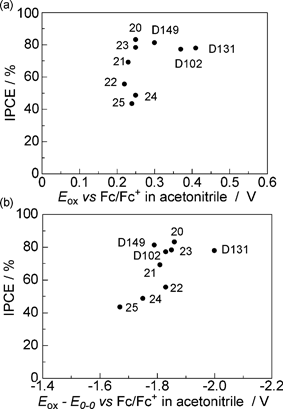 | ||
| Fig. 10 The relationship between IPCE and energy levels: (a) IPCE vs. Eox and (b) IPCE vs. Eox−E0–0. | ||
Conclusion
A series of D131-, D102- and D149-type thiophene-conjugated indoline dyes were examined as sensitizers for zinc oxide solar cells, prepared by the one-step cathode deposition template method. Among the series of thiophene-conjugated indoline dyes, D131-type indoline dyes improved cell performance. This could have been due to their positive Eox levels. In order to improve the performance of D102- and D149-type indoline dyes, it is important to design derivatives of them having more positive Eox levels.Experimental
General
Melting points were measured with a Yanagimoto MP-52 micro-melting-point apparatus. NMR spectra obtained using a JEOL JNM-ECX 400P spectrometer. EI and FAB MS spectra were recorded on a JEOL MStation 700 spectrometer. UV-vis absorption and fluorescence spectra were acquired on Hitachi U-3500 and F-4500 spectrophotometers, respectively. Cyclic voltammetry was carried out using an EG&G Princeton Applied Research Potentiostat/Galvanostat (Model 263A) driven by the M270 software package. One-step cathode electrodeposition was undertaken using a Hokuto-Denko HSV-100 potentiostat system. The photoelectrochemical measurements of solar cells were performed on a Bunko-Keiki CEP-2000 system. The I–V curve measurements of solar cells were performed on an EKO Instruments I–V curve tracer MP-160 and Grating spectroradiometer LS-100.Electrochemical measurements
The electrochemical measurements of indoline dyes D131, 20, 21, 22, 23, D102, 24, D149, 25, ferrocene and potassium iodide, were performed in acetonitrile. The oxidation potential (Eox) was measured by using three small-sized electrodes. Ag/Ag+ was used as a reference electrode. Platinum wire was used as the working and counterelectrode. Acetonitrile solutions (2 ml) of dyes containing tetrabutylammonium perchlorate (0.1 mol dm−3) were prepared. Dry argon gas was introduced into the solution for 10 min. The electrochemical measurements were then performed at a scan rate of 100 mV s−1.Preparation of the zinc oxide solar cell
An aqueous potassium chloride solution (300 ml, 0.1 mol dm−3) was electrolyzed at −1.0 V vs. SCE with bubbling oxygen gas at 70 °C for 30 min. Platinum was used as a counterelectrode. To the pre-electrolyzed film was added an aqueous solution of zinc chloride. The concentration of zinc chloride was adjusted to 5 mmol dm−3. Then, the film was again electrodeposited in the solution at −1.0 V vs. SCE at 70 °C for 20 min with bubbling oxygen gas. To the electrodeposited film was added an aqueous solution of eosin Y (0.050 mmol dm−3). The film was electrodeposited at −1.0 V vs. SCE at 70 °C for 30 min with bubbling oxygen gas. The film was kept in a dilute aqueous potassium hydroxide solution (pH 10.5) for 24 h to remove adsorbed eosin Y. The film was then dried at 100 °C for 1 h. The thin film was immersed in a chloroform solution of dye (1 × 10−4 mol dm−3) and kept at ambient temperature for 1 h to adsorb dyes 20–28 onto the zinc oxide. In the cases of D131, D102 and D149, the film was immersed in an acetonitrile–tert-butyl alcohol 1 : 1 mixed solution (0.5 mmol dm−3). Then, the film was washed with chloroform. In the cases of D131, D102 and D149, the film was washed with an acetonitrile–tert-butyl alcohol 1 : 1 mixed solution. The films were dried under an air atmosphere at ambient temperature. The film was used as the working electrode. A platinum spattered film was used as the counterelectrode. The cell size was 5.0 × 5.0 mm. Thermosetting resin was put around the cell. An acetonitrile–ethylene carbonate (v/v = 1 : 4) mixed solution containing tetrabutylammonium iodide (0.5 mol dm−3) and iodine (0.05 mol dm−3) was used as the electrolyte.Photoelectrochemical measurements
Action spectra were measured under monochromatic light with a constant photon number (5 × 1015 photon cm−2 s−1). I–V characteristics were measured under illumination with AM 1.5 simulated sun light (100 mW cm−2) through a shading mask (5.0 × 4.0 mm).Synthesis of dyes
8. Yield 72%, mp 204–206 °C. δH (400 MHz, CDCl3, Me4Si): 1.46–1.51 (1 H, m), 1.62–1.67 (1 H, m), 1.79–1.94 (3 H, m), 2.00–2.07 (1 H, m), 3.81–3.86 (1 H, m), 4.70–4.73 (1 H, m), 6.93 (1 H, s), 6.98–7.06 (6 H, m), 7.15 (1 H, s), 7.16–7.17 (1 H, m) and 7.26–7.39 (12 H, m). m/z (EI) = 495 (M+, 100), 466 (24) and 248 (8).
9. Yield 49%, mp 105–107 °C. δH (400 MHz, CDCl3, Me4Si): 1.39–1.47 (1 H, m), 1.55–1.59 (1 H, m), 1.69–1.84 (3 H, m), 1.91–2.00 (1 H, m), 3.69–3.73 (1 H, m), 4.58–4.61 (1 H, m), 6.91–7.03 (8 H, m), 7.08–7.10 (3 H, m) and 7.21–7.34 (12 H, m). m/z (FAB) = 578 (MH+).
10. Yield 53%, mp 106–109 °C. δH (400 MHz, CDCl3, Me4Si): 1.48–1.59 (1 H, m), 1.62–1.66 (1 H, m), 1.82–1.93 (3 H, m), 2.03–2.06 (1 H, m), 3.80–3.88 (1 H, m), 4.67–4.78 (1 H, m), 6.94 (1 H, s), 6.98–7.11 (10 H, m), 7.17 (1 H, d, J = 3.4 Hz), 7.21 (1 H, d, J = 4.8 Hz) and 7.27–7.41 (12 H, m). m/z (FAB) = 660 (MH+).
11. Yield 56%, mp 57–61 °C. δH (400 MHz, CDCl3, Me4Si): 0.89 (3 H, t, J = 6.8 Hz), 1.30–1.34 (6 H, m), 1.42–1.52 (1 H, m), 1.59–1.64 (3 H, m), 1.78–1.90 (3 H, m), 1.97–2.07 (1 H, m), 2.58 (2 H, t, J = 7.6 Hz), 3.78–3.82 (1 H, m), 4.66–4.69 (1 H, m), 6.73 (1 H, s), 6.92–7.04 (7 H, m) and 7.22–7.40 (12 H, m). m/z (FAB) = 580 (MH+).
12. Yield 90%, mp 112–114 °C. δH (400 MHz, CDCl3, Me4Si): 1.47–1.53 (1 H, m), 1.65–1.67 (1 H, m), 1.82–1.89 (3 H, m), 2.07–2.11 (1 H, m), 3.81–3.85 (1 H, m), 4.76–4.79 (1 H, m), 6.94 (1 H, s), 6.96 (1 H, d, J = 8.5 Hz), 7.01 (2 H, d, J = 8.8 Hz), 7.05 (2 H, d, J = 8.8 Hz), 7.23–7.39 (13 H, m), 7.68 (1 H, d, J = 4.1 Hz) and 9.80 (1 H, s). m/z (EI) = 523 (M+, 100), 495 (56), 373 (52) and 344 (31).
13. Yield 74%, mp 115–117 °C. δH (400 MHz, CDCl3, Me4Si): 1.44–1.53 (1 H, m), 1.59–1.66 (1 H, m), 1.77–1.91 (3 H, m), 2.00–2.09 (1 H, m), 3.79–3.81 (1 H, m), 4.70–4.73 (1 H, m), 6.93 (1 H, s), 6.95 (1 H, d, J = 8.5 Hz), 6.99 (2 H, d, J = 8.9 Hz), 7.03 (2 H, d, J = 8.9 Hz), 7.08 (1 H, d, J = 3.9 Hz), 7.18 (1 H, d, J = 3.9 Hz), 7.24–7.40 (13 H, m), 7.62 (1 H, d, J = 4.1 Hz) and 9.82 (1 H, s). m/z (FAB) = 606 (MH+).
14. Yield 26%, mp 230–232 °C. δH (400 MHz, CDCl3, Me4Si): 1.46–1.54 (1 H, m), 1.62–1.68 (1 H, m), 1.79–1.86 (3 H, m), 1.98–2.02 (1 H, m), 3.77–3.99 (1 H, m), 4.46–4.69 (1 H, m), 6.38–7.40 (23 H, m), 7.57–7.58 (1 H, m) and 9.78 (1 H, s). m/z (FAB) = 688 (MH+).
15. Yield 96%, mp 66–68 °C. δH (400 MHz, CDCl3, Me4Si): 0.89 (3 H, t, J = 7.0 Hz), 1.30–1.39 (6 H, m), 1.45–1.48 (1 H, m), 1.61–1.73 (3 H, m), 1.79–1.90 (3 H, m), 2.02–2.05 (1 H, m), 2.91 (2 H, t, J = 7.0 Hz), 3.79–3.83 (1 H, m), 4.73–4.77 (1 H, m), 6.93 (1 H, s), 6.94 (1 H, d, J = 8.2 Hz), 7.00 (2 H, d, J = 8.9 Hz), 7.04 (2 H, d, J = 8.9 Hz), 7.06 (1 H, s), 7.23–7.40 (12 H, m) and 9.95 (1 H, s). m/z (FAB) = 608 (MH+).
20. Yield 64%, mp 268–271 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.23–1.32 (1 H, m), 1.59–1.68 (2 H, m), 1.79–1.84 (2 H, m), 1.99–2.08 (1 H, m), 3.82–3.86 (1 H, m), 4.86–4.90 (1 H, m), 6.97 (1 H, d, J = 8.7 Hz), 7.02 (2 H, d, J = 8.5 Hz), 7.07 (1 H, s), 7.11 (2 H, d, J = 8.5 Hz), 7.19–7.22 (2 H, m), 7.28–7.36 (5 H, m), 7.41–7.48 (4 H, m), 7.57–7.59 (2 H, m), 7.95 (1 H, d, J = 3.4 Hz) and 8.43 (1 H, s). m/z (FAB) = 591.2106 (MH+, C39H31N2O2S requires 591.2106).
21. Yield 62%, mp 194–198 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.28–1.35 (1 H, m), 1.58–1.69 (2 H, m), 1.81–1.86 (2 H, m), 2.00–2.07 (1 H, m), 3.81–3.85 (1 H, m), 4.81–4.84 (1 H, m), 6.98 (1 H, d, J = 9.2 Hz), 7.00 (2 H, d, J = 9.1 Hz), 7.06 (1 H, s), 7.09 (2 H, d, J = 9.1 Hz), 7.20–7.21 (2 H, m), 7.28–7.53 (13 H, m), 7.82 (1 H, br s) and 8.28 (1 H, br s). m/z (FAB) = 673.1974 (MH+, C43H33N2O2S2 requires 673.1983).
22. Yield 79%, mp 266–269 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.29–1.36 (1 H, m), 1.61–1.70 (2 H, m), 1.84–1.87 (2 H, m), 2.00–2.05 (1 H, m), 3.81–3.85 (1 H, m), 4.80–4.83 (1 H, m), 6.98 (1 H, d, J = 6.3 Hz), 7.00 (2 H, d, J = 7.8 Hz), 7.06 (1 H, s), 7.08 (2 H, d, J = 7.8 Hz), 7.20–7.22 (2 H, m), 7.28–7.47 (13 H, m), 7.60–7.62 (2 H, m), 7.98 (1 H, d, J = 3.4 Hz) and 8.49 (1 H, s). m/z (FAB) = 755.1831 (MH+, C47H35N2O2S3 requires 755.1861).
23. Yield 93%, mp 240–243 °C. δH (400 MHz, DMSO-d6, Me4Si): 0.84 (3 H, t, J = 6.0 Hz), 1.20–1.30 (7 H, m), 1.57–1.65 (4 H, m), 1.76–1.84 (2 H, m), 1.97–2.06 (1 H, m), 2.74 (2 H, t, J = 7.2 Hz), 3.78–3.82 (1 H, m), 4.82–4.86 (1 H, m), 6.93 (1 H, d, J = 8.2 Hz), 6.99 (2 H, d, J = 8.6 Hz), 7.04 (1 H, s), 7.07 (2 H, d, J = 8.6 Hz), 7.17–7.19 (2 H, m), 7.26–7.34 (5 H, m), 7.38–7.45 (4 H, m), 7.50 (1 H, s), 7.54 (1 H, s) and 8.25 (1 H, s). m/z (FAB) = 675.3117 (MH+, C45H43N2O2S requires 675.3045).
24. Yield 89%, mp 175–178 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.29–1.33 (1 H, m), 1.59–1.68 (2 H, m), 1.81–1.84 (2 H, m), 2.00–2.05 (1 H, m), 3.81–3.85 (1 H, m), 4.69 (2 H, s), 4.84–4.88 (1 H, m), 6.95 (1 H, d, J = 8.5 Hz), 7.01 (2 H, d, J = 8.3 Hz), 7.06 (1 H, s), 7.09 (2 H, d, J = 8.3 Hz), 7.19–7.21 (2 H, m), 7.28–7.34 (5 H, m), 7.39–7.49 (4 H, m), 7.57–7.58 (2 H, m), 7.77 (1 H, d, J = 3.9 Hz) and 8.10 (1 H, s). m/z (FAB) = 697.1647 (MH+, C41H33N2O3S3 requires 697.1653).
25. Yield 31%, mp > 300 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.14 (3 H, t, J = 6.9 Hz), 1.25–1.37 (1 H, m), 1.60–1.68 (2 H, m), 1.80–1.84 (2 H, m), 2.01–2.08 (1 H, m), 3.82–3.86 (1 H, m), 3.98–4.00 (2 H, m), 4.69 (2 H, s), 4.84–4.87 (1 H, m), 6.96 (1 H, d, J = 8.5 Hz), 7.00 (2 H, d, J = 9.3 Hz), 7.07 (1 H, s), 7.08 (2 H, d, J = 9.3 Hz), 7.20–7.55 (13 H, m), 7.68 (1 H, d, J = 3.9 Hz) and 8.02 (1 H, s). m/z (FAB) = 824.1757 (MH+, C46H38N3O4S4 requires 824.1745).
26. Yield 81%, mp > 300 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.24–1.29 (1 H, m), 1.25 (18 H, s), 1.59–1.87 (2 H, m), 1.85–1.87 (2 H, m), 2.01–2.09 (1 H, m), 3.83–3.87 (1 H, m), 4.61 (2 H, s), 4.85–4.88 (1 H, m), 5.19 (2 H, s), 6.98 (1 H, d, J = 8.5 Hz), 7.00 (2 H, d, J = 8.8 Hz), 7.07 (1 H, m), 7.10 (2 H, d, J = 8.8 Hz), 7.19–7.48 (14 H, m), 7.53 (1 H, s), 7.57 (1 H, d, J = 3.9 Hz), 7.72 (1 H, d, J = 4.1 Hz) and 8.07 (1 H, s). m/z (FAB) = 998.3133 (MH+, C59H56N3O4S4 requires 998.3154).
27. Yield 95%, mp > 300 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.22–1.35 (1 H, m), 1.23 (18 H, s), 1.57–1.66 (2 H, m), 1.78–1.85 (2 H, m), 1.95–2.05 (1 H, m), 3.73–3.79 (1 H, m), 4.33 (2 H, s), 4.74–4.78 (1 H, m), 5.10 (2 H, s), 6.91 (1 H, d, J = 8.5 Hz), 6.99 (2 H, d, J = 8.3 Hz), 7.03 (1 H, s), 7.04 (2 H, d, J = 8.3 Hz), 7.17–7.54 (18 H, m), 7.62 (1 H, s) and 7.94 (1 H, s). m/z (FAB) = 1080.3016 (MH+, C63H58N3O4S5 requires 1080.3031).
28. Yield 32%, mp > 300 °C. δH (400 MHz, DMSO-d6, Me4Si): 1.22–1.33 (1 H, m), 1.26 (18 H, s), 1.59–1.68 (2 H, m), 1.80–1.84 (2 H, m), 1.97–2.03 (1 H, m), 3.78–3.83 (1 H, m), 4.46 (2 H, s), 4.77–4.81 (1 H, m), 5.17 (2 H, s), 6.96 (1 H, d, J = 8.5 Hz), 6.98 (2 H, d, J = 9.1 Hz), 7.05 (1 H, s), 7.06 (2 H, d, J = 9.1 Hz), 7.19–7.54 (20 H, m), 7.71 (1 H, s) and 8.02 (1 H, s). m/z (FAB) = 1162.2795 (MH+, C67H60N3O4S6 requires 1162.2908).
Acknowledgements
This work was financially supported in part by Grants-in-Aid for Science Research (no. 19550185) from the Japan Society for the Promotion of Science (JSPS).References
- (a) Z.-S. Wang, K. Hara, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, H. Arakawa and H. Sugihara, J. Phys. Chem. B, 2005, 109, 3907 CrossRef CAS; (b) K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC; (c) K. Hara, K. Sayama, Y. Ohga, A. Shinpo, S. Suga and H. Arakawa, Chem. Commun., 2001, 569 RSC.
- (a) T. Dentani, K. Nagasaka, K. Funabiki, J.-Y. Jin, T. Yoshida, H. Minoura and M. Matsui, Dyes Pigm., 2008, 77, 59 CrossRef CAS; (b) Q.-H. Yao, L. Shan, F.-Y. Li, D.-D. Yin and C.-H. Huang, New J. Chem., 2003, 27, 1277 RSC; (c) Z.-S. Wang, F.-Y. Li and C.-H. Huang, J. Phys. Chem. B, 2001, 105, 9210 CrossRef CAS; (d) Z.-S. Wang, F.-Y. Li, C.-H. Huang, L. Wang, M. Wei, L.-P. Jim and N.-Q. Li, J. Phys. Chem. B, 2000, 104, 9676 CrossRef CAS; (e) Z.-S. Wang, F.-Y. Li and C.-H. Huang, Chem. Commun., 2000, 2063 RSC.
- (a) R. Chen, X. Yang, H. Tian and L. Sun, J. Photochem. Photobiol., A, 2007, 189, 295 CrossRef CAS; (b) M. Liang, W. Xu, F. Cai, P. Chen, B. Peng, J. Chen and Z. Li, J. Phys. Chem. C, 2007, 111, 4465 CrossRef CAS; (c) H. Choi, J.-K. Lee, K.-J. Song, K. Song, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 1553 CrossRef CAS; (d) N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256 CrossRef CAS; (e) S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. D. Angelis, D. D. Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS; (f) D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245 RSC; (g) K. Hara, T. Sato, R. Katoh, A. Furube, T. Yoshihara, M. Murai, M. Kurashige, S. Ito, A. Shinpo, S. Suga and H. Arakawa, Adv. Funct. Mater., 2005, 15, 246 CrossRef CAS; (h) T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. A. Anderson, X. Ai, T.-Q. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806 CrossRef CAS; (i) K. Hara, M. Kurashige, S. Ito, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Chem. Commun., 2003, 252 RSC.
- (a) D. Kim, J.-K. Lee, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 1913 CrossRef CAS; (b) I. Jung, J.-K. Lee, K.-H. Song, K. Song, S. O. Kang and J. Ko, J. Org. Chem., 2007, 72, 3652 CrossRef CAS; (c) S. Kim, J.-K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. D. Angelis, D. D. Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS.
- (a) S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Pechy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202 CrossRef CAS; (b) L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813 CrossRef; (c) T. Horiuchi, H. Miura and S. Uchida, J. Photochem. Photobiol., A, 2004, 164, 29 CrossRef CAS; (d) T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218 CrossRef CAS; (e) T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036 RSC; (f) W. H. Howie, F. Claeyssens, H. Miura and L. M. Peter, J. Am. Chem. Soc., 2008, 130, 1367 CrossRef CAS.
- A. Otsuka, K. Funabiki, N. Sugiyama, T. Yoshida, H. Minoura and M. Matsui, Chem. Lett., 2006, 35, 666 CrossRef.
- P. Y. Reddy, L. Giribabu, C. Lyness, H. J. Snaith, C. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J.-H. Yum, K. Kalyanasundaram, M. Grätzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373 CrossRef CAS.
- M. Matsui, Y. Hashimoto, K. Funabiki, J. Y. Jin, T. Yoshida and H. Minoura, Synth. Met., 2005, 148, 147 CrossRef CAS.
- K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597 CrossRef CAS.
- T. Yoshida, M. Iwaya, H. Ando, T. Oekermann, K. Nonomura, D. Schlettwein, D. Wöhrle and H. Minoura, Chem. Commun., 2004, 400 RSC.
- R. Katoh, A. Furube, T. Yoshihara, K. Hara, G. Fujihashi, S. Takano, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 4818 CrossRef CAS.
- J. D. Mee, US Pat., 5679795, 1997 Search PubMed.
- G. Bidan, A. De Nicola, V. Enée and S. Guilerez, Chem. Mater., 1998, 10, 1052 CrossRef CAS.
- M. Matsui, M. Wang, K. Funabiki, Y. Hayakawa and T. Kitaguchi, Dyes Pigm., 2007, 74, 169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |