Probing multivalency for the inhibition of an enzyme: glycogen phosphorylase as a case study†
Samy
Cecioni
a,
Oana-Andreea
Argintaru
a,
Tibor
Docsa
b,
Pál
Gergely
b,
Jean-Pierre
Praly
a and
Sébastien
Vidal
*a
aUniversité de Lyon, Lyon, Université Lyon 1, Villeurbanne, CNRS, UMR5246, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires, Laboratoire de Chimie Organique 2 - Glycochimie, 43 Boulevard du 11 Novembre 1918, F-69622, Villeurbanne, France. E-mail: sebastien.vidal@univ-lyon1.fr; Fax: +33 472 448 349; Tel: +33 472 448 349
bCell Biology and Signalling Research Group of the Hungarian Academy of Sciences, Department of Medical Chemistry, Research Centre for Molecular Medicine, University of Debrecen, Nagyerdei krt. 98, Debrecen, H-4032, Hungary. E-mail: gpal@dote.hu; Fax: +36 52 412 566; Tel: +36 52 412 345
First published on 23rd October 2008
Abstract
Glycogen phosphorylase is involved in the hepatic glucose production and appears an emerging biological target for the treatment of type 2 diabetes. Two distinct trivalent inhibitors of GP were synthesized either through Cu(I)-assisted 1,3-dipolar cycloaddition or through formation of a tris-oxadiazole derivative. A biological study of the inhibiting properties of these trivalent inhibitors of GP have shown that the valency of the molecules influences slightly the inhibition of the enzyme whereas the presence of a spacer arm between the core and the pharmacophore moieties does not. The possible modes of binding of these multivalent inhibitors to the enzyme are discussed.
Introduction
Glycogen phosphorylase and diabetes
Diabetus mellitus affects about 3% of the world population and up to 6% for the adult population of developed countries. Diabetes, one of the major causes of death worldwide, is characterized by elevated glycaemia causing heart and kidney failures as well as visual impairment problems.1 Type 2 diabetes is non-insulino-dependent and arises from insulin signalling inefficiency resulting in insufficient or even late insulin secretion. A series of biological targets have been identified for anti-diabetic therapy2 such as peroxisome proliferator-activated receptors α/γ (PPARs α/γ),3 glucagon-like peptide-1 (GLP-1),4dipeptidyl peptidase IV (DPP-IV)5 or protein tyrosine phosphatase 1B (PTP 1B).6Glycogen phosphorylase (GP) has recently appeared as an enzyme of interest for the treatment of type 2 diabetes.7 This enzyme catalyses glycogen depolymerisation to release glucose-1-phosphate according to the schematic equation:| (Glucose)n→ (Glucose)n−1 + Glucose-1-phosphate |
Multivalency and inhibition of enzymes
Another approach for the inhibition of GP could take advantage of multivalency. This strategy may provide additional opportunities in the field of drug discovery for the design of potent enzyme inhibitors particularly by reaching higher affinities and probably better selectivities. A few examples of multivalent inhibition of an enzyme are reported in the literature where multimeric species of a specific drug are capable of improving the inhibition in comparison to the monomeric molecule.11 The binding of one ligand subunit from the multivalent molecule to the enzyme generates an increase in local concentration of ligands, thus creating an apparent cooperativity causing an enhancement in inhibition. Dimeric inhibitors of influenza virus neuraminidase have been developed by MacDonald et al.12 and displayed up to a 100-fold increase in inhibition along with improved pharmacokinetic properties. In an additional study of the same group,13 a set of trimeric and tetrameric inhibitors displayed improved antiviral activities and long-lasting protective activities against influenza virus. More recently, the group of J. Gervay-Hague has described the synthesis of a series of trivalent zanamivir derivatives via click chemistry although no biological activity has been reported yet.14 Inhibition of acetylcholinesterase by dimeric molecules resulted in up to 3000-fold increases in potency and selectivity compared to the monomeric inhibitor.15 Glycosidases inhibition16 with tethered dimeric azasugars was investigated and the molecules displayed interesting inhibitions of these enzymes but more importantly a selectivity for two enzymes out of the seven tested.16a However, tetravalent 1-azafagomine inhibitors displayed no improved inhibition compared to the monovalent structure but rather a strong decrease in activity.16b Finally, bivalent inhibitors of tetrameric β-tryptase constructed on a cyclodextrin scaffold have shown increased inhibitions for this enzyme.17Inspired by the above mentioned results for the multivalent neuraminidase inhibition and by the fact that a multimeric enzyme offers additional possibilities for improved inhibition through multivalency, we have designed synthetic routes to trivalent carbohydrate-based GP inhibitors. A single case of a dimeric inhibitor of GP has been reported with a bis(5-chloroindole-2-carboxamide) derivative inhibiting human liver GPa (HLGPa) with an IC50 value of 6 nM compared to 12.5 μM for the parent monovalent inhibitor (2000-fold increase).18 This result highlights a productive and cooperative binding of both ends of the bivalent inhibitor to two binding sites. The co-crystallization of HLGPa with the divalent inhibitor demonstrated that a single molecule of inhibitor was capable of interacting with each monomeric unit of GP by linking the two binding sites across the interface of GP homodimeric structure. This result encouraged us to further investigate this approach for the design of multivalent inhibitors of GP.
A closer look at the modes of binding of multivalent inhibitors to an enzyme reveals several possibilities as depicted in Fig. 1. A dimeric enzyme such as GP can interact with two monomeric molecules of inhibitor in a 1:1 complex (2:2 at the molecular level) providing a reference IC50mono value for monovalent inhibitors. When considering the same inhibitor repeated three times on a molecular scaffold, four main possible cases can then be envisaged. The inhibition of a trivalent inhibitor must be divided by three in order to consider the contribution of each residue in its comparison to a monovalent inhibitor. If a 1:1 complex is formed in solution, a simple statistical effect can be invoked if no positive effect is observed and the IC50 value observed will be similar to 1/3 IC50mono (Fig. 1, Case 1). Nevertheless, the IC50 measured can be improved with a lower value in comparison to 1/3 IC50mo (Fig. 1, Case 2). If each binding site of the enzyme is occupied by a ligand of the multivalent inhibitor, the formation of 3:1 complexes would afford aggregates of proteins. If the size of the aggregates remains small enough to maintain a good solubility of the complex, the IC50 observed will be similar to 1/3 IC50mono (Fig. 1, Case 3). Nevertheless, if the size of the multivalent inhibitor-enzyme clusters becomes large enough to cause their precipitation, the quantity of enzyme present in the solution will diminish and therefore the IC50 value measured will be lower than 1/3 IC50mono (Fig. 1, Case 4). In this case, the IC50 value measured will be a “virtual” value because of the lower quantity of the enzyme available in solution.
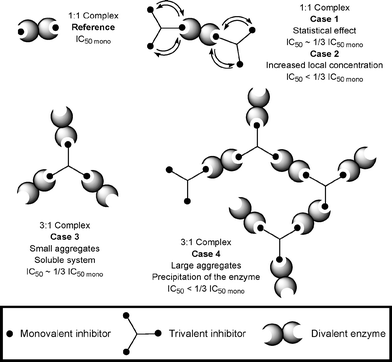 | ||
| Fig. 1 Possible modes of binding for a mono- and trivalent inhibitors with a dimeric enzyme. | ||
Results and discussion
Synthesis of trivalent inhibitors
We have recently reported the preparation of 3-C-glycosyl-5-aryl-1,2,4-oxadiazoles which displayed good inhibition towards GP.10b In this context, we synthesized an alkyne-terminated 3-C-glycosylated 1,2,4-oxadiazole which could then be involved in a 1,3-dipolar cycloaddition with a tris-azido-functionalized derivative to obtain a multivalent GP inhibitor candidate. A more condensed trimeric inhibitor was also prepared by direct coupling of an amidoxime with a tris-acyl chloride and subsequent dehydrative cyclization to the corresponding trivalent C-glycosylated oxadiazole. These inhibitors were designed in order to determine the influence of a spacer arm between the core and the pharmocophore ligands on the inhibition of the enzyme.Synthesis of the trivalent inhibitor with a spacer arm
The perbenzoylated glucosyl cyanide 119 was reacted with hydroxylamine hydrochloride in pyridine to afford the desired amidoxime 2 (Scheme 1). In our previous work,10b the crude product obtained was rather difficult to purify by silica gel column chromatography. The amidoxime 2 could be obtained pure without chromatography simply by diluting the crude product in ethyl acetate and then washing the organic layer with 1 M aqueous HCl to remove pyridine and excess of hydroxylamine, followed by saturated aqueous NaHCO3 and brine. This simple chromatography-free purification process afforded the expected amidoxime 2 in 99% yield and high purity. The formation of the O-acyl-amidoxime 3 was achieved with 4-pentynoic acid in the presence of EDCI/HOBt as coupling agents.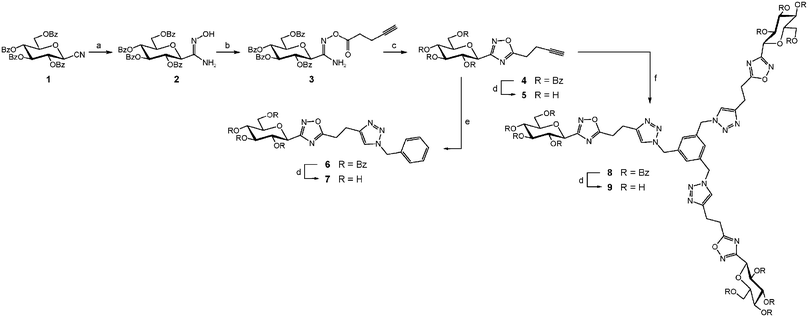 | ||
Scheme 1
Reagents and conditions: (a) NH2OH·HCl, C5H5N, 50 °C, 5 h, 99%; (b) HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(CH2)2CO2H, EDCI, HOBt, CH2Cl2/DMF (9:1), −8 °C then r.t., 16 h, 67%; (c) PhMe, TBAF 10 mol%, μW (150 °C, 200 W, 5 min), 97%; (d) NaOMe, MeOH then Amberlite IR-120 (H+ form); (e) PhCH2N3, CuI, Et3N, μW (110 °C, 150 W, 15 min), 88%; (f) C6H3(CH2N3)3, CuI, Et3N, μW (110 °C, 150 W, 15 min), 98%. C(CH2)2CO2H, EDCI, HOBt, CH2Cl2/DMF (9:1), −8 °C then r.t., 16 h, 67%; (c) PhMe, TBAF 10 mol%, μW (150 °C, 200 W, 5 min), 97%; (d) NaOMe, MeOH then Amberlite IR-120 (H+ form); (e) PhCH2N3, CuI, Et3N, μW (110 °C, 150 W, 15 min), 88%; (f) C6H3(CH2N3)3, CuI, Et3N, μW (110 °C, 150 W, 15 min), 98%. | ||
We previously observed that reactions times lasting from a few hours to a few days were required for the thermal cyclodehydration of O-acyl-amidoximes. In order to optimize both time and yield, we performed this reaction under TBAF catalysis20 and/or microwaves activation21 (Table 1). We observed that the use of TBAF catalysis at room temperature provided the cyclic oxadiazole 4 within 1 day (entry 1) while thermal activation combined with TBAF catalysis drastically shortened the reaction time to 10 minutes (entry 2). No reaction or decomposition of the starting material was observed when applying microwaves activation without TBAF catalysis (entries 3 and 4). Nevertheless, the association of TBAF catalysis and microwaves activation performed on a short timescale (entry 5 and 6) provided a result comparable to that observed under conventional heating (entry 2). These results underline the beneficial influence of TBAF catalysis.
| Entry | Catalyst | T/°C | Microwave condition | Time | Yield (%) |
|---|---|---|---|---|---|
| 1 | 10% TBAF | 25 | None | 24 h | 99 |
| 2 | 10% TBAF | 110 | None | 10 min | 97 |
| 3 | None | 150 | 100 W | 1 h | No reaction |
| 4 | None | 175 | 200 W | 2 h | Decomposition |
| 5 | 10% TBAF | 150 | 200 W | 5 min | 97 |
| 6 | 10% TBAF | 150 | 200 W | 30 min | 66 |
The alkyne-terminated oxadiazole 4 was then engaged in a Huisgen’s Cu(I)-catalyzed 1,3-dipolar cycloaddition22 reaction under microwaves activation with benzyl azide to afford the desired 1,4-disubstituted 1,2,3-triazole 6 in excellent yield. Debenzoylation of compounds 4 and 6 afforded two hydroxylated GP inhibitor candidates 5 and 7. Similarly, the reaction of 1,3,5-tris(azidomethyl)benzene23 with the alkyne derivative 4 under microwaves activation and Cu(I) catalysis afforded the cycloadduct 8. The saponification of the benzoate esters provided the fully hydroxylated macromolecule 9.
Synthesis of the trivalent inhibitor without spacer arm
We next prepared a more condensed trifunctional macromolecule were the C-glucosyl-oxadiazole moiety was directly attached to a benzene ring (Scheme 2). Condensation of amidoxime 2 with 1,3,5-benzenetricarbonyl trichloride afforded the corresponding triester 10 in 73% yield. At first, compound 10 was subjected to cyclodehydration under thermal conditions (reflux in 1,4-dioxane). The product obtained was not the expected tris-oxadiazole 13 but the bis-oxadiazole 11 with one unreacted O-acyl amidoxime moiety as evidenced by mass spectrometry (m/z = 2035.4 [M + H]+). Interestingly, molecular ions could be observed neither for compound 13 nor the mono-oxadiazole intermediate. Saponification of the ester groups of 11 resulted in the concomitant cleavage of the O-acyl amidoxime function and afforded the benzoic acid derivative 12 whose structure was clearly demonstrated by mass spectrometry (m/z = 581 [M − H]−) and NMR analyses. The triple thermal cyclodehydration of 10 was then performed under microwaves activation and TBAF catalysis for 40 minutes. The tris-oxadiazole derivative 13 was isolated in 72% yield as the only product of the reaction highlighting again the positive influence of TBAF catalysis and microwaves activation for this cyclodehydration process. Deprotection under Zemplén conditions afforded the expected hydroxylated trivalent GP inhibitor candidate 14.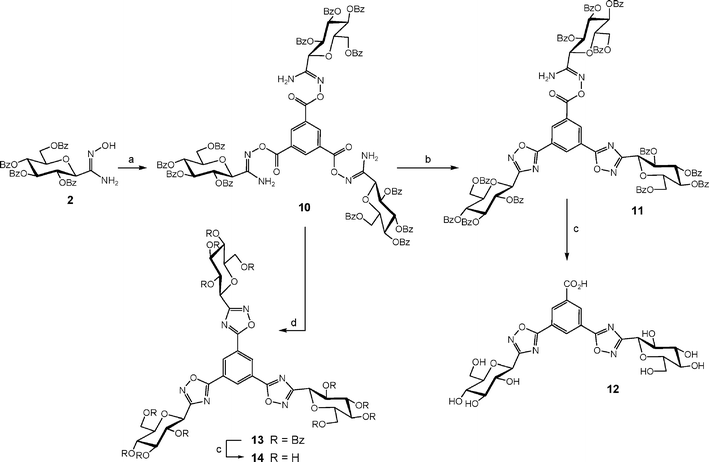 | ||
| Scheme 2 Reagents and conditions: (a) C6H3(COCl)3, 1,4-dioxane, r.t., 24 h, 73%; (b) 1,4-dioxane, 100 °C, 4 days; (c) NaOMe, MeOH then Amberlite IR-120 (H+ form); (d) PhMe, TBAF 30 mol%, μW (150 °C, 200 W, 40 min), 72%. | ||
Inhibition of glycogen phosphorylase
The inhibition of GP was determined, as previously reported,10b for the three monovalent C-glycosylated oxadiazoles (5, 7 and 1510b) and the two trivalent derivatives 9 and 14 (Fig. 2, Table 2). The enzymatic assays were performed at two concentrations and most molecules displayed poor inhibition properties at a concentration of 625 μM and moderate to good inhibition at higher concentration (2.5 mM). In addition, Ki values could be estimated only for trivalent derivatives 9 and 14 (see ESI†).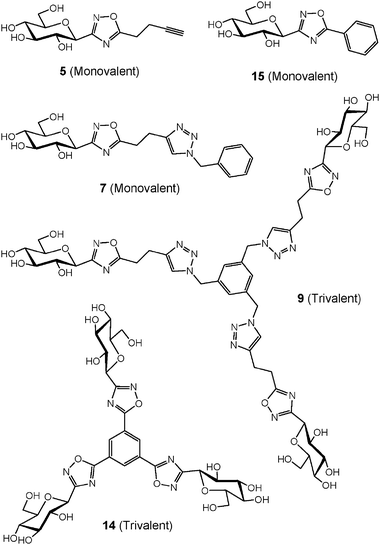 | ||
| Fig. 2 Structure of monovalent and trivalent GP inhibitors tested. | ||
The alkyne-terminated C-glycosylated oxadiazole derivative 5 displayed no inhibition at 625 μM and poor activity at 2.5 mM. The inhibition properties disappeared completely when a spacer arm was added such as in the structure of 7. Interestingly, the trivalent analogue 9 of the non-active derivative 7 was now inhibiting GP with values of 30% at 625 μM and 56% at 2.5 mM. The valency of the molecule is therefore responsible for an increase in inhibition from 0 to 56% when comparing 7 and 9 at 2.5 mM. The C-glycosylated oxadiazole derivative 15 bearing a phenyl group on the 5-position of the oxadiazole ring displayed 10% inhibition at 625 μM.10b The inhibition was again increased to 35% at 625 μM for trivalent analogue 14. We anticipated that the distance between the core and the carbohydrate moiety would influence for the binding to the enzyme. Nevertheless, this was not the case based on the inhibition measured for 9 and 14.
The increase of valency from monovalent to trivalent species is responsible for an increase in inhibition of GP. The inhibition per residue for trivalent molecules is always similar the corresponding monovalent analogue in accordance with the statistical effect model proposed (Fig. 1, Case 1). The 3:1 complex non soluble mode of binding (Fig. 1, Case 4) can be ruled out for these trivalent inhibitors of GP to the dimeric enzyme since no precipitate was observed under the concentrations of trivalent inhibitors and enzyme used for the inhibition studies. The inhibition observed for the trivalent species was never better than 1/3 of the inhibition (in %) observed for the corresponding monovalent molecules. In conclusion, two modes of binding are possible with either 1:1 or 3:1 complexes (Fig. 1, Case 1 or Case 3, respectively) resulting in an observed inhibition close to 1/3 of the inhibition for the parent monovalent inhibitor.
In the present study, the expected binding sites of these glucose-based multivalent inhibitors are the catalytic site of GP homodimer which are separated from each other by a long distance and pointing into opposite directions.8d The structure of the trivalent inhibitors tested did not permit such an intramolecular interaction with both catalytic sites on the same GP dimer, but rather an interaction with two independent GP dimers. In comparison, the bis(5-chloroindole-2-carboxamide) derivative is binding simultaneously at each indole binding site near the interface between the monomeric units of the GP dimer.17 The linker is composed of 12 atoms between two indole aromatic units which are therefore available for interacting with the binding site of each monomer of GP.
The designed trivalent molecules could also bind to the enzyme on a different site. The large aromatic appendage present in the aglycon, composed of oxadiazole, phenyl and triazole rings, might interact with the surface of the protein through hydrophobic interactions. The stability of the inhibitor-protein complex would therefore be lower than complex involving an internal binding site such as the catalytic site. The observed inhibitions would therefore be weak as currently observed in the present study. Nevertheless, we do not possess any experimental data confirming or denying such a mode of interaction.
Conclusions
In conclusion, we have designed two kinds of multivalent inhibitors of GP based on the acylation of an amidoxime intermediate followed by thermal dehydrative cyclization to the corresponding oxadiazole. The introduction of an alkyne residue at the 5-position of the oxadiazole ring allowed the coupling to a trivalent azido-functionalized benzene ring leading to an extended trivalent inhibitor candidate. The enzyme inhibition assays revealed poor to moderate inhibitory effect of these analogues. But, more important was the fact that multivalent inhibitors were always superior to their monovalent counterparts. This study provides one of the few examples of multivalent inhibition for an enzyme, even though the inhibitions observed remain modest.Experimental
General methods
Thin-layer chromatography (TLC) was carried out on aluminum sheets coated with silica gel 60 F254 (Merck). TLC plates were inspected by UV light (λ = 254 nm) and developed by treatment with a mixture of 10% H2SO4 in EtOH/H2O (1:1 v/v) followed by heating. Silica gel column chromatography was performed with Geduran® silica gel Si 60 (40–63 μm) purchased from Merck (Darmstadt, Germany). Reactions under microwave activation were performed on a CEM Discover system. HRMS (LSIMS) mass spectra were recorded in the positive mode using a Thermo Finnigan Mat 95 XL spectrometer. MS (ESI) mass spectra were recorded in the positive mode using a Thermo Finnigan LCQ spectrometer. 1H and 13C NMR spectra were recorded at 23 °C using Brüker Advance DRX300 or DRX500 spectrometers with the residual solvent as the internal standard. The following abbreviations are used to explain the observed multiplicities: s, singlet; d, doublet; dd, doublet of doublet; ddd, doublet of doublet of doublet; t, triplet; td, triplet of doublet; q, quadruplet; m, multiplet; br, broad; p, pseudo. Structure elucidation was deduced from 1D and 2D NMR spectroscopy which allowed, in most cases, complete signal assignments based on COSY, HSQC, and HMBC correlations. NMR solvents were purchased from Euriso-Top (Saint Aubin, France). Atom numbering of the molecules is presented in the ESI.†Syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR–NH2), 165.3, 165.6, 165.7, 166.3 (4s, 4C, COPh), 169.0 (C-1′). ESI-MS (positive mode) m/z: 719.2 [M + H]+, 741.3 [M + Na]+, 786.9 [M + HCOOH + Na]+, 1436.9 [2M + H]+, 1458.9 [2M + Na]+, 1504.5 [2M + HCOOH + Na]+. HR-ESI-MS (positive mode) m/z: calcd. for C40H34N2O11 [M + H]+ 719.2241, found 719.2244.
NO), 161.5 (NOCO), 165.2, 165.58, 165.63, 166.1 (4s, 4C, COPh). LSIMS (positive mode, thioglycerol) m/z: 2071.6 [M + H]+. HR-ESI-MS (positive mode) m/z: calcd. for C114H91N6O33 [M + H]+ 2071.5627, found 2071.5651.
CR–NH2), 165.3, 165.6, 165.7, 166.3 (4s, 4C, COPh), 169.0 (C-1′). ESI-MS (positive mode) m/z: 719.2 [M + H]+, 741.3 [M + Na]+, 786.9 [M + HCOOH + Na]+, 1436.9 [2M + H]+, 1458.9 [2M + Na]+, 1504.5 [2M + HCOOH + Na]+. HR-ESI-MS (positive mode) m/z: calcd. for C40H34N2O11 [M + H]+ 719.2241, found 719.2244.
NO), 161.5 (NOCO), 165.2, 165.58, 165.63, 166.1 (4s, 4C, COPh). LSIMS (positive mode, thioglycerol) m/z: 2071.6 [M + H]+. HR-ESI-MS (positive mode) m/z: calcd. for C114H91N6O33 [M + H]+ 2071.5627, found 2071.5651.
Glycogen phosphorylase inhibition measurements
Glycogen phosphorylase b was prepared from rabbit skeletal muscle according to the method of Fischer and Krebs,24 using dithiothreitol instead of L-cysteine, and recrystallized at least three times before use. Kinetic experiments were performed in the direction of glycogen synthesis as described previously.25 Kinetic data for the inhibition of rabbit skeletal muscle glycogen phosphorylase were collected using different concentrations of α-D-glucose-1-phosphate (2–20 mM), constant concentrations of glycogen (1% w/v) and AMP (1 mM), and various concentrations of inhibitors. Inhibitors were dissolved in dimethyl sulfoxide (DMSO) and diluted in the assay buffer (50 mM triethanolamine, 1 mM EDTA and 1 mM dithiothreitol) so that the DMSO concentration in the assay should be lower than 5%. The enzymatic activities were presented in the form of double-reciprocal plots (Lineweaver–Burk) applying a nonlinear data analysis program. The inhibitor constants (Ki) were determined by Dixon plots, by replotting the slopes from the Lineweaver–Burk plots against the inhibitor concentrations.26,27 The means of standard errors for all calculated kinetic parameters averaged to less than 10%. Ki values for compounds 9 and 14 were also estimated and found to be 490 ± 45 μM and 535 ± 50 μM, respectively. The poor solubility of inhibitors 5, 7 and 15 limited the concentrations used in the kinetic studies. The inhibition of glycogen phosphorylase was therefore determined at 625 μM and 2.5 mM concentrations of these inhibitors and given in Table 2.Acknowledgements
The authors wish to thank Université Claude Bernard Lyon 1 and CNRS for financial support. A stipend to S. C. and financial support from Région Rhône-Alpes «Cluster de Recherche Chimie» are gratefully acknowledged. CNRS is also thanked for additional funding through «Programme Interdisciplinaire: Chimie pour le Développement Durable». This work was also supported by grants from the Hungarian Science Research Fund (OTKA K60620) and the Hungarian Ministry of Health (ETT 083/2006).References
- (a) S. Wild, G. Roglic, A. Green, R. Sicree and H. King, Diabetes Care, 2004, 27, 1047 CrossRef; (b) P. Zimmet, K. G. M. M. Alberti and J. Shaw, Nature, 2001, 414, 782 CrossRef CAS.
- (a) D. E. Moller, Nature, 2001, 414, 821 CrossRef; (b) N. Morral, Trends Endocrinol. Metab., 2003, 14, 169 CrossRef CAS; (c) L. Agius, Best Pract. Res. Clin. Metab., 2007, 21, 587 Search PubMed.
- (a) B. R. Henke, J. Med. Chem., 2004, 47, 4118 CrossRef CAS; (b) C. Fievet, J.-C. Fruchart and B. Staels, Curr. Opin. Pharmacol., 2006, 6, 606 CrossRef CAS.
- (a) L. B. Knudsen, J. Med. Chem., 2004, 47, 4128 CrossRef CAS; (b) M. A. Nauck and J. J. Meier, Regul. Pept., 2005, 128, 135 CrossRef CAS.
- (a) A. E. Weber, J. Med. Chem., 2004, 47, 4135 CrossRef CAS; (b) A. Barnett, Int. J. Clin. Pract., 2006, 60, 1454 Search PubMed; (c) D. J. Drucker, Diabetes Care, 2007, 30, 1335 CrossRef CAS.
- (a) B. J. Goldstein, J. Clin. Endocrinol. Metab., 2002, 87, 2474 CrossRef CAS; (b) R. Hooft van Huijsduijnen, W. H. B. Sauer, A. Bombrun and D. Swinnen, J. Med. Chem., 2004, 47, 4142 CrossRef CAS; (c) L. Bialy and H. Waldmann, Angew. Chem., Int. Ed., 2005, 44, 3814 CrossRef CAS.
- (a) J. L. Treadway, P. Mendys and D. J. Hoover, Exp. Opin. Invest. Drugs, 2001, 10, 439 Search PubMed; (b) N. G. Oikonomakos, Curr. Protein Pept. Sci., 2002, 3, 561 CrossRef CAS; (c) L. Somsak, V. Nagy, Z. Hadady, T. Docsa and P. Gergely, Curr. Pharm. Des., 2003, 9, 1177 CrossRef CAS; (d) D. J. Baker, J. A. Timmons and P. L. Greenhaff, Diabetes, 2005, 54, 2453 CrossRef CAS; (e) L. Somsák, V. Nagy, Z. Hadady, N. Felfoldi, T. Docsa and P. Gergely, Front. Med. Chem., 2005, 2, 253 Search PubMed; (f) N. G. Oikonomakos and L. Somsák, Curr. Opin. Invest. Drugs, 2008, 9, 379 Search PubMed.
- (a) R. J. Fletterick and S. R. Sprang, Acc. Chem. Res., 1982, 15, 361 CrossRef CAS; (b) S. R. Sprang, K. R. Acharya, E. J. Goldsmith, D. I. Stuart, K. Varvill, R. J. Fletterick, N. B. Madsen and L. N. Johnson, Nature, 1988, 336, 215 CrossRef CAS; (c) D. Barford and L. N. Johnson, Nature, 1989, 340, 609 CrossRef CAS; (d) N. G. Oikonomakos, V. T. Skamnaki, K. E. Tsitsanou, N. G Gavalas and L. N. Johnson, Structure, 2000, 8, 575 CrossRef CAS.
- (a) N. G. Oikonomakos, J. B. Schnier, S. E. Zographos, V. T. Skamnaki, K. E. Tsitsanou and L. N. Johnson, J. Biol. Chem., 2000, 275, 34566 CrossRef CAS; (b) M. N. Kosmopoulou, D. D. Leonidas, E. D. Chrysina, N. Bischler, G. Eisenbrand, C. E. Sakarellos, R. Pauptit and N. G. Oikonomakos, Eur. J. Biochem., 2004, 271, 2280 CrossRef; (c) T. Klabunde, K. U. Wendt, D. Kadereit, V. Brachvogel, H. J. Burger, A. W. Herling, N. G. Oikonomakos, M. N. Kosmopoulou, D. Schmoll, E. Sarubbi, E. von Roedern, K. Schonafinger and E. Defossa, J. Med. Chem., 2005, 48, 6178 CrossRef CAS; (d) B. R. Henke and S. M. Sparks, Mini-Rev. Med. Chem., 2006, 6, 845 CrossRef CAS; (e) D. J. Baker, P. L. Greenhaff and J. A. Timmons, Exp. Opin. Ther. Pat., 2006, 16, 459 Search PubMed; (f) A. M. Birch, P. W. Kenny, N. G. Oikonomakos, L. Otterbein, P. Schofield, P. R. O. Whittamore and D. P. Whalley, Bioorg. Med. Chem. Lett., 2007, 17, 394 CrossRef CAS.
- (a) N. G. Oikonomakos, M. N. Kosmopoulou, S. E. Zographos, D. D. Leonidas, E. D. Chrysina, L. Somsak, V. Nagy, J.-P. Praly, T. Docsa, B. Toth and P. Gergely, Eur. J. Biochem., 2002, 269, 1684 CrossRef CAS; (b) M. Benltifa, S. Vidal, B. Fenet, M. Msaddek, P. G. Goekjian, J.-P. Praly, A. Brunyánszki, T. Docsa and P. Gergely, Eur. J. Org. Chem., 2006, 4242 CrossRef CAS; (c) M. Benltifa, S. Vidal, D. Gueyrard, P. G. Goekjian, M. Msaddek and J.-P. Praly, Tetrahedron Lett., 2006, 47, 6143 CrossRef CAS; (d) L. He, Y. Z. Zhang, M. Tanoh, G.-R. Chen, J.-P. Praly, E. D. Chrysina, C. Tiraidis, M. Kosmopoulou, D. D. Leonidas and N. G. Oikonomakos, Eur. J. Org. Chem., 2007, 596 CrossRef CAS.
- H. L. Handl, J. Vagner, H. Han, E. Mash, V. J. Hruby and R. J. Gillies, Exp. Opin. Ther. Targets, 2004, 8, 565–586 Search PubMed.
- (a) S. J. F. Macdonald, K. G. Watson, R. Cameron, D. K. Chalmers, D. A. Demaine, R. J. Fenton, D. Gower, J. N. Hamblin, S. Hamilton, G. J. Hart, G. G. A. Inglis, B. Jin, H. T. Jones, D. B. McConnell, A. M. Mason, V. Nguyen, I. J. Owens, N. Parry, P. A. Reece, S. E. Shanahan, D. Smith, W.-Y. Wu and S. P. Tucker, Antimicrob. Agents Chemother., 2004, 48, 4542 CrossRef CAS; (b) S. J. F. MacDonald, R. Cameron, D. A. Demaine, R. J. Fenton, G. Foster, D. Gower, J. N. Hamblin, S. Hamilton, G. J. Hart, A. P. Hill, G. G. A. Inglis, B. Jin, H. T. Jones, D. McConnell, J. McKimm-Breschkin, G. Mills, V. Nguyen, I. J. Owens, N. Parry, S. E. Shanahan, D. Smith, K. G. Watson, W.-Y. Wu and S. P. Tucker, J. Med. Chem., 2005, 48, 2694.
- K. G. Watson, R. Cameron, R. J. Fenton, D. Gower, S. Hamilton, B. Jin, G. Y. Krippner, A. Luttick, D. McConnell, S. J. F. MacDonald, A. M. Mason, V. Nguyen, S. P. Tucker and W.-Y. Wu, Bioorg. Med. Chem. Lett., 2004, 14, 1589–1592 CrossRef CAS.
- Y. Lu and J. Gervay-Hague, Carbohydr. Res., 2007, 342, 1636–1650 CrossRef CAS.
- Y. F. Han, C. P.-L. Li, E. Chow, H. Wang, Y.-P. Pang and P. R. Carlier, Bioorg. Med. Chem., 1999, 7, 2569–2575 CrossRef CAS.
- (a) B. A. Johns and C. R. Johnson, Tetrahedron Lett., 1998, 39, 749 CrossRef CAS; (b) A. Lohse, K. B. Jensen, K. Lundgren and M. Bols, Bioorg. Med. Chem., 1999, 7, 1965 CrossRef CAS.
- N. Schaschke, G. Matschinerb, F. Zettlb, U. Marquardta, A. Bergnera, W. Bodea, C. P. Sommerhoff and L. Moroder, Chem. Biol., 2001, 8, 313–327 CrossRef CAS.
- V. L. Rath, M. Ammirati, D. E. Danley, J. L. Ekstrom, E. M. Gibbs, T. R. Hynes, A. M. Mathiowetz, R. K. McPherson, T. V. Olson, J. L. Treadway and D. J. Hoover, Chem. Biol., 2000, 7, 677–682 CrossRef CAS.
- L. Somsak and V. Nagy, Tetrahedron: Asymmetry, 2000, 11, 1719–1727 CrossRef CAS.
- A. R. Gangloff, J. Litvak, E. J. Shelton, D. Sperandio, V. R. Wang and K. D. Rice, Tetrahedron Lett., 2001, 42, 1441–1443 CrossRef CAS.
- Y. Wang, R. L. Miller, D. R. Sauer and S. W. Djuric, Org. Lett., 2005, 7, 925–928 CrossRef CAS.
- (a) R. Huisgen, R. Knorr, L. Möbius and G. Szeimies, Chem. Ber., 1965, 98, 4014 CrossRef CAS; (b) R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494 CrossRef CAS; (c) R. Huisgen, K. Fraunberg and H. J. Sturm, Tetrahedron Lett., 1969, 10, 2589 CrossRef; (d) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (e) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- T. M. Garrett, T. J. McMurry, M. W. Hosseini, Z. E. Reyes, F. E. Hahn and K. N. Raymond, J. Am. Chem. Soc., 1991, 113, 2965–2977 CrossRef CAS.
- E. H. Fischer and E. G. Krebs, Methods Enzymol., 1962, 5, 369 Search PubMed.
- N. G. Oikonomakos, V. T. Skamnaki, E. Ősz, L. Szilágyi, L. Somsák, T. Docsa, B. Tóth and P. Gergely, Bioorg. Med. Chem., 2002, 10, 261 CrossRef CAS.
- E. Ősz, L. Somsák, L. Szilágyi, L. Kovács, T. Docsa, B. Tóth and P. Gergely, Bioorg. Med. Chem. Lett., 1999, 9, 1385 CrossRef CAS.
- L. Somsák, L. Kovács, M. Tóth, E. Ősz, L. Szilágyi, Z. Györgydeák, Z. Dinya, T. Docsa, B. Tóth and P. Gergely, J. Med. Chem., 2001, 44, 2843 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of Ki values and detailed atom numbering of molecules. See DOI: 10.1039/b812540f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |