DOI:
10.1039/B812529E
(Paper)
New J. Chem., 2009,
33, 136-147
Neutral 5-nitrotetrazoles: easy initiation with low pollution†
Received (in Durham. UK) 21st July 2008, Accepted 8th September 2008
First published on 24th October 2008
Abstract
5-Nitro-2H-tetrazole (1), 1-methyl-5-nitrotetrazole (2) and 2-methyl-5-nitrotetrazole (3) were synthesized starting from the corresponding 5-amino-substituted tetrazoles in good yields and purities. The compounds were fully characterized by analytical and spectroscopic methods and their solid state structures were determined by low temperature X-ray diffraction techniques. Due to the potential of tetrazoles as energetic materials an extensive computational study (CBS-4M) was performed in order to estimate the energies of formation (ΔfU°) of the molecules, which are highly endothermic (1, 2527 kJ kg−1; 2, 2253 kJ kg−1 and 3, 2006 kJ kg−1). The EXPLO5 software was used to calculated the corresponding detonation velocities (Ddet) and detonation pressures (pdet) (1, Ddet = 9457 m s−1 and pdet = 390 kbar; 2, Ddet = 8085 m s−1 and pdet = 257 kbar and 3, Ddet = 8109 m s−1 and pdet = 262 kbar) by combining the ΔfU° values of the materials with the (X-ray calculated) densities and molecular formulas, giving performances comparable to commonly used secondary explosives (e.g., RDX). Lastly, all three neutral compounds can be easily initiated by impact (<2 J) and with high detonation velocities and excellent combined oxygen and nitrogen contents offer a more powerful and environmentally friendly alternative to commonly used primary explosives in initiating devices.
Introduction
In the continuous search for novel green energetic materials1 with high nitrogen but low carbon content,2,3 several groups around the world are currently investigating HEDMs (High Energy Dense Materials) based on tetrazoles.4 These energetic materials have variable application such as in low-smoke producing pyrotechnic compositions,5 gas generators,6 propellants,7 high explosives8 and primers in primer charges (PC).9 Tetrazole derivatives,10,11 tetrazolate12,13 and tetrazolium14,15 salts are of special interest. One of the most promising class of molecules in this regard are 5-substituted tetrazoles16 (Fig. 1) due to the fact that their properties can be controlled by selection of the substituent at the carbon atom. While electron donating groups (EDGs) such as NH217 or OH18 yield rather stable compounds, electron withdrawing groups (EWGs) such as NO219 and CN20 destabilize the ring system and increase the sensitivity of the materials. Also protonation/alkylation of the tetrazole ring is directed by the electronegativity of the substituent. While EWGs direct the protonation/alkylation to the nitrogen atom labelled as N2 of the tetrazole ring (see crystal structure labels),21 EDGs favor substitution at N1.22 However, there are other factors that contribute to the explosivity of tetrazoles. For example, the high sensitivity of 5-azidotetrazole (C)23 and 5-nitriminotetrazole (D)24,25 is better explained due to the energetic nature of the azide and nitramine groups rather than based on the electronic influence of these groups on the ring system. The combination of a tetrazole ring with energetic groups containing oxygen such as nitro groups (R–NO2),26 nitrate esters (R–O–NO2)27 or nitramines (R2N–NO2)28 is of particular interest. Energetic materials based on tetrazoles show the desirable compromise in properties with high nitrogen contents on the one hand, and surprising kinetic and thermal stabilities due to aromaticity on the other.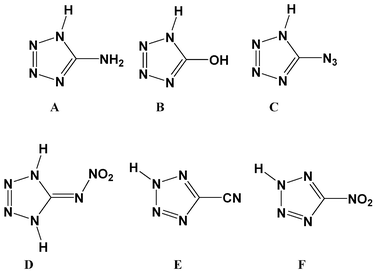 |
| | Fig. 1 Structural formulas of neutral 5-substituted tetrazoles. | |
The interesting energetic properties of tetrazole-based energetic materials have been mainly investigated in view of the properties of such compounds for use as propellants and/or secondary explosives1,29 and it has only been until recent times that metal salts with 5-substituted tetrazole ligands have been studied as prospective primary explosives.30,31 Primary explosives are characterized by easy initiation when submitted to heat or shock and have the ability to transmit the detonation to less sensitive (secondary) explosives. For these reasons, they are used in initiating devices. Typical detonation velocities for this class of compounds are in the range 3500–5500 m s−1, much lower than those of secondary explosives (5500–9000 m s−1).32 Commonly used primary explosives are based on lead (e.g., lead azide or styphnate) with the accompanying environmental impact and thus, recent efforts have focused on the development of more environmentally friendly metal-based alternatives.30,31
In this work we would like to present the syntheses, full analytical, spectroscopic and structural characterization of neutral 5-nitrotetrazoles. In addition, the energetic properties of the compounds were assessed revealing easy initiation by impact and detonation velocities, which are almost twice as high as those of commonly used primary explosives.5
Results and discussion
Syntheses
5-Nitro-2H-tetrazole (1) was prepared starting from 5-amino-1H-tetrazole (5-At) by a modified literature procedure according to Scheme 1.33 5-At was diazotized according to a previously published procedure in our group34 to yield ammonium 5-nitrotetrazolate. “In situ” formation of the potassium salt by reaction with potassium hydroxide in ethanol and subsequent treatment with diluted hydrochloric acid and extraction with ether yields the desired compound. The solvent needs to be removed using vacuum since the product (1) absorbs water on time and the compound needs to be stored under nitrogen. On the other hand, methylation of sodium 5-aminotetrazolate using dimethyl sulfate22 yields a separable mixture of the two (1-methyl and 2-methyl) isomers.35 Both compounds can be treated similarly and diazotization with two equivalents of sodium nitrite in the presence of a non-nucleophilic acid (e.g., sulfuric acid) yields 1-methyl-5-nitrotetrazole (2) and 2-methyl-5-nitrotetrazole (3) as crystalline compounds. Similar reactions are also found in the literature by using N2O5.362 and 3 are extracted from the reaction mixture using CH2Cl2. The selection of the acid for the diazotation process is of utmost importance since it affects the yield of the nitro-compound. For example using hydrochloric acid 1- or 2-methyl-5-chlorotetrazoles are obtained as the main product.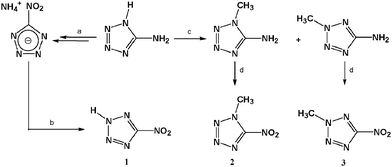 |
| | Scheme 1 Syntheses of neutral 5-nitrotetrazoles: a = ref. 34; b = (i) KOH, (ii) HCl (2 M); (c) (i) NaOH, (ii) Me2SO4; d = 2 eq. NaNO2, H2SO4. | |
Lastly, 1 is readily soluble in most common solvents such as ether, THF, MeCN, acetone, water, DMSO and DMF whereas 2 and 3 show also good solubility in MeOH, EtOH, acetone, MeCN, ethyl acetate, THF, CH2Cl2 and DMSO and DMF.
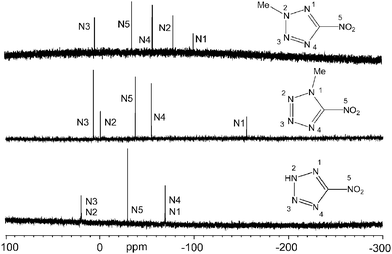
All three neutral 5-nitrotetrazoles were characterized by analytical and spectroscopic methods. The elemental analysis of 1 was omitted due to the risk of explosion found in similar compounds with a high sensitivity30 on the one hand and to the hygroscopicity of the material on the other. The 1H NMR spectra of 1–3 measured in DMSO-d6 show two signals corresponding to the ring proton (1, broad, δ = 6.29 ppm) and to the methyl group protons (2, δ = 3.68 ppm and 3, δ = 4.50 ppm). The electron withdrawing character of the –NO2 group shifts the proton resonances to low field in comparison to 5-amino-1H-tetrazole and 1-methyl- and 2-methyl-5-amino-1H-tetrazole (i.e., –NH2 group).37Table 1 contains summarized the 13C and 15N NMR chemical shifts and the 15N–1H coupling constants for all three compounds. The proton coupled as well as the proton decoupled 15N NMR spectra (with full NOE) were also recorded. As already observed in the 1H NMR spectra, the methyl group resonance (in this case carbon resonance) of the 1-methyl isomer (2, δ = 33.1 ppm) is shifted to higher field in comparison with that of the 2-methyl isomer (3, δ = 41.9 ppm). Similarly, the ring carbon atom signal is also to be found at higher field for 2 (δ = 157.6 ppm) than for 3 (δ = 166.4 ppm). This carbon atom shows the lowest field resonance for 1 (δ = 168.4 ppm), which is in keeping with salts containing the 5-nitrotetrazolate anion.30,34
Table 1 15N and 13C NMR resonances of compounds 1–3 with protonation (1, PIS) and methylation (2 and 3, MIS) induced shiftsa and coupling constantsb
| Compound | N1 | N2 | N3 | N4 | N5 | C1 | C2 |
|---|
| PIS and MIS values are shown in square [] brackets and given in ppm. Coupling constants (J) are given in Hz. NaNT = Sodium 5-nitrotetrazolate (see ref. 30). |
|---|
| 1 | −69.6 [−3.4] | 19.6 [5.2] | 19.6 [5.2] | −69.6 [−3.4] | −29.8 [−4.5] | 168.4 | — |
| 2 | −155.7 [−89.5] | −0.7 [−15.1] | 6.7 [7.7] | −54.8 [11.4] | −37.6 [−12.3] | 157.6 | 33.1 |
| | 2J(N–H) = 2.1 | 3J(N−H) = 1.8 | | | | | |
| 3 | −97.9 [−31.7] | −76.6 [−91.0] | 5.3 [9.1] | −55.1 [11.1] | −33.5 [−8.2] | 166.4 | 41.9 |
| | 3J(N–H) = 1.7 | 2J(N–H) = 2.1 | 3J(N−H) = 1.7 | | | | |
| NaNTc | −66.2 | 14.4 | 14.4 | −66.2 | −25.3 | 169.2 | — |
The quadrupolar moment of the 14N nucleus results in signals at approximately +14 (N2/3), −24 (NO2) and −66 (N1/4) in the 14N NMR spectrum of compound 1, which are broad (ν1/2∼ 300 Hz, ∼60 Hz and ∼320 Hz, respectively). The 15N NMR spectra show comparable resonances to those observed in the 14N NMR spectra but much sharper (Fig. 2). The proton induced shifts (PIS, 1) and methyl induced shifts (MIS, 2 and 3)37 are useful for identifying the protonation/alkylation site as well as assigning the resonances of the nitrogen atoms and are also tabulated in Table 1. Comparison of the resonances observed for the 5-nitrotetrazolate anion in sodium 5-nitrotetrazolate with those of the compounds in this study shows unexpected shifts. The nitrogen atoms labelled as N2 and N3, which are equivalent due to fast exchange in the NMR in solution of 1, show the largest (positive) PIS value, indicative of protonation taking place on these two nitrogen atoms as observed (in the solid state) in the crystal structure of the compound (see X-ray discussion). The remainder of the PIS values are small in value and negative. The MIS effect in compounds 2 and 3 is much more unexpected and the methylated nitrogen atoms (N1 for 2 and N2 for 3) show the largest (negative) MIS values (∼−90 ppm for both). The next nitrogen atom close to the methyl group (i.e., at two bonds) feels the effect of the alkyl group much more weakly (MIS = −31.7 ppm for N1 in 3) but can still be used to assign the resonances of this atom. Lastly, the fast exchange of the proton in 1 results in broadening of the resonance corresponding to the protonated nitrogen atom, whereas 2 and 3 show coupling constants to the methyl groups, which are slightly larger for the nitrogen atoms directly attached to the methyl group (2J(N−H) = 2.1 Hz) than for those at three bonds of the methyl group protons (3J(N−H) = 1.7–1.8 Hz).
Molecular structures
Suitable single crystals of 1 and 2 were picked from the crystallization mixture and mounted in Kel-F oil and transferred to the N2 stream of an Oxford Xcalibur3 diffractometer with a Spellman generator (voltage 50 kV, current 40 mA) and a KappaCCD detector. The data collections were performed using the CrysAlis CCD software,38 the data reduction with the CrysAlis RED software.39 The data for compound 3 were collected on a Nonius Kappa CCD diffractometer under an N2 stream as well. Data collection and reduction was done by the Bruker “Collect” and the “HKL Denzo and Scalepack” software.40 The structures were solved with SIR-92 (2, 3),41 and SHELXS-97 (1),42 refined with SHELXL-9743 and finally checked using the PLATON software.44 The non-hydrogen atoms were refined anisotropically and the hydrogen atoms were located and freely refined. The absorptions of 1 and 2 were corrected by a SCALE3 ABSPACK multi-scan method.45 All relevant data and parameters of the X-ray measurements and refinements are given in Table 2.46
| | 1 | 2 | 3 |
|---|
| Formula | CHN5O2 | C2H3N5O2 | C2H3N5O2 |
| Mr/g mol−1 | 115.07 | 129.09 | 129.09 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21 (No. 4) | P21/n (No. 14) | P21/c (No. 14) |
| Color/habit | Colorless plates | Colorless rods | Colorless rods |
| Size/mm | 0.03 × 0.18 × 0.27 | 0.04 × 0.08 × 0.10 | 0.03 × 0.18 × 0.27 |
| a/Å | 5.3358(4) | 10.0578(4) | 6.331(1) |
| b/Å | 9.4799(7) | 9.7055(4) | 4.993(1) |
| c/Å | 8.3190(8) | 16.5331(6) | 16.388(3) |
| β/° | 106.989(9) | 101.701(4) | 97.13(3) |
| V/Å3 | 402.44(6) | 1580.36(11) | 514.0(2) |
| Z | 4 | 12 | 4 |
| Dc/g cm−3 | 1.899 | 1.628 | 1.668 |
| μ/mm−1 | 0.174 | 0.143 | 0.146 |
| F(000) | 232 | 792 | 264 |
| λMoKα/Å | 0.71073 | 0.71073 | 0.71073 |
| T/K | 123 | 200 | 200 |
| θ Range/° | 4.0, 32.5 | 3.9, 25.0 | 3.2, 26.0 |
| Data set | −7: 7; −14: 14; −12:12 | −11: 10; −11: 11; −19: 15 | −7: 7; −6: 6; −20: 19 |
| Reflections collected | 5975 | 7250 | 3454 |
| Independent reflections | 1452 | 2766 | 1003 |
| Rint | 0.059 | 0.064 | 0.037 |
| Observed reflections | 742 | 1139 | 862 |
| No. parameters | 153 | 258 | 94 |
| R1 (obs) | 0.0274 | 0.0664 | 0.0364 |
| wR2 (all data) | 0.0497 | 0.2102 | 0.0946 |
| GooF | 0.83 | 0.91 | 1.08 |
| Δρ/e Å−3 | −0.23, 0.21 | −0.27, 0.85 | −0.22, 0.14 |
| CCDC | 689202 | 689201 | 689203 |
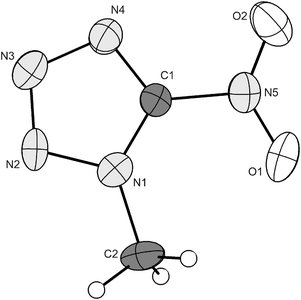
One of the two crystallographically independent formula units found in the crystal structure of compound 1 is represented in Fig. 3. Protonation of the NT− anion occurs at N3, which is in contrast with 5-amino-46 or 5-azidotetrazole.23 In Table 3 there are summarized the angles and distances of the two 5-nitrotetrazolate rings, which, within the limits of error, are not significantly different. The main difference is observed in the twist of the nitro groups in respect to the tetrazole rings. Whereas in one of the two molecules the nitro group is almost coplanar to the ring (O4–N10–C1–N6 = −178.2(3)°), in the other, this is significantly deviated (O2–N5–C1–N1 = −164.9(3)°). So, one of the formula units is similar to metal NT salts, which show small torsion angles between 2 and 5°,30 whereas the other one is more similar to NT salts with nitrogen-rich bases (0–10°).34 A plausible explanation for this could be that in NT salts, there is a negative charge, which is delocalized all around the tetrazole ring and over the nitro group, making them virtually coplanar. Proof for this is the relatively longer C1–N1 distances (∼1.445(4) Å) in 1. Therefore one would expect larger torsion angles due to the lack of delocalization in both formula units. The smaller torsion angle for one of the two units can be explained by hydrogen-bonding effects (see discussion below).
Table 3 Selected bond lengths [Å] and angles [°] for compounds 1–3
| | 1 (A) | 1 (B) | 2 (A) | 2 (B) | 2 (C) | 3 |
|---|
| O1–N5 | 1.229(3) | 1.227(3) | 1.208(6) | 1.194(6) | 1.196(5) | 1.223(2) |
| O2–N5 | 1.221(3) | 1.215(3) | 1.191(6) | 1.202(5) | 1.185(5) | 1.222(2) |
| N5–C1 | 1.440(4) | 1.450(4) | 1.450(7) | 1.481(7) | 1.438(6) | 1.445(2) |
| N1–C1 | 1.319(4) | 1.310(4) | 1.326(6) | 1.316(6) | 1.342(6) | 1.321(2) |
| N1–N2 | 1.323(4) | 1.319(4) | 1.336(6) | 1.345(5) | 1.354(6) | 1.319(2) |
| N2–N3 | 1.318(3) | 1.322(3) | 1.297(6) | 1.317(6) | 1.325(6) | 1.329(2) |
| N3–N4 | 1.324(4) | 1.318(3) | 1.356(7) | 1.363(6) | 1.317(7) | 1.317(2) |
| N4–C1 | 1.333(4) | 1.336(4) | 1.303(6) | 1.291(6) | 1.295(6) | 1.331(2) |
| N1(2)–C2 | | | 1.461(6) | 1.478(7) | 1.487(6) | 1.461(2) |
| |
| O2–N5–O1 | 125.5(3) | 126.0(3) | 127.6(6) | 127.2(5) | 125.6(5) | 125.1(1) |
| O1–N5–C1 | 117.9(3) | 117.2(3) | 115.8(5) | 117.2(5) | 117.4(5) | 117.4(1) |
| O2–N5–C1 | 116.6(3) | 116.7(3) | 116.6(5) | 115.7(5) | 117.0(5) | 117.5(1) |
| N1–C1–N5 | 123.2(3) | 122.3(3) | 125.9(5) | 123.2(5) | 124.8(4) | 121.9(1) |
| C1–N1–N2 | 100.1(3) | 99.0(3) | 106.8(4) | 106.5(4) | 107.3(4) | 99.9(1) |
| N3–N2–N1 | 114.7(3) | 115.4(3) | 106.6(5) | 106.2(4) | 104.9(4) | 114.3(1) |
| N2–N3–N4 | 105.7(2) | 105.5(2) | 111.4(5) | 110.8(4) | 111.5(5) | 106.1(1) |
| N3–N4–C1 | 105.0(2) | 104.3(3) | 103.6(5) | 103.5(4) | 106.6(5) | 104.6(1) |
| N4–C1–N5 | 122.1(3) | 121.9(3) | 122.5(5) | 123.5(5) | 125.4(5) | 123.0(1) |
| N1–C1–N4 | 114.5(3) | 115.8(3) | 111.6(5) | 113.1(5) | 109.7(5) | 115.1(1) |
| C1–N1–C2 | | | 106.8(4) | 131.9(5) | 131.4(5) | |
| N1–N2–C2 | | | | | | 123.6(1) |
| N2–N1–C2 | | | 121.1(5) | 121.5(5) | 121.3(5) | |
| N3–N2–C2 | | | | | | 122.1(1) |
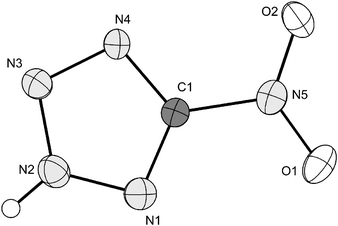 |
| | Fig. 3 Formula unit of 1 with the labelling scheme. Hydrogen atoms shown as spheres of arbitrary radius and thermal displacements set at 50% probability. | |
Regardless of the expected planarity of 1 the presence of a proton surrounded by many electronegative atoms forces the compound to form hydrogen bonds, which prohibits layering. These hydrogen bonds are formed by the protonated nitrogen atom (N2 or N7) as the donor atom and either tetrazole ring nitrogen atoms or, in one instance with one of the nitro groups oxygen atoms (O3). A report of hydrogen-bridges is given in Table 4. The interaction between N7 and O3 (N7⋯O3i = 3.015(4) Å; symmetry code: (i) −x, 0.5 + y, −z) “fixes” the nitro group in such a way that it is coplanar to the tetrazole ring and forms the C1,1(6) motifs represented in Fig. 4 (at the primary level) (Table 5). Similarly, the dimer pairs formed by two crystallographically related rings (N7⋯N3ii = 3.057(4) Å; symmetry code: (ii) 1 −x, 0.5 + y, 1 −z) yield a C1,1(4) graph-set. Lastly, the third hydrogen bond found in the structure forms only finite patterns of the type D1,1(2) at the primary level, which combine with the other two hydrogen bonds yielding larger dimeric interactions with the label D3,3(X) (X = 7, 9) at the secondary level. This results in a highly efficient packing as can be deduced from the high density of the compound (1.899 g cm−3).
Table 4 Geometry for selected hydrogen bonds in the structure of 1
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|
| Symmetry codes for 1: −x, 0.5 + y, −z. 1 −x, 0.5 + y, 1 −z. |
|---|
| 1 | | | | |
| N2–H1⋯N4 | 0.97(4) | 1.88(4) | 2.837(4) | 171.(3) |
| N7–H2⋯O3a | 0.85(4) | 2.22(4) | 3.015(4) | 158.(3) |
| N7–H2⋯N3b | 0.85(4) | 2.53(4) | 3.057(4) | 121.(3) |
Table 5 Graph-set matrix for medium to strong hydrogen bonds in the crystal structure of 1. First level motifs on-diagonal and second level graph sets off-diagonal
| H-Bond | N2–H1⋯N4 | N7–H2⋯O3a | N7–H2⋯N3b |
|---|
| Symmetry codes for 1: −x, 0.5 + y, −z. 1 −x, 0.5 + y, 1 −z. |
|---|
| N2–H1⋯N4 | D1,1(2) | | |
| N7–H2⋯O3a | D3,3(9) | C1,1(6) | |
| N7–H2⋯N3b | D3,3(7) | | C1,1(4) |
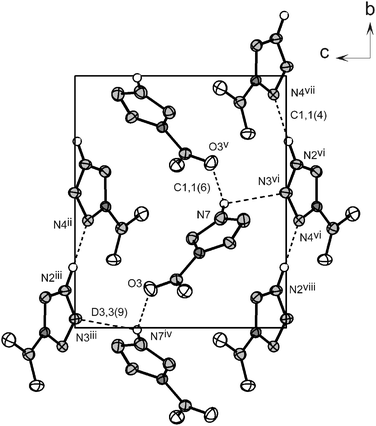 |
| | Fig. 4 View of the unit cell of 1 along the a-axis showing the graph-sets in the structure (dotted lines). Symmetry codes: (ii) 1 −x, 0.5 + y, 1 −z; (iii) 1 + x, y, 1 + z; (iv) 1 −x, −0.5 + y, 1 −z; (v) 2 −x, 0.5 + y, 1 −z; (vi) 1 −x, 0.5 + y, −z; (vii) x, 1 + y, z; (viii) 1 + x, y, z. | |
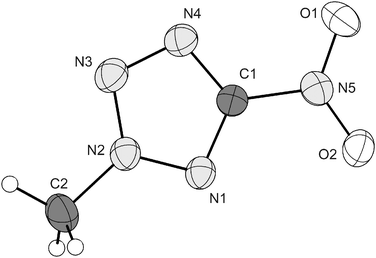
The unit cell of 2, which crystallizes in the monoclinic space group P21/c contains twelve molecules. For better clearness only one molecule of the asymmetric unit is shown in Fig. 5. Since hydrogen bonds are not present in the structures of 2 and 3, the densities (2: 1.628, 3: 1.668 g cm−3) are significantly lower than that observed for 1 (1.899 g cm−3). 3, (in Fig. 6), crystallizes monoclinic in the space group P21/c with four molecules in the unit cell. The molecular geometry of 2 as well as of 3 is particularly comparable to that of 1 and other 5-substituted tetrazoles. All C–N and N–N bond lengths lie between single and double bonds, whereby the shortest distance (1.30–1.33 Å) is observed between the nitrogen atoms N2 and N3. In both cases the NO2 group is co-planar with the tetrazole ring, which confirms previously published assumptions.47 The distances between the atoms C1 and N5 are between 1.43 and 1.48 Å, which are in the range of typical C–N single bonds. The same trend can be found for the N1–C2 and N2–C2 bond lengths (1.46–1.49 Å).
 |
| | Fig. 5 Formula unit of 2 with the labelling scheme. Hydrogen atoms shown as spheres of arbitrary radius and thermal displacements set at 30% probability. | |
 |
| | Fig. 6 Formula unit of 3 with the labelling scheme. Hydrogen atoms shown as spheres of arbitrary radius and thermal displacements set at 50% probability. | |
Thermal and energetic properties
In order to assess the thermal and energetic properties of neutral 5-nitrotetrazoles 1–3 the thermal stability (decomposition points from DSC measurements), as well as the sensitivities to friction, impact, electrostatic discharge and thermal shock of all three compounds were experimentally assessed (Tables 6 and 7) using standard BAM tests.50–55 In addition, for all three CHNO compounds the constant volume energies of combustion were calculated using quantum chemical methods (see Computational Methods section). Initially, we measured experimentally the combustion data for 3 using oxygen bomb calorimetry, however the high sensitivity of the compound did not allow reproducible values to be obtained. The material explodes rather than burning in the aerobic conditions of the measurements leading to erroneous values. The heats and energies of formation of 1–3 were back-calculated from the combustion data and subsequently used in conjunction with the molecular formula and density (from X-ray) to predict the performance (detonation pressure and velocity) for each compound using the EXPLO5 computer code.61
Table 6 Physico-chemical properties, initial safety data and predicted performance of compounds 1–3
| | 1 | 2 | 3 |
|---|
| BAM methods, see ref. 50–55. BAM methods, see ref. 50–55. OZM electric spark tester, see ref. 57–59. Nitrogen content. Nitrogen + oxygen content. Oxygen balance.60 Fast heating behavior. Decomposition temperature from DSC (β = 5 °C). Estimated from X-ray diffraction. Calculated molar enthalpy of formation. Energy of formation. Energy of explosion, EXPLO5 V5.02. Explosion temperature. Detonation pressure. Detonation velocity. Assuming only gaseous products. |
|---|
| Formula | CHN5O2 | C2H3N5O2 | C2H3N5O2 |
| Molecular mass/g mol−1 | 115.05 | 129.08 | 129.08 |
| Impact sensitivitya/J | < 1 | 2 | 1 |
| Friction sensitivityb/N | < 5 | 82 | 40 |
| Electrical dischargec/J | — | 0.50 | 0.20 |
| N (%)d | 60.9 | 54.3 | 54.3 |
| N + O (%)e | 88.6 | 79.0 | 79.0 |
| Ω (%)f | −7.0 | −43.4 | −43.4 |
| Thermal shockg | Deflagration | Combustion | Combustion |
| Combustion | Very good | Good | Good |
| Smokeless | + | + | + |
| DSCh/°C | 98 (mp), 130 (decomp.) | 45 (mp), 155 (decomp.) | 75 (mp), 150 (decomp.) |
| Densityi/g cm−3 | 1.899 | 1.628 | 1.668 |
ΔfHm°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j/kJ mol−1 j/kJ mol−1 | 281 | 278 | 247 |
| ΔfU°k/kJ kg−1 | +2527 | +2253 | +2006 |
| |
| Calculated values using EXPLO5 |
| −ΔEUm°l/J g−1 | −5744 | −5588 | −5368 |
| TEm/K | 4804 | 4226 | 4071 |
| pn/kbar | 390 | 257 | 262 |
| Do/m s−1 | 9457 | 8085 | 8109 |
| Gas vol.p/L kg−1 | 779 | 766 | 763 |
Table 7 Comparison of energetic properties of compounds 1–3 with RDX
| | | 1 | 2 | 3 | RDX |
|---|
| Density/g cm−3 | ρ | 1.899 | 1.630 | 1.670 | 1.820 |
| Oxygen balance (%) | Ω | −7.0 | −43.4 | −43.4 | −21.6 |
| Energy of formation/kJ kg−1 | ΔfU | +2527 | +2253 | +2006 | +67 |
| Heat of detonation/kJ kg−1 | Qv | −5744 | −5588 | −5368 | −5902 |
| Detonation temperature/K | Tex. | 4804 | 4226 | 4071 | 3986 |
| Detonation pressure/kbar | P | 390 | 257 | 262 | 299 |
| Detonation velocity/m s−1 | D | 9457 | 8085 | 8109 | 8796 |
| Volume of detonation gases/L kg−1 | V0 | 779 | 766 | 763 | 932 |
Fig. 7 shows typical DSC thermographs of compounds 1–3. Slow heating in a DSC apparatus (β = 5 °C min−1) of samples of ∼1.5 mg of each energetic material gives rapid decomposition at temperatures above 130 °C for all three compounds. All three materials show highly exothermic decompositions following to endothermic peaks at 98 (1), 45 (2) and 75 (3) °C corresponding to the melting of the compounds. The difference in area between endothermic and exothermic peaks gives a feeling for the energy released upon decomposition. For all 1–3 the decomposition releases much more energy than that required for melting. Particularly, compound 1 shows only a small melting endotherm followed by highly energetic decomposition. It is interesting to note the effect of the substituent in the tetrazole ring in the melting and decomposition points. The presence of the ring proton in 1 results in a higher melting point than for 2 and 3 due to the possibility of forming classical hydrogen bonds in 1, however, the compound is much more sensitive (i.e., less stable) and decomposes at lower temperatures. The substitution pattern of the methyl group in 2 and 3 also accounts for the lower melting point of 2 in comparison to 3 due to the formation of a less effective packing, as suggested by the lower crystal density of 2 (1.628 g cm−3) in comparison to 3 (1.668 g cm−3). Furthermore, the presence of the methyl group results in an increase (20–25 K) of the decomposition temperatures (∼150 °C) as observed for methylated 5-aminotetrazoles.37 Further studies on the decomposition of 3 can be found in literature.48 In addition to DSC analysis, the response to thermal shock of 1–3 was tested by placing a small sample (∼0.5–1.0 mg) of compound in the flame. This resulted in an vigorous reaction (deflagration) in the case of 1 and normal burning in the case of 2 and 3, in all cases smokeless. By comparison with typical primary explosives such as lead azide or styphnate, which both explode in the flame, the compounds studied here are less sensitive to thermal shock and show a similar response to classical secondary explosives such as TNT or RDX.
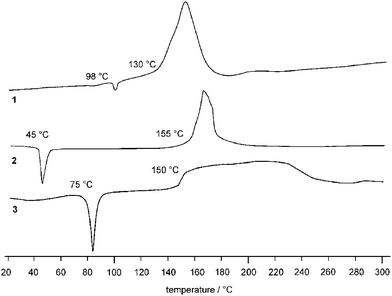 |
| | Fig. 7 DSC thermographs of 5-nitrotetrazoles 1–3 at a heating rate β = 5 °C min−1. | |
Data collected for initial safety testing of compounds 1–3 are summarized in Table 6. The impact and friction sensitivities as well as the electrostatic sensitivity were determined.49 The impact sensitivity tests were carried out according to STANAG 448950 modified according to instruction51 using a BAM (Bundesanstalt für Materialforschung)52 drophammer.53 The friction sensitivity tests were carried out according to STANAG 448754 modified according to instruction55 using the BAM friction tester. Compound 1 is very sensitive towards impact (<1 J) and extremely friction sensitive (<5 N). 2 and 3 are also very sensitive towards impact (2, 2 J and 3, 1 J) but less sensitive towards friction (2, 82 N and 3, 40 N). Grinding of the compounds in a mortar results in rattling and (in some instances) a loud explosion. According to the “UN Recommendations on the transport of dangerous goods”,56 compounds 1–3 are classified as “very sensitive” regarding the impact sensitivity values. The compounds in this study are significantly more sensitive to friction and impact than nitrogen-rich salts of 5-nitro-2H-tetrazole34 and the impact sensitivity approaches that of alkali metal salts of 5-nitro-2H-tetrazole.30 Comparison of the energetic compounds of the materials in this study with those of commonly used high explosives are useful to assess the potential of the materials described here. All three materials have impact sensitivity values, which are comparable to lead azide (2.5–4.0 J, pure product). As for the friction sensitivity, 1 has a value between that of the primary explosives lead azide (0.1–1.0 N, pure product) and tetrazene (7 N), whereas 2 and 3 have similar sensitivity to the secondary explosive PETN (60 N).5 In addition, the sensitivity towards electrostatic discharge of 2 and 3 was tested using an electric spark tester ESD 2010EN (OZM Research) operating with the “Winspark 1.15 software package”.57 Due to the hygroscopicity of compound 1, this compound was omitted from this study. The electrical spark sensitivities of microcrystalline materials (5–100 μm)58 were determined to be 0.50 ± 0.05 (2) and 0.20 (3) ± 0.04 J. These values can be compared to those of commonly used secondary explosives, e.g. RDX (0.15 J), PETN (0.19) and TNT (0.57). However, the ESD sensitivities determined are higher than those of modern insensitive explosives such as TATB (1,3,5-triamino-2,4,6-trinitrobenzene).59 Lastly, the high sensitivities of 1–3 can be attributed not only to the high endothermicity of the materials (see discussion below) but also to the only slightly negative oxygen balance, in particular in the case of compound 1.
In addition to safety considerations, performance of HEDMs is of utmost importance. Using the molecular formula, density (from X-ray) and energy of formation, the EXPLO5 computer code61 can be used to calculate the detonation velocity and pressure of CHNO-based explosive materials. The program is based on the chemical equilibrium, steady-state model of detonation. It uses the Becker–Kistiakowsky–Wilson’s equation of state (BKW EOS) for gaseous detonation products and Cowan–Fickett’s equation of state for solid carbon.61,62 The calculation of the equilibrium composition of the detonation products is done by applying modified White, Johnson and Dantzig’s free energy minimization technique. The program is designed to enable the calculation of detonation parameters at the CJ point. The BKW equation in the following form was used with the BKWN set of parameters (α, β, κ, θ) as stated below the equations and Xi being the mol fraction of ith gaseous product, ki is the molar covolume of the ith gaseous product.40,41 The results of the EXPLO5 calculations for neutral 5-nitrotetrazoles 1–3 are presented in Tables 6 and 7, with the corresponding values for commonly used RDX for comparison purposes.
| pV/RT = 1 + xeβxx = (κ∑Xiki)/[V (T + θ)]α; α = 0.5, β = 0.176, κ = 14.71, θ = 6620. |
Further physico-chemical properties of all three compounds are tabulated in
Table 7. Compounds
1–3 have high
nitrogen contents in the range between ∼50 and 60%, excellent combined oxygen and
nitrogen balances in the range between ∼80 and 90% and slightly negative oxygen balances approximately in the range between that of dinitroglycol (C
2H
4N
2O
6,
Ω = ±0.0%) and that of
nitromethane (CH
3NO
2,
Ω = −39.3%). As expected, the densities, calculated from the X-ray measurements, are lower in the case of the methylated derivatives
2 and
3 (∼1.65 g cm
−3) but still comparable to
TNT (1.654 g cm
−3), while
1 has an exceptionally high density of 1.899 g cm
−3 comparable to β-HMX (1.900 g cm
−3).
5 The energies of formation of
1–3 were back-calculated from the energies of
combustion on the basis of their
combustion equations (see below), Hess’s Law, the known standard heats of formation for
water and carbon dioxide and a correction for change in gas volume during
combustion. No corrections for the non-ideal formation of
nitric acid (typically ∼5% of the
nitrogen content reacts to form HNO
3) were made. As pointed out above, all three compounds are highly endothermic with energies of formation above +2000 kJ kg
−1.
1 has the most positive value of all at +2527 kJ kg
−1 and all three compounds show similar endothermicities to
nitrogen-rich salts with the 5,5′-azotetrazolate anion ([N
4C–N = N–CN
4]
2−).
12,63| | | 1: CHN5O2 (s) + 0.25 O2 (g) → CO2 (g) + 0.5 H2O (l) + 2.5 N2 (g) | (1) |
| | | 2, 3: C2H3N5O2 (s) + 1.75 O2 (g) → 2 CO2 (g) + 1.5 H2O (l) + 2.5 N2 (g) | (2) |
The methylated derivatives
2 and
3 have calculated detonation velocities of ∼8100 m s
−1, higher than
TNT (6900 m s
−1), lower than
RDX (8800 m s
−1) and similar to 5,5′-azotetrazole salts,
12,63 regardless of the high sensitivity of the compounds. On the other hand,
1 although being very sensitive to impact and friction and thus classifying as a primary explosive has an astonishingly high calculated detonation velocity of 9457 m s
−1, which is comparable to some of the highest performing secondary explosives known to date such as HMX (
octogen, 9100 m s
−1),
CL-20 and
octanitrocubane (∼10
000 m s
−1)
5 and also higher than the primary explosive
5-azido-1H-tetrazole regardless of the lower endothermicity of the –NO
2 group in comparison to the –N
3 substituent.
21 Here it is necessary to mention that the previous study of Koldobskii and coworkers on compound
1, reports a experimental density value of 1.73 g cm
−3,
33 which is much lower than our calculated value of 1.899 g cm
−3 and therefore affects strongly the detonation parameters. The detonation pressures have accordingly high values (390 kbar for
1 and ∼260 kbar for
2 and
3), which are comparable to HMX (
octogen) (
pdet. = 384 kbar) and
RDX (
hexogen) (
pdet. = 299 kbar) respectively.
5Using the calculated heats of formation, the calculated density (from X-ray) and the molecular formula the ICT code64 was used to predict the heats of explosion as well as the decomposition gases formed upon explosion/decomposition of compounds 1–3. Table 8 contains tabulated results of these calculations together with the predicted values for two commonly used high explosives, namely lead azide (primary explosive) and RDX (secondary explosive), for comparative purposes.
Table 8 Predicted decomposition gases and heats of explosion of compounds 1–3 and comparison with commonly used high explosives (using the ICT code)
| Compoundab | CO2 | H2O | N2 | CO | H2 | NH3 | CH4 | HCN | C | Pb | ΔHexc |
|---|
| The amount of gases formed at 298 K is given in g kg−1 (i.e., grams of gas per kilogram of energetic compound). —, the decomposition product was not predicted by the code. Heat of explosion in cal g−1. Measured at a density of 1.76 g cm−3. |
|---|
| 1 | 276.61 | 75.54 | 607.47 | 17.11 | 0.05 | 1.34 | — | — | 21.40 | — | 1621 |
| 2 | 100.72 | 185.39 | 533.13 | 17.25 | 0.47 | 11.16 | 0.80 | 0.44 | 150.35 | — | 1558 |
| 3 | 100.45 | 186.28 | 533.22 | 16.21 | 0.43 | 11.06 | 0.66 | 0.43 | 150.97 | — | 1512 |
| Pb(N3)2 | — | — | 288.56 | — | — | — | — | — | — | 711.44 | 391 |
| RDXd | 292.09 | 232.40 | 373.83 | 22.90 | 0.21 | 5.26 | 0.16 | 0.30 | 72.28 | — | 1592 |
As expected from the high nitrogen content of the materials molecular nitrogen is predicted to be the major product of the decomposition of compounds 1–3 (∼500–600 g kg−1). By comparison, the decomposition of RDX is expected to produce much lower amounts of environmentally-friendly nitrogen (374 g kg−1), which is however still the main expected product followed by the formation of carbon dioxide (292 g kg−1). In keeping with the logic that RDX derives its energy from both oxidation of the carbon backbone and the formation of nitrogen. The second main product predicted upon decomposition of 1 is also carbon dioxide (277 g kg−1), which is anticipated to form in larger amounts than for compounds 2 and 3 (∼100 g kg−1) fitting with the better oxygen balance of 1. 2 and 3 are nevertheless expected to generate larger amounts of water than 1 (∼185 vs. 75 g kg−1). Apart from carbon soot, which is predicted to form in relatively large amounts for 2 and 3 (∼150 g kg−1), the rest of the decomposition gases (CO, H2, NH3, CH4 and HCN) are foreseen to form in marginally low quantities (<20 g kg−1). The amount of highly toxic gases (i.e., CO and HCN) expected from the explosion of 1–3 are then comparable to those formed upon explosion of RDX and in contrast with the large amounts of highly toxic lead powder predicted for the explosion of lead azide (711 g kg−1). Lastly, the heats of explosion have all values above 1500 cal g−1, is larger for the more energetic compound 1, are comparable to the secondary explosive RDX and much larger than the primary explosive lead azide (391 cal g−1).
Computational methods. Due to the high sensitivity of all compounds studied here, bomb calorimetric measurements could only be performed with small amounts of the materials and doubtful combustion data was obtained. Therefore we decided to estimate the thermodynamic data by quantum chemical methods. All calculations were carried out using the Gaussian G03W (revision B.03) program package.65 The enthalpies (H) and free energies (G) were calculated using the complete basis set (CBS) method described by Petersson and coworkers in order to obtain very accurate values. The CBS models use the known asymptotic convergence of pair natural orbital expressions to extrapolate from calculations using a finite basis set to the estimated complete basis set limit. CBS-4 begins with a HF/3-21G(d) geometry optimization; the zero point energy is computed at the same level. It then uses a large basis set SCF calculation as a base energy, and a MP2/6-31+G calculation with a CBS extrapolation to correct the energy through second order. A MP4(SDQ)/6-31+(d,p) calculation is used to approximate higher order contributions. In this study we applied the modified CBS-4M method (M referring to the use of minimal population localization) which is a re-parametrized version of the original CBS-4 method and also includes some additional empirical corrections.66,67 The enthalpies of the gas-phase species M were computed according to the atomization energy method (eqn (3)) (Tables 9–12).68| | | ΔfH°(g, M, 298) = H(Molecule, 298)−∑H°(Atoms, 298) + ∑ΔfH°(Atoms, 298) | (3) |
From the gas-phase enthalpies of formation ΔfH°(g) the enthalpies of the solid state were calculated using the enthalpies of sublimation by the equation:| | | ΔfH°(s) = ΔfH°(g) − (ΔsubH) | (4) |
For a solid compound the enthalpy of sublimation (ΔsubH) can be approximated on the basis of TROUTON’s rule 72 if the melting temperature (Tm in K) is known:| | | ΔsubH [J mol−1] = 188 Tm [K] | (5) |
With the known enthalpies of formation of carbon dioxide (ΔfH°298(CO2(g)) = −393.8 kJ mol−1) and water (ΔfH°298(H2O(g)) = −241.9 kJ mol−1) the enthalpies of formation of 1–3 can now be calculated. From these values, the energy of formation (ΔfU°298) can easily be obtained from the combustion eqns (1)–(3) according to eqn (6) with Δn being the change of moles of the gaseous components (Δn: 1 = −3.25; 2, 3 = 2.75) in eqns (7) and (8).| | | 1: C (s) + 0.5 H2 (g) + 2.5 N2 (g) + O2 (g) → CHN5O2 (s) | (7) |
| | | 2, 3: 2 C (s) + 1.5 H2 (g) + 2.5 N2 (g) + O2 (g) → C2H3N5O2 (s) | (8) |
As can be seen from Table 13 compounds 1–3 are formed strongly endothermically (1: 281, 2: 278, 3: 247 kJ mol−1). These values are slightly higher than that of 5-amino-1H-tetrazole (ΔfH°(s) = 208 kJ mol−1) and in the same range observed for 5-nitriminotetrazole (264 kJ mol−1) and 1-methyl-5-nitriminotetrazole (260 kJ mol−1). The enthalpy of formation of energetic materials are governed by the molecular structure of the compound. Therefore, heterocycles with a higher nitrogen content (e.g. imidazole (ΔfH°(s) = 58.6 kJ mol−1),72 1,2,4-triazole (ΔfH°(s) = 109.3 kJ mol−1),73 1H-1,2,3,4-tetrazole (ΔfH°(s) = 237.4 kJ mol−1))74 show trends in increasing heats of formation.
Table 9 CBS-4M results
Table 10 Literature values for atomic ΔH°f298/kcal mol−1
| | ref. 69 | NIST70 |
|---|
| H | 52.6 | 52.1 |
| C | 170.2 | 171.3 |
| N | 113.5 | 113.0 |
| O | 60.0 | 59.6 |
Table 11 Enthalpies of the gas-phase species M
| | Tm/K | ΔHsub/kcal mol−1 |
|---|
| 1 | 374 | 16.8 |
| 2 | 318 | 14.3 |
| 3 | 348 | 15.6 |
Table 13 Solid state enthalpies (ΔfH°) and energies of formation (ΔfU°)
| | ΔfH°(s)/kcal mol−1 | ΔfH°(s)/kJ mol−1 | Δn | ΔfU°(s)/kcal mol−1 | M/g mol−1 | ΔfU°(s)/kJ kg−1 |
|---|
| 1 | +67.2 | +281.4 | −4 | +69.5 | 115.07 | +2527.1 |
| 2 | +66.5 | +278.4 | −5 | +69.5 | 129.08 | +2252.8 |
| 3 | +58.9 | +246.6 | −5 | +61.9 | 129.08 | +2006.4 |
Experimental
CAUTION! The 5-nitrotetrazoles described here are energetic compounds, which are sensitive towards heat, impact, friction and electrostatic discharge. Although we experienced no difficulties in the synthesis of these materials, proper protective measures (safety glasses, face shield, leather coat, earthened equipment and shoes, Kevlar® gloves and ear plugs) should be used when undertaking work involving 1–3 on small and in particular on larger scales.General method
All reagents and solvents were used as received (Sigma-Aldrich, Fluka, Acros Organics) unless stated otherwise. Melting points were measured with a Linseis PT10 DSC75 and checked with a Büchi Melting Point B-450 apparatus (uncorrected). DSC measurements were performed at a heating rate of 5 °C min−1 in closed aluminum sample pans with a 1 μm hole in the top for gas release under a nitrogen flow of 20 mL min−1 with an empty identical aluminum sample pan as a reference. NMR spectra were recorded with a Jeol Eclipse 270, Jeol EX 400 or a Jeol Eclipse 400 instrument. All chemical shifts are quoted in ppm relative to TMS (1H, 13C) and MeNO2 (14N, 15N). Infrared (IR) spectra were recorded using a Perkin-Elmer Spektrum One FT-IR instrument.76 Transmittance values are qualitatively described as “very strong” (vs), “strong” (s), “medium” (m) and “weak” (w). Raman spectra were measured using a Perkin-Elmer Spektrum 2000R NIR FT-Raman instrument equipped with a Nd:YAG laser (1064 nm). The intensities are reported as percentages of the most intense peak and are given in parentheses. Elemental analyses were performed with a Netsch Simultaneous Thermal Analyzer STA 429.Anhydrous ammonium 5-nitrotetrazolate (0.44 g, 3.32 mmol) and potassium hydroxide (0.19 g, 3.32 mmol) were dissolved in 3.7 mL water. The solution was stirred at reflux until no more ammonia gas was evolved. At this point, the reaction mixture was cooled by means of an ice-bath and cold ∼25% sulfuric acid (2 mL) was added dropwise using a plastic syringe. The solution was then extracted with ether (4 × 6 mL) and the ether extracts were combined and washed to remove the excess of acid with water (6 mL). The organic phase was then dried with magnesium sulfate and filtered and the solvent was stripped under high vacuum (∼10−3 mbar) yielding the pure product as a slightly yellow semicrystalline solid (0.27 g, 72%), which was carefully (!!) scratched out using a plastic spatula and analyzed. DSC (5 °C min−1, °C): 98 (mp), >130 (decomp.); IR ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 (KBr, rel. int.): 3443 (s), 2013 (w), 1629 (m), 1565 (vs), 1443 (m), 1401 (m), 1320 (s), 1262 (vw), 1192 (w), 1103 (w), 1047 (m), 1022 (m), 840 (s), 666 (w), 534 (vw); Raman /cm−1 (rel. int.): 3316 (2), 3261 (2), 1572 (14), 1492 (9), 1446 (100), 1433 (90), 1396 (15), 1358 (7), 1317 (13), 1200 (13), 1186 (14), 1142 (30), 1094 (42), 1069 (13), 1043 (11), 1027 (26), 837 (22), 775 (13), 736 (4), 592 (3), 532 (11), 444 (22), 256 (17), 240 (16), 154 (6); 1H NMR (DMSO-d6, 400.18 MHz, 25 °C, TMS) δ/ppm: 6.29 (1H, NH); 13C{1H} NMR (DMSO-d6, 100.63 MHz, 25 °C, TMS) δ/ppm: 168.4 (1C, C–NO2); 14N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: +14 (2 N, ν1/2∼300 Hz, N2/3), −24 (1 N, ν1/2∼60 Hz, NO2), −66 (2 N, ν1/2∼320 Hz, N1/4); 15N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: +19.6 (2 N, s, N2/N3), −29.8 (1 N, s, NO2), −69.6 (2 N, s, N1/4); m/z (FAB−, xenon, 6 keV, m-NBA matrix): 113.9 (100, CN5O2−); EA (CHN5O2, 115.07): calc. C 10.44, H 0.88, N 60.87; found: not determinable due to high sensitivity; BAM drophammer: <1 J, friction tester: <5 N, flame: deflagration.A suspension of 1-methyl-5-aminotetrazole (2.00 g, 20 mmol) in 1 M sulfuric acid (10 mL) and 30 mL of water was added at 0 °C to 30 mL water containing sodium nitrite (2.76 g, 40 mmol). After stirring at room temperature for 12 h and filtration of the precipitated bis(1-methyltetrazolyl)triazene, the solvent was evaporated. Dry acetone (80 mL) was added to this and the precipitated Na2SO4 was removed by filtration. After evaporating the acetone the crude product was recrystallized from a small amount of ethanol (1.52 g, yield 59%); DSC (5 °C min−1, °C): 45 °C (mp), 155 °C (decomp.); IR (KBr, cm−1): = 3038 (w), 2860 (w), 1550 (vs), 1481 (s), 1467 (m), 1408 (s), 1364 (s), 1328 (vs), 1280 (w), 1209 (m), 1073 (m), 1025 (w), 846 (s), 720 (s), 535 (w), 430 (m); Raman (1064 nm, 200 mW, 25 °C, cm−1): = 3054 )2), 2978 (6), 2964 (6), 1530 (20), 1508 (9), 1469 (100), 1463 (44), 1447 (44), 1425 (15), 1412 (11), 1329 (24), 1261 (10), 1208 (13), 1103 (11), 1085 (23), 1028 (21), 923 (7), 779 (2), 740 (2), 709 (3), 679 (4); 1H NMR ([d6]-DMSO, 25 °C, ppm) δ: 3.68 (s, 3H, CH3); 13C NMR ([d6]-DMSO, 25 °C, ppm) δ: 157.6 (CN4), 33.1 (CH3); 14N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: −37 (1 N, ν1/2∼60 Hz, NO2), 15N NMR ([d6]-DMSO, 25 °C) δ = 4. 5 (N3), −14.1 (N6), −18.29 (N2, t, 3JNH = 1.9 Hz), −71.15 (N4), −157.16 (N5), −168.38 (N1, d, 2JNH = 2.2 Hz), –289.13 (N7, 1JNH = 102.7 Hz), −329.66 (N8, 1JNH = 69.4 Hz); m/z (DEI+): 130 (19) [M + H]+, 129 (65) [M]+, 100 (1), 83 (8), 55 (15), 54 (17), 53 (100), 46 (38), 43 (28), 40 (7), 39 (5), 28 (45), 18 (8), 15 (4); EA (C2H3N5O2, 129.08): calcd.: C 18.61, H 2.34, N 54.26; found: C 18.39, H 2.28, N 52.80; BAM drophammer: 2 J; friction tester: 82 N, ESD: 0.50 ± 0.05 J, flame: combustion.To 20 mL of an aqueous sodium nitrite (2.76 g, 0.04 mol) solution, a solution of 2-methyl-5-aminotetrazole (2.00 g, 0.02 mol) in 20 mL 1N sulfuric acid was added at 0 °C. The reaction mixture was stirred for 8 h and the precipitated bis(2-methyltetrazolyl)triazene precipitated was removed by filtration. Afterwards the product was extracted three times with 20 mL of CH2Cl2. The organic phases were combined, dried over MgSO4 and evaporated. The crude product was recrystallized from acetone yielding single crystals suitable for XRD analysis. (1.68 g, yield 65%); DSC (5 °C min−1, °C): 75 °C (mp), 150 °C (decomp.); IR (KBr, cm−1): = 3022 (m), 1610 (m), 1565 (s), 1510 (m), 1468 (m), 1412 (s), 1285 (s), 1160 (m), 1001 (w), 880 (m), 788 (m), 750 (m), 670 (w), 610 (w), 530 (w); Raman (1064 nm, 200 mW, 25 °C, cm−1): = 3052 (12), 2967 (60), 1555 (28), 1486 (40), 1468 (26), 1418 (100), 1369 (11), 1335 (8), 1322 (14), 1287 (12), 1209 (30), 1075 (12), 1043 (40), 1026 (44), 841 (16), 776 (17), 715 (46), 547 (10), 436 (30), 378 (18), 307 (16), 218 (16); 1H NMR ([d6]-DMSO, 25 °C, ppm) δ: 4.50 (s, 3H, CH3); 13C NMR ([d6]-DMSO, 25 °C, ppm) δ: 166.4 (CN4), 41.9 (CH3); 14N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: −34 (1 N, ν1/2∼50 Hz, NO2); 15N NMR ([d6]-DMSO, 25 °C) δ = 5.3 (N3), −33.5 (N5), −55.1 (N4), −76.6 (N2, 2JNH = 2.1 Hz), –97.9 (N1, 3JNH = 1.7 Hz); m/z (DEI+): 130 (2) [M + H]+, 129 (2) [M]+, 115 (1) [M + H − CH 3]+, 101 (15), 58 (89), 43 (100) [HN3]+, 42 (6), 28 (3) [N2]+, 18 (29); EA (C2H3N5O2, 129.08): calcd.: C 18.61, H 2.34, N 54.26; found: C 18.88, H 2.35, N 52.99; BAM drophammer: 1 J; friction tester: 40 N, ESD: 0.20 ± 0.04 J, flame: combustion.
/cm−1 (KBr, rel. int.): 3443 (s), 2013 (w), 1629 (m), 1565 (vs), 1443 (m), 1401 (m), 1320 (s), 1262 (vw), 1192 (w), 1103 (w), 1047 (m), 1022 (m), 840 (s), 666 (w), 534 (vw); Raman /cm−1 (rel. int.): 3316 (2), 3261 (2), 1572 (14), 1492 (9), 1446 (100), 1433 (90), 1396 (15), 1358 (7), 1317 (13), 1200 (13), 1186 (14), 1142 (30), 1094 (42), 1069 (13), 1043 (11), 1027 (26), 837 (22), 775 (13), 736 (4), 592 (3), 532 (11), 444 (22), 256 (17), 240 (16), 154 (6); 1H NMR (DMSO-d6, 400.18 MHz, 25 °C, TMS) δ/ppm: 6.29 (1H, NH); 13C{1H} NMR (DMSO-d6, 100.63 MHz, 25 °C, TMS) δ/ppm: 168.4 (1C, C–NO2); 14N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: +14 (2 N, ν1/2∼300 Hz, N2/3), −24 (1 N, ν1/2∼60 Hz, NO2), −66 (2 N, ν1/2∼320 Hz, N1/4); 15N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: +19.6 (2 N, s, N2/N3), −29.8 (1 N, s, NO2), −69.6 (2 N, s, N1/4); m/z (FAB−, xenon, 6 keV, m-NBA matrix): 113.9 (100, CN5O2−); EA (CHN5O2, 115.07): calc. C 10.44, H 0.88, N 60.87; found: not determinable due to high sensitivity; BAM drophammer: <1 J, friction tester: <5 N, flame: deflagration.A suspension of 1-methyl-5-aminotetrazole (2.00 g, 20 mmol) in 1 M sulfuric acid (10 mL) and 30 mL of water was added at 0 °C to 30 mL water containing sodium nitrite (2.76 g, 40 mmol). After stirring at room temperature for 12 h and filtration of the precipitated bis(1-methyltetrazolyl)triazene, the solvent was evaporated. Dry acetone (80 mL) was added to this and the precipitated Na2SO4 was removed by filtration. After evaporating the acetone the crude product was recrystallized from a small amount of ethanol (1.52 g, yield 59%); DSC (5 °C min−1, °C): 45 °C (mp), 155 °C (decomp.); IR (KBr, cm−1): = 3038 (w), 2860 (w), 1550 (vs), 1481 (s), 1467 (m), 1408 (s), 1364 (s), 1328 (vs), 1280 (w), 1209 (m), 1073 (m), 1025 (w), 846 (s), 720 (s), 535 (w), 430 (m); Raman (1064 nm, 200 mW, 25 °C, cm−1): = 3054 )2), 2978 (6), 2964 (6), 1530 (20), 1508 (9), 1469 (100), 1463 (44), 1447 (44), 1425 (15), 1412 (11), 1329 (24), 1261 (10), 1208 (13), 1103 (11), 1085 (23), 1028 (21), 923 (7), 779 (2), 740 (2), 709 (3), 679 (4); 1H NMR ([d6]-DMSO, 25 °C, ppm) δ: 3.68 (s, 3H, CH3); 13C NMR ([d6]-DMSO, 25 °C, ppm) δ: 157.6 (CN4), 33.1 (CH3); 14N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: −37 (1 N, ν1/2∼60 Hz, NO2), 15N NMR ([d6]-DMSO, 25 °C) δ = 4. 5 (N3), −14.1 (N6), −18.29 (N2, t, 3JNH = 1.9 Hz), −71.15 (N4), −157.16 (N5), −168.38 (N1, d, 2JNH = 2.2 Hz), –289.13 (N7, 1JNH = 102.7 Hz), −329.66 (N8, 1JNH = 69.4 Hz); m/z (DEI+): 130 (19) [M + H]+, 129 (65) [M]+, 100 (1), 83 (8), 55 (15), 54 (17), 53 (100), 46 (38), 43 (28), 40 (7), 39 (5), 28 (45), 18 (8), 15 (4); EA (C2H3N5O2, 129.08): calcd.: C 18.61, H 2.34, N 54.26; found: C 18.39, H 2.28, N 52.80; BAM drophammer: 2 J; friction tester: 82 N, ESD: 0.50 ± 0.05 J, flame: combustion.To 20 mL of an aqueous sodium nitrite (2.76 g, 0.04 mol) solution, a solution of 2-methyl-5-aminotetrazole (2.00 g, 0.02 mol) in 20 mL 1N sulfuric acid was added at 0 °C. The reaction mixture was stirred for 8 h and the precipitated bis(2-methyltetrazolyl)triazene precipitated was removed by filtration. Afterwards the product was extracted three times with 20 mL of CH2Cl2. The organic phases were combined, dried over MgSO4 and evaporated. The crude product was recrystallized from acetone yielding single crystals suitable for XRD analysis. (1.68 g, yield 65%); DSC (5 °C min−1, °C): 75 °C (mp), 150 °C (decomp.); IR (KBr, cm−1): = 3022 (m), 1610 (m), 1565 (s), 1510 (m), 1468 (m), 1412 (s), 1285 (s), 1160 (m), 1001 (w), 880 (m), 788 (m), 750 (m), 670 (w), 610 (w), 530 (w); Raman (1064 nm, 200 mW, 25 °C, cm−1): = 3052 (12), 2967 (60), 1555 (28), 1486 (40), 1468 (26), 1418 (100), 1369 (11), 1335 (8), 1322 (14), 1287 (12), 1209 (30), 1075 (12), 1043 (40), 1026 (44), 841 (16), 776 (17), 715 (46), 547 (10), 436 (30), 378 (18), 307 (16), 218 (16); 1H NMR ([d6]-DMSO, 25 °C, ppm) δ: 4.50 (s, 3H, CH3); 13C NMR ([d6]-DMSO, 25 °C, ppm) δ: 166.4 (CN4), 41.9 (CH3); 14N NMR (DMSO-d6, 40.55 MHz, 25 °C, MeNO2) δ/ppm: −34 (1 N, ν1/2∼50 Hz, NO2); 15N NMR ([d6]-DMSO, 25 °C) δ = 5.3 (N3), −33.5 (N5), −55.1 (N4), −76.6 (N2, 2JNH = 2.1 Hz), –97.9 (N1, 3JNH = 1.7 Hz); m/z (DEI+): 130 (2) [M + H]+, 129 (2) [M]+, 115 (1) [M + H − CH 3]+, 101 (15), 58 (89), 43 (100) [HN3]+, 42 (6), 28 (3) [N2]+, 18 (29); EA (C2H3N5O2, 129.08): calcd.: C 18.61, H 2.34, N 54.26; found: C 18.88, H 2.35, N 52.99; BAM drophammer: 1 J; friction tester: 40 N, ESD: 0.20 ± 0.04 J, flame: combustion.Conclusions
From this combined experimental and theoretical study the following conclusions can be drawn:Convenient procedures for the synthesis of three highly energetic neutral 5-nitrotetrazoles (1–3) are presented, which allow the materials to be obtained at low cost using facile routes, good yields and excellent purities. The full characterization of the compounds by analytical and spectroscopic methods is described in detail. In addition we determined the molecular structure of the compounds in the solid state by X-ray diffraction methods. The energetic properties of the compounds were assessed by means of standard tests and quantum chemical calculations (CBS-4M). 1–3 are calculated to be strongly endothermic with heats of formation between 247 and 282 kJ mol−1. The compounds show highly exothermic decomposition peaks (DSC) and are easily initiated by impact. Furthermore, 1–3 have high performances (EXPLO5 code) comparable to commonly used secondary explosives regardless of their ease of initiation and 1 classifies as a primary explosive in regard to its impact and friction sensitivity values and still has a calculated detonation velocity, which is almost twice as large as that of common primary explosives (e.g., lead azide) and puts it first in the list of high performing primers. Unfortunately, 1 absorbs water, which limits its application. Furthermore 1–3 are thermally too unstable for use as conventional high explosives. The predicted products (ICT code) formed upon explosion of 1–3 are expected to be less harmful than those expected from the decomposition of commonly used high explosives, which suggest their potential (2 and 3) as environmentally friendly alternatives with high performance and low initiation barriers (by impact) for use in energetic applications (e.g., as ingredients for energetic fillers in high explosive compositions).
Acknowledgements
Financial support of this work by the Ludwig-Maximilian University of Munich (LMU), the Fonds der Chemischen Industrie (FCI), the European Research Office (ERO) of the U.S. Army Research Laboratory (ARL) under contract nos. N-62558-05-C-0027, 9939-AN-01 and W911NF-07-1-0569 and the Bundeswehr Research Institute for Materials, Explosives, Fuels and Lubricants (WIWEB) under contract nos. E/E210/4D004/X5143 and E/E210/7D002/4F088 is gratefully acknowledged. The authors are indebted to and thank Dr Betsy Rice and Dr Gary Chen for many helpful discussions and support of our work. We also acknowledge Mr. Stefan Huber for help with the sensitivity tests. The authors acknowledge collaborations with Dr M. Krupka (OZM Research, Czech Republic) in the development of new testing and evaluation methods for energetic materials and with Dr M. Sućesca (Brodarski Institute, Croatia) in the development of new computational codes to predict the detonation parameters of high-nitrogen explosives.References
- T. M. Klapötke, in ‘Moderne Anorganische Chemie’, ed. E. Riedel (Hrsg.), Walter de Gruyter, Berlin, New York, 2003, 2nd edn, pp. 95–100 Search PubMed; T. M. Klapötke, in High Energy Density Materials, ed. T. M. Klapötke (Hrsg.), Springer, Berlin, Heidelberg, 2007, pp. 85–122 Search PubMed.
- A. Hammerl, G. Holl, T. M. Klapötke, P. Mayer, H. Nöth, H. Piotrowski and M. Warchhold, Eur. J. Inorg. Chem., 2002, 4, 834 CrossRef.
- T. M. Klapötke, P. Mayer, A. Schulz and J. J. Weigand, J. Am. Chem. Soc., 2005, 127, 2032 CrossRef.
- R. P. Singh, H. Gao, D. T. Meshri and J. M. Shreeve, in High Energy Density Materials, ed. T. M. Klapötke (Hrsg.), Springer, Berlin, Heidelberg, 2007, pp. 35–84 Search PubMed.
- A. Köhler and R. Mayer, ‘Explosivstoffe’, Wiley-VCH, D-Weinheim, 9nd edn, 1998 Search PubMed; D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 Search PubMed.
- S. Oga, Jpn. Kokai Tokkyo Koho, JP2006249061, p. A20060921, 2006 Search PubMed.
- T. M. Klapötke, G. Holl. J. Geith, A. Hammerl and J. J. Weigand, New Trends in Research of Energetic Materials, Proceedings of the Seminar, Pardubice, 7th, Czech Republic, 2004 Search PubMed.
- T. M. Stierstorfer and J. Stierstorfer, Phys. Chem. Chem. Phys., 2008, 10, 4340–4346 RSC; T. M. Klapötke and J. Stierstorfer, Eur. J. Inorg. Chem., 2008, 26, 4055–4062 CrossRef; T. M. Klapötke, J. Stierstorfer and A. U. Wallek, Chem. Mater., 2008, 20(13), 4519–4530 CrossRef.
- G. Geisberger, T. M. Klapötke and J. Stierstorfer, Eur. J. Inorg. Chem., 2007, 30, 4743–4750 CrossRef.
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45(22), 3584 CrossRef CAS.
- T. M. Klapötke, P. Mayer, A. Schulz and J. J. Weigand, Propellants, Explos., Pyrotech., 2004, 29(6), 325 CrossRef.
- A. Hammerl, M. A. Hiskey, G. Holl, T. M. Klapötke, K. Polborn, J. Stierstorfer and J. J. Weigand, Chem. Mater., 2005, 17, 3784 CrossRef CAS.
- C. Ye, J. C. Xiao, B. Twamley and J. M. Shreeve, Chem. Commun., 2005, 2750 RSC.
- J. C. Galvez-Ruiz, G. Holl, K. Karaghiosoff, T. M. Klapötke, K. Loehnwitz, P. Mayer, H. Noeth, K. Polborn, C. J. Rohbogner, M. Suter and J. J. Weigand, Inorg. Chem., 2005, 44(14), 5192 CrossRef CAS.
- H. Xue, B. Twamley and J. M. Shreeve, Inorg. Chem., 2004, 43(25), 7972 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66(24), 7945–7950 CrossRef CAS.
- J. Thiele, Liebigs Ann., 1892, 270, 1–2 Search PubMed.
- K. Hattori, E. Lieber and J. P. Horwitz, J. Am. Chem. Soc., 1956, 78, 411–415 CrossRef.
- G. I. Koldobskii, D. S. Soldatenko, E. S. Gerasimova, N. R. Khokhryakova, M. B. Shcherbinin, V. P. Lebedev and V. A. Ostrovskii, Russ. J. Org. Chem. (Engl. Transl.), 1997, 33(12), 1771–1783 CAS.
- E. Oliveri-Mandala and T. Passalaqua, Gazz. Chim. Ital., 1912, 41(II), 430–435 CAS.
- G. F. Holland and J. N. Pereira, J. Med. Chem., 1967, 10(2), 149–154 CAS; S. V. Voitekhovich, P. N. Gaponik and A. O. Koren, Mendeleev Commun., 1997, 1, 41–42 Search PubMed; P. N. Gaponik, S. V. Voitekhovich and B. G. Klyaus, Russ. J. Org. Chem., 2004, 40(4), 598–600 CrossRef CAS; M. D. Cullen, B. L. Deng, T. L. Hartman, K. M. Watson, R. W. Buckheit, Jr, C. Pannecouque, E. De Clercq and M. Cushman, J. Med. Chem., 2007, 50(20), 4854–4867 CrossRef CAS.
- R. A. Henry, W. G. Finnegan and E. Lieber, J. Am. Chem. Soc., 1954, 76, 2894–2898 CrossRef CAS; C. Stadler, J. Daub, J. Köhler, R. W. Saalfrank, V. Coropceanu, V. Schuenemann, C. Ober, A. X. Trautwein, S. F. Parker, M. Poyraz, T. Inomata and R. D. Cannon, Dalton Trans., 2001, 3373–3383 RSC; S. Achamlale, A. Elachqar, A. El Hallaoui, A. Alami, S. Elhajji, M. L. Roumestant and P. Viallefont, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 140, 103–111 Search PubMed; A. O. Koren, P. N. Gaponik, O. A. Ivashkevich and T. B. Kovalyova, Mendeleev Commun., 1995, 1, 10–11 Search PubMed.
- J. Stierstorfer, T. M. Klapötke, A. Hammerl and B. Chapman, Z. Anorg. Allg. Chem., 2008, 634, 1051–1057 CrossRef CAS; A. Hammerl and T. M. Klapötke, Inorg. Chem., 2002, 41(4), 906–912 CrossRef CAS; A. Hammerl, T. M. Klapötke, P. Mayer and J. J. Weigand, Propellants, Explos., Pyrotech., 2005, 30(1), 17–26 CrossRef CAS.
- T. M. Klapötke and J. Stierstorfer, Helv. Chim. Acta, 2007, 90, 2132–2150 CrossRef; T. M. Klapötke and J. Stierstorfer, New Trends in Research of Energetic Materials, Proceedings of the Seminar, Pardubice, Czech Republic, 2007, 10th, vol. 2, pp. 674–690 Search PubMed.
- A. M. Astakhov, R. S. Stepanov, L. A. Kruglyakova and A. A. Nefedov, Russ. J. Org. Chem., 2001, 37(4), 577–582 CrossRef CAS.
- D. Adam, K. Karaghiosoff, G. Holl, M. Kaiser and T. M. Klapötke, Propellants, Explos., Pyrotech., 2002, 27(1), 7 CrossRef CAS.
- K. Karaghiosoff, T. M. Klapötke, A. Michailovski, H. Nöth and M. Suter, Propellants, Explos., Pyrotech., 2003, 28(1), 1 CrossRef CAS.
- T. M. Klapötke, P. Mayer and V. Verma, Propellants, Explos., Pyrotech., 2006, 31(4), 263 CrossRef.
- H. Xue, S. W. Arritt, B. Twamley and J. M. Shreeve, Inorg. Chem., 2004, 43, 7972–7977 CrossRef CAS; H. Xue, Y. Gao, B. Twamley and J. M. Shreeve, Chem. Mater., 2005, 17, 191–198 CrossRef CAS; H. Xue, Y. Gao, B. Twamley and J. M. Shreeve, Inorg. Chem., 2005, 44, 5068–5072 CrossRef CAS; Y. Gao, B. Twamley and J. M. Shreeve, Chem. Eur. J., 2006, 12, 9010–9018 CrossRef CAS; J. C. Gálvez-Ruiz, G. Holl, K. Karaghiosoff, T. M. Klapötke, K. Löhnwitz, P. Mayer, H. Nöth, K. Polborn, C. J. Rohbogner, M. Suter and J. J. Weigand, Inorg. Chem., 2005, 44(12), 4237–4253 CrossRef CAS; T. M. Klapötke and C. Miró Sabaté, Z. Anorg. Allg. Chem., 2007, 633, 2671–2677 CrossRef; C. Darwich, T. M. Klapötke and C. Miró Sabaté, Chem. Eur. J., 2008, 14, 5756–5771 CrossRef CAS.
- T. M. Klapötke, C. Miró Sabaté and J. M. Welch, Dalton Trans., 2008 10.1039/b811410b.
- M. H. V. Huynh, M. D. Coburn, T. J. Meyer and M. Wetzler, Proc. Natl. Acad. Sci. USA, 2006, 103(27), 10322–10327 CrossRef CAS; M. H. V. Huynh, M. A. Hiskey, T. J. Meyer and M. Wetzler, Proc. Natl. Acad. Sci. USA, 2006, 103(14), 5409–5412 CrossRef CAS.
- J. Akhavan, in The Chemistry of Explosives, RSC Paperbacks, Cambridge, UK, 2nd edn, 2004 Search PubMed.
- G. I. Koldobskii, D. S. Solyearnko, E. S. Gerasimova, N. R. Khokhryakova, M. B. Shcherbinin, V. P. Lebedev and V. A. Ostrovskii, Russ. J. Org. Chem. (Engl. Transl.), 1997, 33, 1771–1783 CAS.
- T. M. Klapötke, P. Mayer, C. Miró Sabaté, J. M. Welch and N. Wiegand, Inorg. Chem., 2008, 47, 6014–6027 CrossRef.
- R. A. Henry and W. G. Finnegan, J. Am. Chem. Soc., 1954, 76, 923–926 CrossRef CAS.
- A. M. Churakov, S. E. Semenov, S. L. loffe, Y. A. Strelenko and V. A. Tartakovskii, Mendeleev Commun., 1995, 5(3), 102–103 Search PubMed.
- K. Karaghiosoff, T. M. Klapötke, P. Mayer, C. Miró Sabaté, A. Penger and J. M. Welch, Inorg. Chem., 2008, 47, 1007–1019 CrossRef CAS.
- CrysAlis CCD, Oxford Diffraction Ltd., Version 1.171.27p5 beta (release 01-04-2005 CrysAlis171 .NET) (compiled Apr 1 2005,17:53:34).
- CrysAlis RED, Oxford Diffraction Ltd., Version 1.171.27p5 beta (release 01-04-2005 CrysAlis171 .NET) (compiled Apr 1 2005,17:53:34).
- Z. Otwinowski and W. Minor, in “Processing of X-ray Diffraction Data Collected in Oscillation Mode”, Methods in Enzymology, Volume 276 Macromolecular Crystallography, Part A, eds. C. W. Carter Jr and R. M. Sweet, Academic Press, New York, 1997, pp. 307–326, Search PubMed.
- SIR-92, A program for crystal structure solution A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, Universität Göttingen, 1997.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1999.
- SCALE3 ABSPACK-An Oxford Diffraction program (1.0.4,gui:1.0.3), © 2005 Oxford Diffraction Ltd.
- D. D. Bray and J. G. White, Acta Crystallogr., Sect. B, 1979, 35, 3039 CrossRef.
- M. M. Sokolova, V. V. Mel'nikov, A. A. Mel'nikov and B. V. Gidaspov, Chem. Heterocycl. Compd., 1977, 6, 689–692 CrossRef.
- V. I. Kolesov, V. P. Sinditskii, V. Y. Egorshev, B. A. Lurie and M. Y. Soloviev, International Annual Conference of ICT, 32nd, 2001, 117/1-117/12.
- M. Sućeska, Test Methods for Explosives, Springer, New York, 1995, p. 21 (impact), p. 27 (friction) Search PubMed.
- NATO standardization agreement (STANAG) on explosives, impact sensitivity tests, no. 4489, Ed. 1, Sept. 17, 1999.
- WIWEB-Standardarbeitsanweisung 4-5.1.02, Ermittlung der Explosionshier der Schlagempfindlichkeit mit dem Fallhammer, Nov. 8, 2002.
- http://www.bam.de.
- http://www.reichel-partner.de/.
- NATO standardization agreement (STANAG) on explosive, friction sensitivity tests, no. 4487, Ed. 1, Aug. 22, 2002.
- WIWEB-Standardarbeitsanweisung 4-5.1.03, Ermittlung der Explosionsgefährlichoder der Reibeempfindlichkeit mit dem Reibeapparat, Nov. 8, 2002.
- Impact: Insensitive >40 J, less sensitive ≥35 J, sensitive ≥4, very sensitive ≤3 J. Friction: Insensitive > 360 N, less sensitive = 360 N, sensitive <360 N and >80 N, very sensitive ≤80 N, extremely sensitive ≤10 N. According to the UN Recommendations on the Transport of Dangerous Goods.
- http://www.ozm.cz/testing-instruments/small-scale-electrostatic-discharge-tester.htm.
- S. Zeman, V. Pelikán and J. Majzlík, Cent. Eur. J. Energ. Mater., 2006, 3(3), 45–51 Search PubMed.
- D. Skinner, D. Olson and A. Block-Bolten, Propellants, Explos., Pyrotech., 1997, 23, 34–42.
- Calculation of the oxygen balance: Ω (%) = (wO − 2xC − 1/2yH − 2zS)1600/M. (w: number of oxygen atoms, x: number of carbon atoms, y: number of hydrogen atoms, z: number of sulfur atoms, M: molecular weight).
- M. Sućeska, Mater. Sci. Forum, 2004, 465–466, 325–330 CrossRef CAS; M. Sućeska, Propellants, Explos., Pyrotech., 1991, 16, 197–202 CrossRef CAS; M. Sućeska, Propellants, Explos., Pyrotech., 1999, 24, 280–285 CrossRef CAS.
- M. L. Hobbs and M. R. Baer, Calibration of the BKW-EOS With a Large Product Species Data Base and Measured C–J, Properties Proc. of the 10th Symp. (International) on Detonation, ONR 33395-12, Boston, MA, July 12–16, 1993, p. 409.
- T. M. Klapötke and C. Miró Sabaté, Chem. Mater., 2008, 20(5), 1750–1763 CrossRef.
- ICT-Thermodynamic Code, version 1.0, Fraunhofer-Institut für Chemische Technologie (ICT): Pfinztal/Berghausen, Germany, 1988–2000; R. Webb and M. van Rooijen, Proceedings of the 29th International Pyrotechnics Seminar, 2002, 823–828 Search PubMed; H. Bathelt and F. Volk, 27th International Annual Conference of ICT, 1996, 92, 1–16 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision B0.4), Gaussian, Inc., Pittsburgh, PA, 2004 Search PubMed.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery, Jr, J. Chem. Phys., 1996, 104, 2598 CrossRef CAS.
- J. A. Montgomery, Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532 CrossRef.
- E. F. Byrd and B. M. Rice, J. Phys. Chem., 2006, 110(3), 1005–1013 Search PubMed; B. M. Rice, S. V. Pai and J. Hare, Combust. Flame, 1999, 118(3), 445–458 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, J. Chem. Phys., 1997, 106(3), 1063 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg, MD, June 2005, 20899, http://webbook.nist.gov.
- M. S. Westwell, M. S. Searle, D. J. Wales and D. H. Williams, J. Am. Chem. Soc., 1995, 117, 5013 CrossRef CAS.
- R. C. West and S. M. Selby, Handbook of Chemistry and Physics, The Chemical Rubber Co., Cleveland, OH, 48th edn, 1967–1968, pp. D22–D51 Search PubMed.
- V. A. Ostrovskii, M. S. Pevzner, T. P. Kofman and I. V. Tselinskii, Targets Heterocycl. Syst., 1999, 3, 467 Search PubMed.
- W. S. McEwan and M. W. Rigg, J. Am. Chem. Soc., 1951, 73, 4725–4727 CrossRef CAS.
- http://www.linseis.com/.
- http://www.perkin-elmer.com/.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.