The annular tautomerism of the curcuminoid NH-pyrazoles†
Pilar
Cornago
*a,
Pilar
Cabildo
a,
Rosa M.
Claramunt
a,
Latifa
Bouissane
a,
Elena
Pinilla
b,
M. Rosario
Torres
b and
José
Elguero
c
aDepartamento de Química Orgánica y Bio-Orgánica, Facultad de Ciencias, UNED, Senda del Rey 9, E-28040 Madrid, Spain. E-mail: mcornago@ccia.uned.es; Fax: +34 913988372; Tel: +34 913987323
bDepartamento de Química Inorgánica I, Facultad de Ciencias Químicas, Universidad Complutense de Madrid (UCM), 28040 Madrid, Spain
cInstituto de Química Médica, CSIC, Juan de la Cierva 3, E-28006 Madrid, Spain
First published on 2nd December 2008
Abstract
The structures of four NH-pyrazoles, (E)-3,5-bis[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-1H-pyrazole (3), (E)-3(5)-[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-5(3)-methyl-1H-pyrazole (4), (E)-3(5)-[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-4,5(3)-dimethyl-1H-pyrazole (5) and (E)-3(5)-[β-(3,4-dimethoxyphenyl)-ethenyl]-4-methyl-5(3)-phenyl-1H-pyrazole (8), have been determined by X-ray crystallography. Compounds that have a phenol residue crystallize forming sheets that are stabilized by a complex pattern of hydrogen bonds between a unique tautomer (4), or by a 2 : 1 mixture of both tautomers (5) (these tautomers being identical in the case of 3). Pyrazole 8, which lacks OH groups, crystallizes in cyclic dimers that are stabilized by N–H⋯N hydrogen bonds. The tautomerism in solution and in the solid state was determined by 13C and 15N CPMAS NMR spectroscopy. For compounds 4, 5 and 8, the solid state results agree with those observed by crystallography; the most abundant tautomer in solution coincides with the tautomer present in the solid state (4 and 8) or with the most abundant tautomer in the crystal (5).
Introduction
Turmeric is a spice derived from the rhizomes of Curcuma longa, which is a member of the ginger family.1 The bright yellow color of turmeric comes mainly from polyphenolic pigments known as curcuminoids. Curcumin (1) (Scheme 1) is the principal curcuminoid found in turmeric, and is generally considered to be its most active constituent. In addition to its use as a spice and a pigment, turmeric has been used in India for medicinal purposes for centuries. More recently, evidence that 1 may have anti-inflammatory and anti-cancer activities has renewed scientific interest in its potential to prevent and treat disease. 1 is also an effective scavenger of reactive oxygen and nitrogen species in vitro. In addition to its direct antioxidant activity, 1 has been found to inhibit PLA2, COX-2 and 5-LOX activity in cultured cells. It has also been found to inhibit NF-κB-dependent gene transcription, and to inhibit the induction of COX-2 and iNOS in cell culture and animal studies.21 has been found to induce cell cycle arrest and apoptosis in a variety of cancer cell lines grown in cultures. The ability of 1 to induce apoptosis in cultured cancer cells has generated scientific interest in its potential to prevent some types of cancer. Oral administration of 1 has been found to inhibit the development of chemically-induced cancer in animal models of oral, stomach, liver and colon cancer.
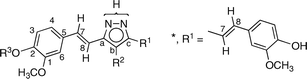
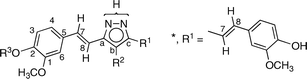
We have devoted a series of papers to the annular tautomerism of NH-pyrazoles 2 (2avs.2b),3,4 and decided to study those derived from 1 and related β-diketones.
Pyrazole 3, which is derived from 1, has been prepared many times since 1991.5–11 It has been described as a pale yellow solid that melts at 211–2145 or 2157 °C.
The activity of the curcuminoid pyrazoles covers domains such as anti-inflammatory (5-lipooxygenase and cyclooxygenase inhibitors)5,8 and anti-tumoral (anti-angiogenic)6–8 agents, and drugs for the treatment of Alzheimer’s disease (AD; potent γ-secretase inhibitors, potent ligands for fibrillar Ab42 aggregates, tau aggregation inhibitors and depolymerizing agents for tau aggregates).10,11 Particularly promising for treating reduced cognitive functions is 4,4′-[(1-phenyl-1H-pyrazole-3,5-diyl)di-(1E)-2,1-ethenediyl]bis(2-methoxyphenol) (CNB-001), the product obtained by reacting 1 with phenylhydrazine.12 In the last of these applications, curcumin-derived pyrazoles were synthesized in order to minimize the metal chelation properties of 1. The reduced rotational freedom and the absence of stereoisomers were anticipated to enhance the inhibition of γ-secretase. Accordingly, the replacement of the 1,3-dicarbonyl moiety by isosteric heterocycles, such as pyrazoles, turned these compounds into very interesting candidates for AD research.
The aim of this paper is to determine and discuss the structure, tautomerism and possible proton transfer in the solid state (SSPT) of six NH-pyrazoles by using a combination of X-ray crystallography and 13C/15N NMR spectroscopy.
The nomenclature used in the text and in the experimental is not in accordance with IUPAC rules. For all of the compounds with phenolic hydroxyl groups, 3–6, the phenol system has the highest priority; however, using IUPAC nomenclature here would be at the expense of comparability and clearness. For instance, compound 4 would be 2-methoxy-4-[(E)-2-(5-methyl-1H-pyrazol-3-yl)vinyl]phenol under IUPAC rules, rather than (E)-3(5)-[β-(4-hydroxy-3-methoxyphenyl)ethenyl]-5(3)-methyl-1H-pyrazole. In order to prioritize comparability over correct nomenclature, we have named all of the compounds as pyrazole derivatives.
Results and discussion
Synthesis
All of the compounds discussed in this work (Scheme 2) are reported in the experimental section. They were prepared by the reaction of hydrazine with the corresponding β-diketone, the most common method of synthesizing pyrazoles,13 which in the case of 3 was 1.14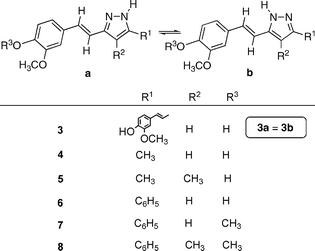 | ||
| Scheme 2 The structures of the NH-pyrazoles. | ||
X-Ray structure determination
The structures of pyrazoles 3 (derived from 1), 4, 5 and 8 have been determined by X-ray crystallography†.Concerning tautomerism, in the case of 3, tautomers 3a and 3b are identical. In the case of 4, the only tautomer present is 3-(3-methoxy)-4-hydroxy-styryl-5-methyl-1H-pyrazole (4a). In the case of 5, there is a 2 : 1 mixture of 3-(3-methoxy)-4-hydroxy-styryl-4,5-dimethyl-1H-pyrazole (5a) and 3,4-dimethyl-5-(3-methoxy)-4-hydroxy-styryl-1H-pyrazole (5b). In the case of 8, the only observed tautomer is 3-phenyl-4-methyl-5-(3-methoxy)-4-hydroxy-styryl-1H-pyrazole (8b). The main data are collected in Table 1 and Table 2. A characteristic feature of the geometry of NH-pyrazoles is that the angle centered at N1 (the atom bearing the NH proton) is always larger than that centered at N2, about 112 and 104°, respectively.15
| 3 | 4 | 5(1) | 5(2) | 5(3) | 8 | |
|---|---|---|---|---|---|---|
| N1–N2 | 1.354(3) | 1.365(3) | 1.349(4) | 1.352(4) | 1.359(3) | 1.351(3) |
| N2–C3 | 1.347(4) | 1.339(3) | 1.341(5) | 1.348(5) | 1.349(5) | 1.339(4) |
| C3–C4 | 1.399(4) | 1.398(4) | 1.388(6) | 1.410(5) | 1.401(6) | 1.415(4) |
| C4–C5 | 1.373(4) | 1.369(3) | 1.381(6) | 1.366(5) | 1.374(6) | 1.379(4) |
| C5–N1 | 1.353(4) | 1.340(3) | 1.332(6) | 1.346(5) | 1.331(5) | 1.360(4) |
| C3–C6 | 1.445(4) | 1.453(3) | — | 1.463(5) | 1.450(6) | 1.377(3) |
| C5–C6 | — | — | 1.446(7) | — | — | 1.444(4) |
| C6–C7 | 1.327(4) | 1.325(3) | 1.309(1) | 1.310(6) | 1.304(4) | 1.333(4) |
| C7–C8 | 1.467(4) | 1.472(3) | 1.460(1) | 1.457(5) | 1.475(6) | 1.460(4) |
| C3–C15 | — | — | 1.484(6) | — | — | — |
| C5–C15 | 1.450(4) | 1.484(2) | — | 1.490(5) | 1.509(6) | — |
| C15–C16 | 1.322(4) | — | — | — | — | — |
| C16–C17 | 1.463(4) | — | — | — | — | — |
| C10–O2 | 1.376(4) | 1.367(3) | 1.359(6) | 1.367(4) | 1.372(5) | 1.372(3) |
| O2–C14 | 1.433(4) | 1.419(3) | 1.424(6) | 1.430(5) | 1.442(5) | 1.416(4) |
| C11–O1 | 1.368(4) | 1.369(3) | 1.372(5) | 1.361(5) | 1.363(5) | 1.371(3) |
| C15–O1 | — | — | — | — | — | 1.418(4) |
| C19–O4 | 1.366(4) | — | — | — | — | — |
| O4–C23 | 1.416(4) | — | — | — | — | — |
| C20–O3 | 1.382(4) | — | — | — | — | — |
| N2–N1–C5 | 112.2(3) | 112.7(2) | 112.2(4) | 111.8(3) | 111.6(3) | 112.4(2) |
| N1–N2–C3 | 105.3(2) | 104.4(2) | 104.4(3) | 105.2(3) | 104.7(3) | 105.3(2) |
| Compound | D–H⋯A | d D–H | d H⋯A | d D⋯A | ∠D–H⋯A |
|---|---|---|---|---|---|
Symmetry transformations used to generate equivalent atoms: a −x + 2, y − ½, −z + ½.b −x + 1, y + ½, z + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) .c x + 1, y, z − 1.d −x + 1, −y + 2, −z + 1.e x, −y + , z − ½.f −x + 4, y − ½, −z + .g −x + 1, y − ½, −z + ½.h −x + 4, y + ½, −z + .i −x + 1, y + ½, −z + ½.j −x + 2, −y, −z + 1. .c x + 1, y, z − 1.d −x + 1, −y + 2, −z + 1.e x, −y + , z − ½.f −x + 4, y − ½, −z + .g −x + 1, y − ½, −z + ½.h −x + 4, y + ½, −z + .i −x + 1, y + ½, −z + ½.j −x + 2, −y, −z + 1. |
|||||
| 3 | O3–H3⋯O4 | 1.10 | 1.98 | 2.647(4) | 115.1 |
| N1–H1B⋯O3a | 1.06 | 1.93 | 2.864(4) | 144.7 | |
| O1–H1A⋯N2b | 1.17 | 1.79 | 2.811(4) | 142.7 | |
| O3–H3⋯O1c | 1.10 | 2.26 | 2.825(4) | 108.7 | |
| 4 | O1–H1A⋯N2d | 0.99 | 1.86 | 2.832(3) | 167.5 |
| N1–H1B⋯O2e | 1.07 | 2.17 | 2.962(3) | 128.6 | |
| 5 | O13–H113⋯N21 | 1.16 | 1.81 | 2.782(5) | 137.3 |
| N12–H12⋯N23 | 1.10 | 1.82 | 2.914(5) | 175.6 | |
| O11–H111⋯O13f | 0.92 | 2.03 | 2.813(4) | 141.3 | |
| O12–H112⋯N22g | 1.14 | 1.57 | 2.673(4) | 159.4 | |
| N11–H11⋯O11g | 1.08 | 2.01 | 2.951(5) | 144.3 | |
| N13–H13⋯O12i | 1.02 | 1.93 | 2.853(4) | 148.6 | |
| 8 | N1–H1⋯N2j | 0.90(4) | 2.07(4) | 2.872(3) | 147(4) |
Crystals of sufficient quality for X-ray diffraction analysis were obtained for compounds 3 (1 : 1 H2O/EtOH), 4 (1 : 1 : 1 CH2Cl2/hexane/EtOH), 5 (1 : 1 : 1 CH2Cl2/hexane/EtOH) and 8 (1 : 1 : 1 CH2Cl2/hexane/EtOH) from their respective solvent mixtures. Table 1 shows selected bond lengths and angles for each of these compounds, and Table 2 shows the distances and angles of the intermolecular hydrogen bonds.
One crystallographically-independent molecule was identified in the structural determination of 3, where the pyrazole and phenyl rings were co-planar, with bond distances and angles within normal ranges (Fig. 1). The intermolecular hydrogen bonds led to layers parallel to (1 0 1), as shown in Fig. 2.
 | ||
| Fig. 1 The X-ray molecular structure of compound 3 (ORTEP plot, 35% probability for the ellipsoids). | ||
 | ||
| Fig. 2 The view along the a axis of 3, showing the formation of layers due the intermolecular hydrogen bonds. | ||
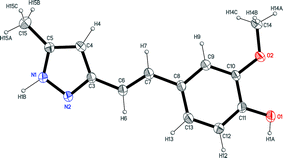
Fig. 3 shows an ORTEP representation of the asymmetric unit of compound 4, a non-planar molecule with a dihedral angle of 19.0(1)° between the pyrazole and phenyl rings. Dimers (O1–H1A–N2) linked by hydrogen bonds (N1–H1B–O2) led to layers parallel to (1 0 0), as shown in Fig. 4.
 | ||
| Fig. 3 The X-ray molecular structure of compound 4 (ORTEP plot, 35% probability for the ellipsoids). | ||
 | ||
| Fig. 4 The view along the b axis of 4, showing the formation of layers due the intermolecular hydrogen bonds. | ||
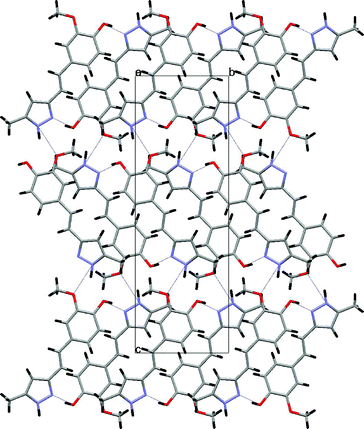
The asymmetric unit of compound 5 is presented in Fig. 5. The crystal consists of three crystallographically-independent, almost planar molecules, held together by hydrogen bonds that form a trimer, which, through additional hydrogen bonding, forms layers parallel to (–1 0 3), as shown in Fig. 6.
 | ||
| Fig. 5 The X-ray molecular structure of compound 5 (ORTEP plot, 40% probability for the ellipsoids). | ||
 | ||
| Fig. 6 The view along the a axis of 5, showing the formation of layers due the intermolecular hydrogen bonds. | ||
Fig. 7 shows the non-planar molecule of compound 8, with a dihedral angle of 15.7(1)° between the pyrazole and the phenyl ring at the 3-position, and 36.5(1)° between the pyrazole and the phenyl ring of the styryl group at the 5-position. Molecules of 8 are centrosymmetrically linked by hydrogen bonds (Table 2), giving rise to dimers, and these species are within van der Waals distances (Fig. 8).
 | ||
| Fig. 7 The X-ray molecular structure of compound 8 (ORTEP plot, 35% probability for the ellipsoids). | ||
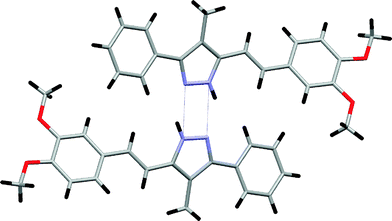 | ||
| Fig. 8 The view along the a axis of 8, showing the formation of dimers. | ||
The cyclic N–H⋯N hydrogen-bonded motifs (cyclamers) of NH-pyrazoles have been studied on several occasions.4d,16,17 These motifs are characteristic of NH-pyrazoles lacking substituents that bear hydrogen bonding functional groups, such as –OH or –CO2H. These groups, as well as solvent molecules like H2O and ROH, participate in the hydrogen bonding network that determines the secondary structure of the crystals, destroying the (N–H⋯N)nhydrogen bonds.18–20 In three of the compounds described in the present paper, those bearing phenol groups (3, 4 and 5) form several hydrogen bonds involving the OH group: 3 (O–H⋯N, N–H⋯O, O–H⋯O), 4 (O–H⋯N, N–H⋯O) and 5 (O–H⋯N, N–H⋯O, O–H⋯O, N–H⋯N; present as two molecules of tautomer 5a and one molecule of tautomer 5b). In the case of 8, which lacks phenol groups, the compound crystallizes as a dimer. This kind of cyclamer is characteristic of NH-pyrazoles that are substituted with phenyl groups at the 3- and 5-positions,16 to which compound 8 is clearly related.
NMR study
We have reported the 1H, 13C and 15N NMR results concerning compounds 3–8 in Table 3, Table 4 and Table 5, respectively. These data have been collected with the aim of determining the tautomeric equilibrium constants by simple integration. Although it has been pointed out that only 1H NMR signal intensities are reliable for the determination of populations, in our experience, 13C and 15N signals can also been used in connection with signals related by tautomerism, i.e.carbon or nitrogen atoms linked to the same substituents.3c The assignments of the signals were based on standard 2D experiments, on the values of coupling constants (auto-consistency) and by comparison with other NH-pyrazoles where tautomerization is blocked.21|
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Solvent | Conc./M | T/K | NH | R2 | H3 | H4 | H6 | OMe | OR3 | H7 | H8 | R1 | Tautomerism |
| a The coupling constants were, on average: 3JH3–H4 = 8.0 Hz, 4JH4–H6 = 2.0 Hz (not always observed) and 3JH7–H8trans = 16.5 Hz. | |||||||||||||||||
| 3 | * | H | H | DMSO | 0.12 | 300 | 12.80 | 6.61 (H) | 6.76 | 6.93 | 7.13 | 3.82 | 9.17 (H) | 7.03 | 6.91 | — | Average |
| 4 | CH3 | H | H | DMSO | 0.07 | 300 | 12.40 | 6.20 (H) | 6.74 | 6.91 | 7.12 | 3.81 | 9.15 (H) | 6.95 | 6.88 | 2.19 (Me) | Average |
| HMPA | 0.07 | 300 | 13.26 | 6.11 (H) | 6.85/7.16 | 3.80 | 10.26 (H) | 6.85/7.16 | 2.23 (Me) | ~50% a | |||||||
| HMPA | 0.07 | 300 | 13.20 | 6.11 (H) | 6.85/7.16 | 3.80 | 10.24 (H) | 6.85/7.16 | 2.15 (Me) | ~50% b | |||||||
| HMPA | 0.10 | 276 | 13.34 | 6.19 (H) | 6.85 | 6.85 | 7.06 | 3.80 | 10.42 (H) | 7.20 | 6.87 | 2.25 (Me) | ~50% a | ||||
| HMPA | 0.10 | 276 | 13.27 | 6.11 (H) | 6.85 | 6.85 | 7.06 | 3.80 | 10.34 (H) | 6.92 | 6.83 | 2.14 (Me) | ~50% b | ||||
| 5 | CH3 | CH3 | H | DMSO | 0.07 | 300 | 12.29 | 2.03 (Me) | 6.75 | 6.91 | 7.13 | 3.83 | 9.08 (H) | 6.95 | 6.86 | 2.10 (Me) | Average |
| HMPA | 0.10 | 300 | 13.16 | 2.04 (Me) | 6.87 | 6.92 | 6.97 | 3.80 | 10.28 (H) | 7.21 | 6.82 | 2.04 (Me) | 35% a | ||||
| HMPA | 0.10 | 300 | 13.10 | 2.04 (Me) | 6.87 | 6.92 | 6.97 | 3.80 | 10.20 (H) | 7.21 | 6.82 | 2.07 (Me) | 65% b | ||||
| HMPA | 0.10 | 268 | 13.25 | 2.05 (Me) | 6.87 | 6.96 | 7.00 | 3.81 | 10.44 (H) | 7.25 | 6.88 | 2.05 (Me) | 35% a | ||||
| HMPA | 0.10 | 268 | 13.20 | 2.05 (Me) | 6.87 | 6.96 | 7.00 | 3.81 | 10.38 (H) | 7.25 | 6.88 | 2.07 (Me) | 65% b | ||||
| 6 | C6H5 | H | H | DMSO | 0.11 | 300 | 12.96 | 6.88 (H) | 6.78 | 6.96 | 7.15 | 3.84 | 9.10 (H) | 7.10 | 6.95 | 7.80 (o) | 36% a |
| 7.43 (m) | |||||||||||||||||
| 7.31 (p) | |||||||||||||||||
| DMSO | 0.11 | 300 | 13.18 | 6.88 (H) | 6.78 | 6.96 | 7.15 | 3.84 | 9.21 (H) | 7.10 | 6.95 | 7.80 (o) | 64% b | ||||
| 7.43 (m) | |||||||||||||||||
| 7.31 (p) | |||||||||||||||||
| 7 | C6H5 | H | CH3 | DMSO | 0.06 | 300 | 13.00 | 6.87 (H) | 6.96 | 7.06 | 7.19 | 3.83 | 3.78 (Me) | 7.14 | 7.03 | 7.80 (o) | 40% a |
| 7.43 (m) | |||||||||||||||||
| 7.32 (p) | |||||||||||||||||
| DMSO | 0.06 | 300 | 13.21 | 6.87 (H) | 6.96 | 7.06 | 7.19 | 3.83 | 3.78 (Me) | 7.14 | 7.03 | 7.80 (o) | 60% b | ||||
| 7.43 (m) | |||||||||||||||||
| 7.32 (p) | |||||||||||||||||
| 8 | C6H5 | CH3 | CH3 | DMSO | 0.05 | 300 | 12.94 | 2.29 (Me) | 6.95 | 7.07 | 7.25 | 3.84 | 3.77 (Me) | 7.14 | 7.06 | 7.65 (o) | Rich in b |
| 7.45 (m) | |||||||||||||||||
| 7.34 (p) | |||||||||||||||||
| HMPA | 0.06 | 268 | 13.94 | 2.34 (Me) | 7.09 | 7.09 | 7.25 | 3.88 | 3.84 (Me) | 7.45 | 7.13 | 7.70 (o) | b | ||||
| 7.45 (m) | |||||||||||||||||
| 7.31 (p) | |||||||||||||||||
We have illustrated with one example the kind of spectra that we obtained (Fig. 9). The spectrum corresponds to compound 5 in HMPA-d18, concentration 0.10 M and temperature 268 K (Table 4). The region of the methyl groups shows two narrow signals corresponding to the most abundant tautomer, and two broad signals corresponding to the less abundant one, as expected by simple consideration of the energy profile.
 | ||
| Fig. 9 The methyl group region of the 13C NMR spectrum of 5. | ||
For compounds whose structure had not been determined by crystallography, we relied on CPMAS NMR results: 6b and 7b were the only tautomers present in the solid state (see Table 4 and Table 5). We are aware that solid state NMR and single crystal X-ray diffraction do not show exactly the same properties, for instance, static vs. dynamic disorder.3b To avoid further complications, we used fine powders for CPMAS NMR, obtained by grinding the same batch of crystals that we used for X-ray crystallography.
|
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Solvent | Conc./M | T/K | Ca | Cb | Cc | R2 | C1 | C2 | C3 | Tautomerism |
| C4 | C5 | C6 | C7 | C8 | OCH3 | R1 | ||||||||
| a The 1J coupling constants are not reported; their average values are: pyrazole C4–Hb = 175 Hz; phenyl CH = 159 Hz except C4–H and C6–H = 156 Hz; olefin C–H = 155 Hz; OCH3 = 144 Hz; C–Me substituents: 126.5 Hz. The other couplings (Hz) are: 2J = 2.2 (C1), 2J = 4.5 (C7), 2J = 5.9 (Cb–Me4); 3J = 8.4 (C1), 3J = 7.3 (C2), 3J = 5.8 (C4), 3J = 6.8 (C5), 3J = 6.0 (C6), 3J = 4.5 (C7), 3J = 2.4 (Cb–H). b Not observed. | ||||||||||||||
| 3 | * | H | H | DMSO | 0.12 | 300 | 151.0 | 99.3 | 142.0 | — (H) | 147.9 | 146.8 | 115.6 | No tautomerism |
| 120.1 | 128.4 | 109.5 | 129.8 | 112.9 (C8) | 55.6 | |||||||||
| 118.4 (C8′) | ||||||||||||||
| CPMAS | — | 300 | 150.2 | 95.5 | 142.8 | — (H) | 147.4 | 145.1 | 114.5 | No tautomerism | ||||
| N.o.b | 127.1 | 106.3 | 129.9 | 111.9 | 53.5 | |||||||||
| 56.5 | ||||||||||||||
| 4 | CH3 | H | H | DMSO | 0.36 | 300 | 149.6 | 101.3 | 140.5 | — (H) | 147.9 | 146.6 | 115.7 | Average |
| 119.9 | 128.6 | 109.5 | 129.0 | 117.4 | 55.6 | 11.6 (Me) | ||||||||
| HMPA | 0.10 | 276 | 150.9 | 100.0 | 142.4 | — (H) | 148.7 | 148.6 | 115.7 | ~50% a | ||||
| 119.4 | 128.4 | 110.5 | 128.7 | 113.2 | 55.9 | 10.8 (Me-5) | ||||||||
| 138.4 | 101.6 | 148.9 | — (H) | 148.7 | 146.9 | 115.7 | ~50% b | |||||||
| 119.6 | 128.4 | 110.5 | 129.4 | 119.3 | 55.9 | 13.9 (Me-3) | ||||||||
| CPMAS | — | 300 | 151.5 | 101.1 | 142.4 | — (H) | 148.8 | 143.3 | 115.3 | a | ||||
| 120.5 | 129.9 | 113.2 | 129.9 | 113.2 | 55.9 | 9.9 (Me-5) | ||||||||
| 115.3 | ||||||||||||||
| 5 | CH3 | CH3 | H | DMSO | 0.07 | 300 | 141.6 | 110.4 | 141.6 | 8.1 (Me) | 147.9 | 146.6 | 115.6 | Average |
| 119.8 | 128.8 | 109.6 | 127.9 | 114.9 | 55.7 | 10.6 | ||||||||
| HMPA | 0.08 | 300 | 147.5 | 109.9 | 135.7 | 8.4 (Me) | 149.0 | 148.7 | 116.1 | 35% a | ||||
| 119.6 | 129.0 | 111.5 | 127.7 | 118.7 | 56.3 | 11.9 (br) | ||||||||
| HMPA | 0.08 | 300 | 138.3 | 109.9 | 145.8 | 8.4 (Me) | 149.0 | 148.7 | 116.1 | 65% b | ||||
| 119.6 | 129.0 | 111.5 | 128.4 | 112.7 | 56.3 | 11.9 (br) | ||||||||
| HMPA | 0.10 | 268 | 147.5 | 110.0 | 135.7 | 8.8 (br, Me) | 148.8 | 148.6 | 115.8 | 35% a | ||||
| 119.5 | 128.8 | 110.7 | 127.6 | 118.5 | 55.9 | 9.1 (br) | ||||||||
| HMPA | 0.10 | 268 | 138.3 | 109.9 | 145.8 | 8.4 (Me) | 148.8 | 148.6 | 115.8 | 65% b | ||||
| 119.5 | 128.8 | 110.7 | 128.3 | 112.4 | 55.9 | 12.1 | ||||||||
| CPMAS | — | 300 | 145.9 | 110.2 | 138.6 | 9.8 (Me) | 148.8 | 146.6 | 121.8 | 66% a | ||||
| 123.4 | 130.9 | 105.5 | 128.9 | 117.0 | 55.3 | 11.2 (br) | ||||||||
| 137.5 | 112.0 | 146.6 | 9.8 (Me) | 148.8 | 146.6 | 121.8 | 34% b | |||||||
| 123.4 | 130.9 | 105.5 | 128.9 | 119.0 | 55.3 | 11.2 (br) | ||||||||
| 6 | C6H5 | H | H | DMSO | 0.11 | 300 | 151.4 | 100.4 | 140.3 | — (H) | 147.9 | 146.6 | 115.3 | 36% a |
| 122.1 | 128.1 | 109.5 | 130.1 | 118.4 | 55.5 | 132.0 (i) | ||||||||
| 125.0 (o) | ||||||||||||||
| 128.7 (m) | ||||||||||||||
| 127.5 (p) | ||||||||||||||
| DMSO | 0.11 | 300 | 142.6 | 99.5 | 151.0 | — (H) | 147.9 | 147.1 | 115.6 | 64% b | ||||
| 120.2 | 128.1 | 109.5 | 130.1 | 112.7 | 55.6 | 133.6 (i) | ||||||||
| 125.1 (o) | ||||||||||||||
| 128.7 (m) | ||||||||||||||
| 127.5 (p) | ||||||||||||||
| 6b | C6H5 | H | H | CPMAS | — | 300 | 144.0 | 103.5 | 152.6 | — (H) | 148.3 | 116.0 | ||
| 116.0 | 129.0 | 112.3 | 129.0 | 113.5 | 54.0 | 133.2 (i) | ||||||||
| 126.4 (o) | ||||||||||||||
| 129.0 (m) | ||||||||||||||
| 129.0 (p) | ||||||||||||||
| 7 | C6H5 | H | CH3 | DMSO | 0.11 | 300 | 151.3 | 99.2 | 142.8 | — (H) | 149.0 | 149.0 | 111.9 | 40% a |
| 119.4 | 129.4 | 108.9 | 128.9 | 113.6 | 55.51 (C1) | 133.7 (i) | ||||||||
| 55.45 (C2) | 125.0 (o) | |||||||||||||
| 128.6 (m) | ||||||||||||||
| 127.4 (p) | ||||||||||||||
| DMSO | 0.11 | 300 | 142.4 | 99.8 | 150.9 | — (H) | 149.0 | 149.0 | 111.9 | 60% b | ||||
| 119.9 | 129.4 | 108.9 | 129.7 | 113.6 | 55.51 (C1) | 133.7 (i) | ||||||||
| 55.45 (C2) | 125.0 (o) | |||||||||||||
| 128.6 (m) | ||||||||||||||
| 127.4 (p) | ||||||||||||||
| 7b | C6H5 | H | CH3 | CPMAS | — | 300 | 143.1 | 96.3 | 149.8 | — (H) | 148.8 | 148.8 | 110.6 | |
| 120.9 | 129.1 | 108.0 | 129.1 | 110.6 | 53.5 (C1*) | 132.1 (i) | ||||||||
| 56.1 (C2*) | 125.2 (o) | |||||||||||||
| 129.1 (m) | ||||||||||||||
| 126.2 (p) | ||||||||||||||
| 8 | C6H5 | CH3 | CH3 | DMSO | 0.31 | 300 | 141.7 | 110.7 | 147.1 | 9.4 (Me) | 149.1 | 148.8 | 111.9 | Aver. |
| 119.9 | 130.0 | 109.1 | 128.4 | 114.5 | 55.6 (C1) | 133.2 (i) | Rich in b | |||||||
| 55.5 (C2) | 127.1 (o) | |||||||||||||
| 128.5 (m) | ||||||||||||||
| 127.3 (p) | ||||||||||||||
| HMPA | 0.06 | 268 | 139.7 | 110.4 | 149.5 | 10.1 (Me) | 149.8 | 149.3 | 112.1 | b | ||||
| 120.2 | 130.9 | 109.1 | 128.8 | 113.2 | 55.9 (C1) | 135.9 (i) | ||||||||
| 55.9 (C2) | 127.2 (o) | |||||||||||||
| 128.6 (m) | ||||||||||||||
| 126.9 (p) | ||||||||||||||
| CPMAS | — | 300 | 140.7 | 112.5 | 148.8 | 9.2 (Me) | 148.8 | 148.8 | 112.5 | b | ||||
| 124.7 | 130.2 | 110.7 | 130.2 | 117.1 | 54.7 | 134.6 (i) | ||||||||
| 128.5 (o) | ||||||||||||||
| 130.2 (m) | ||||||||||||||
| 126.7 (p) | ||||||||||||||
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Solvent | Conc./M | T/K | N–H | –N= | % a | % b | PT a |
| a Proton transfer b Not observed | |||||||||||
| 3 | * | H | H | CPMAS | — | 300 | −180.8 | −100.6 | 50 | 50 | No |
| 4 | CH3 | H | H | HMPA | 0.10 | 276 | −180.5 | N.o.b | ~50 | ~50 | No |
| −173.6 | |||||||||||
| 4a | CH3 | H | H | CPMAS | — | 300 | −177.7 | −100.9 | 100 | 0 | No |
| 5 | CH3 | CH3 | H | HMPA | 0.08 | 300 | −185.6 (major) | N.o. | 30 | 70 | No |
| −175.9 | |||||||||||
| CPMAS | — | 300 | −187.8 | −111.2 | 66 | 34 | No | ||||
| −172.0 (major) | −103.6 (major) | ||||||||||
| 6b | C6H5 | H | H | CPMAS | — | 300 | –181.5 | −105.3 | 0 | 100 | No |
| 8b | C6H5 | CH3 | CH3 | HMPA | 0.06 | 268 | –182.2 | N.o. | 0 | 100 | No |
| CPMAS | — | 300 | –181.3 | −98.7 | 0 | 100 | No | ||||

Percentages of tautomers and equilibrium constants
Although some exceptions are known, the assumption of the identical nature of the most stable tautomer in solution and the tautomer present in the crystal is one of the most basic tenets in tautomerism.3b,3c,17b The results in Table 6 confirm this principle for compounds 4, 5 and 8, and allow us to conclude that in the solid state, 6 should crystallize as 6b and 7 as 7b, or at least in cyclamers where 6b and 7b are predominant.Compound 5 exists in the solid state as a 66% 5a/34% 5b mixture and in HMPA as a 35% 5a/65% 5b mixture, thus being an exception to the rule of similarity between solution and solid state. However, the difference in energy at 300 K between the two situations is only of 3.2 kJ mol−1.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–)a
CH–)a
| Compound | Tautomers | X-Ray | CPMAS | DMSO | HMPA |
|---|---|---|---|---|---|
| a N. M. means not measured. b Proton transfer | |||||
| 3 | a, b: 3,5-BisSty | 3a = 3b | 3a = 3b | 3a = 3b No PTb | N. M. |
| 4 | a: 3-Sty-5-Me | 4a | 4a | Average rich in 4a | ~50% 4a |
| b: 3-Me-5-Sty | ~50% 4b | ||||
| 5 | a: 3-Sty-5-Me | 66% 5a | 66% 5a | Average rich in 5b | 35% 5a |
| b: 3-Me-5-Sty | 34% 5b | 34% 5b | 65% 5b | ||
| 6 | a: 3-Sty-5-Ph | N. M. | 6b | 36% 6a | N. M. |
| b: 3-Ph-5-Sty | 64% 6b | ||||
| 7 | a: 3-Sty-5-Ph | N. M. | 7b | 40% 7a | N. M. |
| b: 3-Ph-5-Sty | 60% 7b | ||||
| 8 | a: 3-Sty-5-Ph | 8b | 8b | Average rich in 8b | 8b |
| b: 3-Ph-5-Sty |
Conclusions
The structure, tautomerism and absence of SSPT have been determined for six NH-pyrazoles by a combination of X-ray crystallography and 13C/15N NMR spectroscopy. Two of the conditions required to observe SSPT in NH-pyrazoles are the identity (or, at least, strong similarity) of the substituents at the 3- and 5-positions, and the formation of cyclic structures, cyclamers, linked by N–H⋯N hydrogen bonds. Compound 3 has the same substituent at both positions (tautomer 3a is identical to tautomer 3b), but crystallizes in a complex network of hydrogen bonds involving the OH groups. Compound 8 crystallizes as a dimer, but with only one tautomer present (8a). Thus, none of the compounds of Table 6 display SSPT. Finally, compound 5 is the only known example of an NH-pyrazole that crystallizes as a 2 : 1 mixture of two tautomers (there are examples of 2 : 2 and 3 : 1 mixtures, but in cyclic tetramers17b,22).Experimental
The melting points of pyrazoles 3–8 were determined by differential scanning calorimetry (DSC) on a Seiko DSC 220C connected to a Model SSC5200H Disk Station; for the other compounds, a hot stage microscope was used. Thermograms (sample size 0.003–0.0010 g) were recorded at a scanning rate of 2.0 °C min−1. Thin-layer chromatography (TLC) was performed using Merck silica gel (60 F254) and compounds were detected with a 254 nm UV lamp. Silica gel (60–320 mesh) was employed for routine column chromatography separations. Elemental analyses for carbon, hydrogen and nitrogen were carried out by the Microanalytical Service of the Universidad Complutense of Madrid on a Perkin-Elmer 240 analyzer.General procedure for the preparation of pyrazole derivatives (3–8)
Compounds 3–8 were prepared by reacting the corresponding β-diketones23 (1 mmol) with hydrazine hydrate 98% (1.5 mmol) in acetic acid (5 mL). After heating at reflux for 2 h, the reaction mixture was poured into water, and the precipitate filtered off, washed with water and dried. The solid was purified by column chromatography using ethyl acetate as the eluent.(E)-3,5-Bis[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-1H-pyrazole (3)
3 was prepared from purified commercially available 1. The compound was obtained as a colourless solid after recrystallization from H2O/EtOH (1 g, 2.74 mmol, 63%). Mp: 217.1 °C, lit.: 211–214 °C5 or 215 °C.8 Anal. calc. for C21H20N2O4 (364.14): C, 69.22; H, 5.53; N, 7.69; found: C, 68.79; H, 5.53; N, 7.70%.(E)-3(5)-[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-5(3)-methyl-1H-pyrazole (4)
4 was prepared from (E)-6-(4-hydroxy-3-methoxyphenyl)hex-5-ene-2,4-dione.23 The compound was obtained as a colourless solid after recrystallization from CH2Cl2/hexane/EtOH (251 mg, 1.1 mmol, 85%). Mp: 141.6 °C. Anal. calc. for C13H14N2O2 (230.11): C, 67.26; H, 6.44; N, 12.11; found: C, 67.81; H, 6.13; N, 12.17%.(E)-3(5)-[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-4,5(3)-dimethyl-1H-pyrazole (5)
5 was prepared from (E)-6-(4-hydroxy-3-methoxyphenyl)-3-methylhex-5-ene-2,4-dione.23 The compound was obtained as a colourless solid after recrystallization from CH2Cl2/hexane/EtOH (180 mg, 0.73 mmol, 61%). Mp: 176.1 °C. Anal. calc. for C14H16N2O2 (244.12): C, 68.46; H, 6.61; N, 11.35; found: C, 68.83; H, 6.60; N, 11.47%.(E)-3(5)-[β-(4-hydroxy-3-methoxyphenyl)-ethenyl]-5(3)-phenyl-1H-pyrazole (6)
6 was prepared from (E)-5-(4-hydroxy-3-methoxyphenyl)-1-phenylpent-4-ene-1,3-dione.23 The compound was obtained as a colourless solid after recrystallization from CH2Cl2/hexane/EtOH (228 mg, 0.78 mmol, 77%). Mp: 142.9 °C. Anal. calc. for C18H16N2O2 (292.12): C, 73.95; H, 5.54; N, 10.11; found: C, 73.95; H, 5.54; N, 10.11%.(E)-3(5)-[β-(3,4-dimethoxyphenyl)-ethenyl]-5(3)-phenyl-1H-pyrazole (7)
7 was prepared from (E)-5-(3,4-dimethoxyphenyl)-1-phenylpent-4-ene-1,3-dione.23 The compound was obtained as a colourless solid after recrystallization from CH2Cl2/hexane/EtOH (196 mg, 1.27 mmol, 51%). Mp: 173.4 °C. Anal. calc. for C19H18N2O2 (306.37): C, 74.48; H, 5.92; N, 9.14; found: C, 74.21; H, 5.82; N, 9.16%.(E)-3(5)-[β-(3,4-dimethoxyphenyl)-ethenyl]-4-methyl-5(3)-phenyl-1H-pyrazole (8)
8 was prepared from (E)-5-(3,4-dimethoxyphenyl)-2-methyl-1-phenylpent-4-ene-1,3-dione.23 The compound was obtained as a colourless solid after recrystallization from CH2Cl2/hexane/EtOH (170 mg, 0.53 mmol, 58%). Mp: 182.0 °C. Anal. calc. for C20H20N2O2 (320.39): C, 74.97; H, 6.29; N, 8.74; found: C, 74.28; H, 6.14; N, 8.77%.X-Ray data collection and structure refinement (compounds 3, 4, 5 and 8)
Data collection for all of the compounds was carried out at room temperature on a Bruker Smart CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) operating at 50 kV and 30 mA. In all cases, the data were collected over a hemisphere of the reciprocal space by the combination of three exposure sets. Each frame exposure time was either 10 or 20 s, covering 0.3° in ω. The cell parameters were determined and refined by a least-squares fit of all reflections collected. The first 100 frames were re-collected at the end of the data collection to monitor crystal decay, and no appreciable decay was observed. A summary of the fundamental crystal and refinement data is given in Table 7. The structures of all the compounds were solved by direct methods and conventional Fourier synthesis, and refined by full matrix least-squares on F2 (SHELXL-97).24 All non-hydrogen atoms were refined anisotropically.| Crystal data | 3 | 4 | 5 | 8 | |
|---|---|---|---|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = ∑[w(Fo2 − Fc2)2]/∑[w(Fo2). | |||||
| Empirical formula | C21H20N2O4 | C13H14N2O2 | C14H16N2O2 | C20H20N2O2 | |
| Formula weight | 364.39 | 230.26 | 244.29 | 320.38 | |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic | |
| Space group | P2(1)/c | Pbca | P2(1)/c | Pbca | |
| Unit cell dimensions | a/Å | 8.2394(10) | 13.2563(15) | 8.519(2) | 13.2363(13) |
| b/Å | 14.0198(17) | 7.6962(9) | 12.964(4) | 8.2769(8) | |
| c/Å | 16.306(2) | 22.855(3) | 34.615(10) | 30.673(3) | |
| β (°) | 101.060(3) | — | 94.607(7) | — | |
| Volume/Å3 | 1848.7(4) | 2331.7(5) | 3810.6(19) | 3360.4(6) | |
| Z | 4 | 8 | 12 | 8 | |
| Density (calculated)/Mg m−3 | 1.309 | 1.312 | 1.277 | 1.267 | |
| Absorption coefficient/mm−1 | 0.092 | 0.090 | 0.087 | 0.083 | |
| Scan technique | ω and ϕ | ω and ϕ | ω and ϕ | ω and ϕ | |
| F(000) | 768 | 976 | 1560 | 1360 | |
| Range for data collection (°) | 1.93 to 25.00 | 1.78 to 27.00 | 1.18 to 25.00 | 1.33 to 25.00 | |
| Index ranges | −9, −16, −18 to 9, 16, 19 | −13, −9, −29 to 16, 9, 29 | −9, −15, −41 to 10, 15, 41 | −15, −9, −36 to 10, 9, 32 | |
| Reflections collected | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 998 998 |
19397 |
28784 |
16484 |
|
| Independent reflections | 3244 | 2541 | 6720 | 2954 | |
| Observed reflections [I > 2σ(I)] | 1418 | 1248 | 2855 | 1655 | |
| R int | 0.1198 | 0.0889 | 0.0905 | 0.0708 | |
| Completeness to θ (%) | 99.6 | 100.0 | 100.0 | 99.9 | |
| Data/restraints/parameters | 3244/0/245 | 2541/0/156 | 6720/2/497 | 2954/0/224 | |
| Goodness-of-fit on F2 | 0.912 | 1.034 | 0.984 | 1.074 | |
| R1a | 0.0539 | 0.0508 | 0.0769 | 0.0507 | |
| wR2b (all data) | 0.1808 | 0.1768 | 0.2486 | 0.1848 | |
| Largest differential peak and hole/eÅ−3 | 0.232 and −0.278 | 0.214 and −0.247 | 0.950 and −0.377 | 0.193 and −0.192 | |
In all cases, the hydrogen atoms were calculated, included and refined as riding on their respective carbon-bonded atom with a common anisotropic displacement. The rest of the hydrogen atoms, i.e. those bonded to nitrogen or oxygen atoms, were located in a Fourier difference synthesis, and in all cases were included and refined as riding on their respective bonded atoms for 3, 4 and 5, while for 8, its coordinates were refined and the thermal factors kept constant. The longer O–H bond distances in some of the hydroxyl groups are due to the formation of hydrogen bonds.25
The largest peaks and holes in the final difference map were 0.232 and −0.278, 0.214 and −0.247, 0.950 and −0.377, and 0.193 and −0.192 eÅ−3 for 3, 4, 5 and 8, respectively. The final R1 and wR2 values were 0.0539 and 0.1808, 0.0508 and 0.1768, 0.0769 and 0.2486, and 0.0507 and 0.1848 for 3, 4, 5 and 8, respectively.†
NMR spectroscopy
Acknowledgements
This work has been financed by the Spanish MEC (CTQ2006-02586 and CTQ2007-62113).References
- J. Higdon, Curcumin, Linus Pauling Institute, Oregon State University, Oregon, USA (http://lpi.oregonstate.edu/infocenter/phytochemicals/curcumin/) Search PubMed.
- (a) R. M. Claramunt, D. Sanz del Castillo, J. Elguero, P. Nioche, C. S. Raman, P. Martasek and B. S. S. Masters, XVIIth International Symposium on Medicinal Chemistry, Poster P101, Drugs Future, 2002, 27(Suppl. A), 177; (b) C. Pérez-Medina, M. Pérez-Torralba, C. López, R. M. Claramunt, P. Nioche and C. S. Raman, XIV Congreso Nacional de la Sociedad Española de Química Terapeútica, Bilbao, Spain, 2005 Search PubMed.
- (a) J. Elguero, C. Marzin, A. R. Katritzky and P. Linda, The Tautomerism of Heterocycles, Academic Press, New York, 1976, pp. 655 Search PubMed; (b) V. I. Minkin, A. D. Garnosvskii, J. Elguero, A. R. Katritzky and O. V. Denisko, Adv. Heterocycl. Chem., 2000, 76, 157–323 CrossRef CAS; (c) R. M. Claramunt, C. López, M. D. Santa María, D. Sanz and J. Elguero, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 169–206 CrossRef CAS.
- (a) J. Quiroga Puello, B. Insuasty Obando, C. Foces-Foces, L. Infantes, R. M. Claramunt, P. Cabildo, J. A. Jimenez and J. Elguero, Tetrahedron, 1997, 53, 10783–10802 CrossRef; (b) M. H. Holschbach, D. Sanz, R. M. Claramunt, L. Infantes, S. Motherwell, P. R. Raithby, M. L. Jimeno, D. Herrero, I. Alkorta, N. Jagerovic and J. Elguero, J. Org. Chem., 2003, 68, 8831–8837 CrossRef CAS; (c) R. M. Claramunt, M. Á. García, C. López, S. Trofimenko, G. P. A. Yap, I. Alkorta and J. Elguero, Magn. Reson. Chem., 2005, 43, 89–91 CrossRef CAS; (d) S. Trofimenko, G. P. A. Yap, F. A. Jove, R. M. Claramunt, M. Á. García, M. D. Santa María, I. Alkorta and J. Elguero, Tetrahedron, 2007, 63, 8104–8111 CrossRef CAS.
- D. L. Flynn, T. R. Belliotti, A. M. Boctor, D. Connor, C. Kostlan, D. E. Nies, D. F. Ortwine, D. J. Schrier and J. C. Sircar, J. Med. Chem., 1991, 34, 518–525 CrossRef CAS.
- J. S. Shim, D. H. Kim, H. J. Jung, J. H. Kim, D. Lim, S.-K. Lee, K.-W. Kim, J. W. Ahn, J.-S. Yoo, J.-R. Rho, J. Shin and H. J. Kwon, Bioorg. Med. Chem., 2002, 10, 2439–2444 CrossRef.
- J. Ishida, H. Ohtsu, Y. Tachibana, Y. Nakanishi, K. F. Bastow, M. Nagai, H.-K. Wang, H. Itokawa and K.-H. Lee, Bioorg. Med. Chem., 2002, 10, 3481–3487 CrossRef CAS.
- H. Ohtsu, Z. Xiao, J. Ishida, M. Nagai, H.-K. Wang, H. Itogawa, C.-Y. Su, C. Shih, T. Chiang, E. Chang, Y. Lee, M.-Y. Tsai, C. Chang and K.-H. Lee, J. Med. Chem., 2002, 45, 5037–5042 CrossRef CAS.
- C. Selvam, S. M. Jachak, R. Thilagavathi and A. K. Chakraborti, Bioorg. Med. Chem. Lett., 2005, 15, 1793–1797 CrossRef CAS.
- R. Narlawar, K. Baumann, R. Schubenel and B. Schmidt, Neurodegener. Dis., 2007, 4, 88–93 Search PubMed.
- R. Narlawar, M. Pickhardt, S. Leuchtenberger, K. Baumann, S. Krause, T. Dyrks, S. Weggen, E. Mandelkow and B. Schmidt, ChemMedChem, 2008, 3, 165–172 CrossRef CAS.
- P. Maher, T. Akaishi, D. Schubert and K. Abe, Neurobiol. Aging, 2008 DOI:10.1016/j.neurobiolaging.2008.05.020.
- (a) J. Elguero, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, 1984, vol. 5, pp. 167 Search PubMed; (b) J. Elguero, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. Scriven, Pergamon Press, Oxford, 1996, vol. 3, pp. 1 Search PubMed; (c) B. Stanovnik and J. Svete, in Science of Synthesis, ed. R. Neier, Thieme, Stuttgart, 2002, vol. 12, ch. 12.1 Search PubMed.
- B. B. Aggarwal, A. Kumar, M. S. Aggarwal and S. Shishodia, Curcumin Derived from Tumeric (Curcuma longa): a Spice for All Seasons, in Phytopharmaceuticals in Cancer Chemoprevention, ed. D. Bagchi and H. G. Preuss, CRC Press, Boca Raton, 2005, pp. 349–387 Search PubMed.
- I. Alkorta, J. Elguero, B. Donnadieu, M. Etienne, J. Jaffart, D. Schagen and H.-H. Limbach, New J. Chem., 1999, 23, 1231–1237 RSC.
- (a) C. Foces-Foces, I. Alkorta and J. Elguero, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 1018–1028 CrossRef; (b) I. Alkorta, J. Elguero, C. Foces-Foces and L. Infantes, ARKIVOC (Gainesville, FL, U. S.), 2006, ii, 15–30 Search PubMed.
- (a) R. M. Claramunt, P. Cornago, V. Torres, E. Pinilla, M. R. Torres, A. Samat, V. Lokshin, M. Valés and J. Elguero, J. Org. Chem., 2006, 71, 6881–6891 CrossRef CAS; (b) R. M. Claramunt, P. Cornago, M. D. Santa María, V. Torres, E. Pinilla, M. R. Torres and J. Elguero, Supramol. Chem., 2006, 18, 349–356 CrossRef CAS.
- C. Foces-Foces, C. Cativiela, J. L. Serrano, M. M. Zurbano, N. Jagerovic and J. Elguero, J. Chem. Crystallogr., 1996, 26, 127–131 CAS.
- (a) A. M. S. Silva, L. M. P. M. Almeida, J. A. S. Cavaleiro, C. Foces-Foces, A. L. Llamas-Saiz, C. Fontenas, N. Jagerovic and J. Elguero, Tetrahedron, 1997, 53, 11645–11658 CrossRef CAS; (b) D. C. G. A. Pinto, A. M. S. Silva, J. A. S. Cavaleiro, C. Foces-Foces, A. L. Llamas-Saiz, N. Jagerovic and J. Elguero, Tetrahedron, 1999, 55, 10187–10200 CrossRef CAS.
- C. Foces-Foces, A. Echevarría, N. Jagerovic, I. Alkorta, J. Elguero, U. Langer, O. Klein, M. Minguet-Bonvehí and H.-H. Limbach, J. Am. Chem. Soc., 2001, 123, 7898–7906 CrossRef CAS.
- M. Begtrup, G. Boyer, P. Cabildo, C. Cativiela, R. M. Claramunt, J. Elguero, J. I. García, C. Toiron and P. Vedsø, Magn. Reson. Chem., 1993, 31, 107–168 CAS.
- J. Elguero, N. Jagerovic, C. Foces-Foces, F. H. Cano, M. V. Roux, F. Aguilar-Parrilla and H.-H. Limbach, J. Heterocycl. Chem., 1995, 32, 451–456 CrossRef CAS.
- P. Cornago, R. M. Claramunt, L. Bouissane, I. Alkorta and J. Elguero, Tetrahedron, 2008, 64, 8089–8094 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
Footnote |
| † CCDC reference numbers 690489–690492. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b812018h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |