DOI:
10.1039/B814400A
(Paper)
New J. Chem., 2009,
33, 119-124
Synthesis, crystal structures and luminescence properties of lanthanide oxalatophosphonates with a three-dimensional framework structure†
Received
(in Montpellier, France)
20th August 2008
, Accepted 3rd October 2008
First published on 14th November 2008
Abstract
Six new three-dimensional (3D) lanthanide oxalatophosphonates, [Ln(HL)(C2O4)0.5(H2O)2]·H2O (Ln = La (1), Ce (2), Pr (3), Nd (4), Sm (5), Eu (6); H3L = H2O3PCH(OH)CO2H), have been synthesized under hydrothermal conditions and structurally characterized by single-crystal X-ray diffraction as well as by infrared spectroscopy, elemental analysis and thermogravimetric analysis. Compounds 1–6 are isomorphous and they exhibit a complex three-dimensional (3D) open-framework structure with a one-dimensional channel system along the c-axis. The interconnection of the lanthanide(III) ions by phosphonate ligands results in a lanthanide phosphonate layer, and these layers are further bridged by oxalate anions to form a 3D open-framework. Compound 6 shows strong red luminescence in the solid state at room temperature.
Introduction
Metal phosphonates as a class of inorganic–organic hybrid materials have attracted a great deal of research interest as a result of their ability to form interesting structures with potential applications as catalysts, ion exchangers, sorbents, meso-/microporous materials, or intercalation chemistry.1–5 Usually, the metal phosphonates adopt layered or pillared layered structures, with the organic groups filling in between the inorganic layers.6–10 Other structural types have also been observed in some phosphonates, among which the open-framework and porous structures are of particular interest.11–16 The strategy of attaching functional groups such as amine, hydroxyl and carboxylate groups to the phosphonic acid has proven to be effective for the isolation of a variety of metal phosphonates with open-framework and microporosity.17–21 Based on 2-hydroxyphosphonoacetic acid, proline-N-methylphosphonic acid and DL-(α-aminoethyl)phosphonic acid, a series of metal phosphonates with two-dimensional (2D) layer and three-dimensional (3D) open-framework structures have also been isolated in our laboratory.22
Recently, many research activities have focused on the synthesis of inorganic–organic hybrid compounds by incorporating organic ligands in the structures of metal phosphonates.23–26 The direct use of two types of ligands in the preparation, such as a phosphonic acid and a carboxylic acid, has been found to be another effective method for the exploration of hybrid open-frameworks. Among these studies, the oxalate moiety, C2O42−, was found to be a good candidate and has been successfully incorporated into phosphonate frameworks with transition metals and main group elements.27–30 Although some progress has been made in the construction of metal oxalatophosphonates as mentioned above, less progress has been achieved in the synthesis of lanthanide oxalatophosphonates.31–33 Lanthanide phosphonates normally have low solubility in water and other organic solvents, hence introducing a second ligand such as C2O42− into the lanthanide phosphonate system can improve the solubility and crystallinity of the lanthanide phosphonate. In addition, the coordination of two types of ligands with the lanthanide ion may reduce or eliminate water molecules from the coordination sphere of the lanthanide(III) ion, hence increasing the luminescent intensity and lifetime of the materials.34 In this paper, we selected 2-hydroxyphosphonoacetic acid (H3L) as the phosphonate ligands and oxalate as the second metal linker. Hydrothermal reactions of the above two ligands with lanthanide(III) chlorides afforded six new lanthanide oxalatophosphonate hybrids with 3D open-framework structures, namely, [Ln(HL)(C2O4)0.5(H2O)2]·H2O (Ln = La (1), Ce (2), Pr (3), Nd (4), Sm (5), Eu (6); H3L = H2O3PCH(OH)CO2H). The luminescent property of compound 6 has also been studied.
Results and discussion
Synthesis
By using 2-hydroxyphosphonoacetic acid as the phosphonate ligand and oxalate as the second metal linker, six new lanthanide(III) oxalatophosphonates have been synthesized under hydrothermal conditions. The compounds 1–6 were obtained as a pure phase materials by adjusting the synthetic conditions. The molar ratio of the starting materials and the pH of the reaction mixture play an important role in the formation of these six compounds. NaOH was employed as the inorganic base to adjust the pH of the reaction mixture. It was found that pure phases of compounds 1–6 can be obtained with good yields when the molar ratio of LnCl3·6H2O, H3L, H2C2O4·2H2O, NaOH, H2O and the pH are 1 : 4 : 4 : 4 : 888 and 1.5–2.5. In addition, the reaction temperature was very important for the formation of suitable single crystals for X-ray diffraction. The compounds 1–6 were obtained at 120–140 °C under hydrothermal conditions. The powder XRD patterns of compounds 1–6 and the simulated XRD patterns of compound 1 are shown in the supplementary material (Fig. S1, ESI†). The diffraction peaks on the patterns correspond well in position, confirming these six compounds are isomorphous, and showing their phase purity. The differences in reflection intensities are probably due to preferred orientation in the powder samples.
Description of the crystal structures
Compounds 1–6 are isomorphous and feature three-dimensional open frameworks, hence only the structure of 3 will be discussed in detail as a representation. The ORTEP diagram for compound 3 is shown in Fig. 1. Crystallographic data and structural refinements for compounds 1–6 are summarized in Table 1.
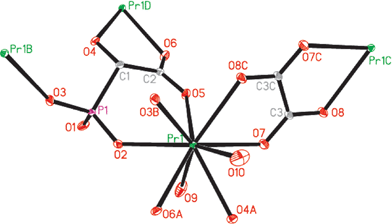 |
| | Fig. 1 ORTEP representation of a selected unit of compound 3. The thermal ellipsoids are drawn at the 30% probability level. All H atoms and lattice water molecules are omitted for clarity: Pr(1)–O(3)B, 2.372(3) Å; Pr(1)–O(2), 2.481(3) Å; Pr(1)–O(7), 2.493(3) Å; Pr(1)–O(6)A, 2.522(3) Å; Pr(1)–O(5), 2.559(3) Å; Pr(1)–O(9), 2.537(4) Å; Pr(1)–O(8)C, 2.571(3) Å; Pr(1)–O(10), 2.542(4) Å; Pr(1)–O(4)A, 2.573(3) Å. Symmetry codes: A: −x + 2, y + 1/2, −z + 3/2; B: −x + 2, −y, −z + 2; C: −x + 1, −y, −z + 1; D: −x + 2, y− 1/2, −z + 3/2. | |
Table 1
Crystal data and structure refinements for compounds 1–6
| |
1
|
2
|
3
|
4
|
5
|
6
|
|
R
1 = ∑(||Fo| − |Fc|)/∑|Fo|; wR2 = [∑w(|Fo| − |Fc|)2/∑wFo2]1/2.
|
| Empirical formula |
C3H9O11PLa |
C3H9O11PCe |
C3H9O11PPr |
C3H9O11PNd |
C3H9O11PSm |
C3H9O11PEu |
|
M
|
390.98 |
392.19 |
392.98 |
396.31 |
402.42 |
404.03 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
P21/c |
P21/c |
P21/c |
|
a/Å |
7.1991(6) |
7.2260(7) |
7.2300(7) |
7.2326(6) |
7.2351(6) |
7.2293(9) |
|
b/Å |
13.3838(11) |
13.2918(13) |
13.2239(13) |
13.1558(11) |
13.0550(10) |
13.0429(16) |
|
c/Å |
10.2926(8) |
10.2745(10) |
10.2535(10) |
10.2337(8) |
10.1999(8) |
10.1980(13) |
|
β/° |
98.8980(10) |
99.4930(10) |
99.8690(10) |
100.1070(10) |
100.4270(10) |
100.541(2) |
|
V/Å3 |
979.77(14) |
973.32(16) |
965.82(16) |
958.63(14) |
947.51(13) |
945.4(2) |
|
Z
|
4 |
4 |
4 |
4 |
4 |
4 |
|
D
c/g cm−3 |
2.651 |
2.676 |
2.703 |
2.746 |
2.821 |
2.839 |
|
μ/mm−1 |
4.576 |
4.894 |
5.263 |
5.636 |
6.420 |
6.858 |
| GOF on F2 |
1.017 |
1.063 |
1.074 |
1.039 |
1.090 |
1.099 |
|
R
1 [I > 2σ(I)]a |
0.0210 |
0.0239 |
0.0278 |
0.0220 |
0.0206 |
0.0243 |
| wR2 [I > 2σ(I)]a |
0.0547 |
0.0526 |
0.0726 |
0.0543 |
0.0512 |
0.0589 |
|
R
1 (All data)a |
0.0233 |
0.0296 |
0.0318 |
0.0242 |
0.0226 |
0.0255 |
| wR2 (All data)a |
0.0560 |
0.0555 |
0.0753 |
0.0557 |
0.0523 |
0.0595 |
As shown in Fig. 1, the Pr(III) ion is nine-coordinated by two phosphonate oxygen atoms, two carboxylate oxygen atoms, and one hydroxyl oxygen atoms from three HL2− anions, two oxygen atoms from one oxalate anion as well as two aqua ligands. The Pr–O distances range from 2.372(3) to 2.573(3) Å, which are comparable to those reported for other praseodymium(III) phosphonates.23,33 The asymmetric unit contains half of an oxalate ion which lies about an inversion centre. The oxalate anion is bidentate, and it chelates with two different Pr(III) ions by using its four carboxylate oxygen atoms. Each oxalate anion forms two Pr–O–C–C–O five-membered chelating rings. The pentadentate HL2− ligand is bidentate with Pr1 and Pr1D and monodentate with Pr1B. Each HL2− anion chelates with Pr1D ion by using its one carboxylate oxygen atom (O6) and one hydroxyl oxygen atom (O4), and one carboxylate oxygen atom (O5) and one phosphonate oxygen atom (O2) chelate with Pr1 ion. One phosphonate oxygen atom (O3) is unidentate, whereas the remaining one (O1) is protonated and noncoordinated. Such configuration is favorable because of the formation of stable six-atom rings (P–O–Pr–O–C–C) and five-atom rings (O–Pr–O–C–C). It is noted that the Ln–O (hydroxyl oxygen) distances are longer than the other Ln–O distances in compounds 1–6, attributed to the presence of the hydroxyl proton (Table S1, ESI†).
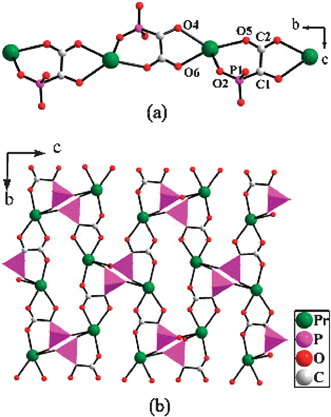
The HL2− anion acts as a bridging ligand to link Pr(III) ions into a 1D chain of {Pr(HL)}+ along the b axis (Fig. 2(a)). The dihedral angle between two chelating rings sharing a common Pr(III) ion is 74.61(10)°. These chains are cross-linked by bridging HL2− anions to form a praseodymium phosphonate layer in the bc plane (Fig. 2(b)), the layers are interconnected by sharing Pr(III) ions into a pillared-layered architecture with the oxalate groups acting as pillars (Fig. 3). The result of connections in this manner is the formation of a 1D channel system along the c axis (Fig. 4(a)). The channel is formed by 21-atom rings composed of five Pr(III) ions, three HL2− anions and two oxalate anions (Fig. 4(b)). The dimensions of the channels are estimated to be 11.2 Å (Pr1a–P1e) × 10.2 Å (Pr1–C3c) based on the structural data. The oxygen atoms from coordinated water molecules are oriented toward the channel center, and lattice water molecules are located inside the channels. The structure of 3 can be viewed as the praseodymium phosphonate layer being connected viaoxalate anions to form a complex 3D open-framework structure.
 |
| | Fig. 2 (a) 1D chain {Pr(HL)}+ and (b) A 2D praseodymium(III) phosphonate layer in compound 3. | |
 |
| | Fig. 3 A ball-and-stick and polyhedral view of compound 3 along the b axis. | |
 |
| | Fig. 4 (a) View of the framework for compound 3 along the c-axis showing the voids in the structure. (b) A 21-atom rings in compound 3. All H atoms and lattice water molecules are omitted for clarity. Symmetry codes: a: −x + 2, y− 1/2, −z + 3/2; b: x, −y− 1/2, z− 1/2; c: −x + 1, y− 1/2, −z + 1/2; d: −x + 1, −y, −z + 1; e: x− 1, y, z− 1. | |
Thermal properties
Except for the final weight loss temperature and total weight losses, the TGA curves of compounds 1–6 are very similar, with three main continuous weight losses. Herein, we use compound 1 as an example to illuminate the weight losses in detail. As shown in Fig. 6, the first step corresponds to the loss of one lattice water molecule and two aqua ligands. The weight loss started at 50 °C and was completed at 145 °C. The observed weight loss of 14.0% is close to the calculated value (13.8%). The second step between 345 and 430 °C can be attributed to decomposition of oxalate and phosphonate units. The third step corresponds to the further decomposition of the phosphonate group. The observed total weight loss at 735 °C is about 41.5%, and the final products are not identified. However, we suspect they are mainly LaPO4. The total weight loss of 41.5% is close to the calculated value (40.2%) if the final product is assumed to be LaPO4. The observed total weight losses of compounds 2–6 are 41.4, 39.0, 37.0, 38.6, 40.3%, respectively. Considering the thermal stability of the compounds, X-ray powder diffraction studies were performed for the as-synthesized compound 1 and the samples calcined at 150 and 180 °C. The XRD patterns for the calcined samples fit well with that of the as-synthesized samples, indicating that the structure of these six compounds can be kept after dehydration process (Fig. S2, ESI†).
 |
| | Fig. 6
TGA curves of compounds 1–6. | |
Photoluminescent properties
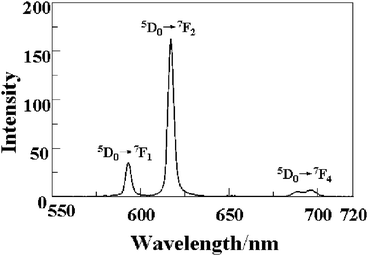
It is well-known that the lanthanides, especially europium and terbium, can absorb ultraviolet radiation efficiently through an allowed electronic transition to convert to the excited state 5D4, and these excited states are deactivated to the multiplet 7FJ states radiatively via emission of visible radiation as luminescence. The solid-state luminescence property of compound 6 was investigated at room temperature. Compound 6 emits red light upon excitation at 396 nm, and its luminescence spectrum is depicted in Fig. 7. These emission bands arise from 5D0→7FJ (J = 1, 2 and 4) transitions, typical of Eu(III) ions.34,37 The 5D0→7F1 transition (593 nm) corresponds to a magnetic dipole transition, and the intensity of this emission for 6 is medium-strong. The most intense emission in the luminescent spectrum is the 5D0→7F2 transitions at 617 nm, which are the so-called hypersensitive transitions and are responsible for the brilliant-red emission of compound 6.38 The emission spectrum of 6 shows a weak emission band at 695 nm, which can be attributed to the 5D0→7F4 transition. The results indicate that compound 6 is a good candidate as a red-light luminescent material.
Conclusions
By using 2-hydroxyphosphonoacetic acid as the phosphonate ligand and oxalate as the second metal linker, six new lanthanide(III) oxalatophosphonates with a general formula [Ln(HL)(C2O4)0.5(H2O)2]·H2O (Ln = La (1), Ce (2), Pr (3), Nd (4), Sm (5), Eu (6); H3L = H2O3PCH(OH)CO2H) have been synthesized and structurally characterized. Compounds 1–6 are isomorphous and the structure of these compounds features a 3D open-framework with a one-dimensional channel system along the c-axis. The interconnection of the lanthanide(III) ions by phosphonate ligands results in a lanthanide phosphonate layer, and these layers are further bridged by oxalate anions to form 3D open-frameworks. Compound 6 is a new example of luminescent rare-earth oxalatophosphonates characterized by a significant red luminescence. The results of our study indicate that by introduction of oxalate as the second ligand, we can obtain lanthanide oxalatophosphonates with well characterized crystal structures as well as strong luminescence.
Experimental
Materials
2-Hydroxylphosphonoacetric acid (H3L) solution was obtained from Taihe Chemical Factory (48.0 wt%). The lanthanide(III) chlorides were prepared by the dissolving corresponding lanthanide oxides (General Research Institute for Nonferrous Metals, 99.99%) in hydrochloric acid followed by recrystallization and drying. All other chemicals were used as received without further purification.
Physical measurements
Elemental analyses (carbon and hydrogen) were performed using a PE-2400 elemental analyzer. La, Ce, Pr, Nd, Sm, Eu and P were determined by using an inductively coupled plasma (ICP) atomic absorption spectrometer. IR spectra were recorded on a Bruker AXSTENSOR-27 FT-IR spectrometer with KBr pellets in the range 4000–400 cm−1. The X-ray powder diffraction data were collected on a Bruker AXS D8 Advance diffractometer using Cu-Kα radiation (λ = 1.5418 Å) in the 2θ range of 9–60° with a step size of 0.02°. The luminescence analysis was performed on a JASCO FP-6500 spectrofluorimeter (solid). TG analysis was performed on a Perkin–Elmer Pyris Diamond TG–DTA thermal analysis system in static air with a heating rate of 10 K min−1 from 50 to 800 °C.
Synthesis
[La(HL)(C2O4)0.5(H2O)2]·H2O (1).
A mixture of LaCl3·6H2O (0.18 g, 0.50 mmol), H3L (0.50 ml, 2.00 mmol), H2C2O4·2H2O (0.25 g, 2.00 mmol), and NaOH (0.08 g, 2.00 mmol) was dissolved in 8 mL distilled water. The resulting solution was stirred for about 1 h at room temperature, sealed in a 20 mL Teflon-lined stainless steel autoclave, and heated at 140 °C for 4 days under autogenous pressure. After the mixture was cooled slowly to room temperature, colorless block crystals were obtained in ca. 40.0% yield based on La. C3H9O11PLa (390.98): calc.: C 9.21, H 2.30, P 7.93, La 35.53; found: C 9.28, H 2.22, P 7.85, La 35.45%. IR (KBr) data: 3540 (br), 1644 (m), 1581 (s), 1432 (w), 1373 (w), 1309 (w), 1209 (s), 1178 (w), 1070 (s), 935 (m), 781 (w), 719 (w), 619 (w), 580 (w), 526 (w) cm−1.
[Ce(HL)(C2O4)0.5(H2O)2]·H2O (2).
The procedure was the same as that for 1 except that LaCl3·6H2O was replaced by CeCl3·7H2O (0.19 g, 0.50 mmol). Yield: 81.0% (based on Ce). C3H9O11PCe (392.19): calc.: C 9.18, H 2.29, P 7.90, Ce, 35.73; found: C 9.11, H 2.21, P 7.95, Ce 35.65%. IR (KBr) data: 3465 (br), 2221 (w), 1648 (m), 1583 (s), 1425 (w), 1373 (w), 1311 (w), 1213 (s), 1074 (s), 937 (w), 781 (w), 721 (w), 615 (w), 588 (w), 524 (w) cm−1.
[Pr(HL)(C2O4)0.5(H2O)2]·H2O (3).
The procedure was the same as that for 1 except that LaCl3·6H2O was replaced by PrCl3·6H2O (0.18 g, 0.50 mmol). Yield: 76.0% (based on Pr). C3H9O11PPr (392.98): calc.: C 9.16, H 2.29, P 7.89, Pr 35.86; found: C 9.23, H 2.20, P 7.96, Pr 35.94%. IR (KBr) data: 3473 (br), 2915 (w), 1650 (m), 1575 (s), 1432 (m), 1371 (m), 1363 (m), 1317 (m), 1214 (s), 1064 (s), 927 (m), 831 (w), 777 (w), 696 (w), 617 (w), 572 (w), 516 (w) cm−1.
[Nd(HL)(C2O4)0.5(H2O)2]·H2O (4).
The procedure was the same as that for 1 except that LaCl3·6H2O was replaced by NdCl3·6H2O (0.18 g, 0.50 mmol). Yield: 55.0% (based on Nd). C3H9O11PNd (396.31): calc.: C 9.08, H 2.27, P 7.82, Nd 36.40; found: C 9.15, H 2.35, P 7.91, Nd 36.49%. IR (KBr) data: 3484 (br), 2915 (w), 1652 (s), 1577 (s), 1432 (m), 1369 (m), 1319 (m), 1209 (s), 1064 (s), 968 (w), 931 (m), 835 (w), 786 (w), 781 (w), 694 (w), 619 (m), 580 (m), 520 (m) cm−1.
[Sm(HL)(C2O4)0.5(H2O)2]·H2O (5).
A mixture of SmCl3·6H2O (0.19 g, 0.50 mmol), H3L (0.50 ml, 2.00 mmol), H2C2O4·2H2O (0.25 g, 2.00 mmol), and NaOH (0.08 g, 2.00 mmol) in 8 mL distilled water was sealed in an autoclave equipped with a 20 mL Teflon liner, and then heated at 120 °C for 4 days. After the mixture was cooled slowly to room temperature, pale yellow block crystals were obtained in ca. 86.0% yield based on Sm. C3H9O11PSm (402.42): calc.: C 8.95, H 2.25, P 7.69, Sm 37.36; found: C 9.03, H 2.33, P 7.63, Sm 37.28%. IR (KBr) data: 3477 (br), 3290 (br), 2925 (w), 1660 (s), 1575 (s), 1433 (m), 1366 (m), 1318 (m), 1212 (s), 1072 (s), 930 (m), 783 (w), 704 (w), 617 (m), 524 (m) cm−1.
[Eu(HL)(C2O4)0.5(H2O)2]·H2O (6).
The procedure was the same as that for 5 except that SmCl3·6H2O was replaced by EuCl3·6H2O (0.19 g, 0.50 mmol). Yield: 45.0% (based on Eu). C3H9O11PEu (404.03): calc.: C 8.92, H 2.24, P 7.67, Eu 37.61; found: C 8.98, H 2.31, P 7.58, Eu 37.69%. IR (KBr) data: 3475 (br), 3292 (br), 2920 (w), 1670 (s), 1579 (s), 1435 (m), 1361 (m), 1317 (m), 1217 (s), 1180 (m), 1070 (s), 974 (w), 921 (m), 783 (w), 707 (w), 621 (m), 580 (w), 526 (w), 482 (m) cm−1.
Crystallographic determinations
Data collections for compounds 1–6 were performed on the Bruker Smart APEX II X-diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 ± 2 K. An empirical absorption correction was applied using the SADABS program. All structures were solved by direct methods and refined by full-matrix least squares fitting on F2 by SHELXS-97.39 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of organic ligands were generated geometrically with fixed isotropic thermal parameters, and included in the structure factor calculations.
Acknowledgements
This research was supported by grants from the Natural Science Foundation of Liaoning Province of China (20062140).
References
-
(a) S. Cheng, G.-Z. Peng and A. Clearfield, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 2
 ;
(b) G. Cao, H. G. Hong and T. E. Mallouk, Acc. Chem. Res., 1992, 25, 420 CrossRef CAS ;
(c) G. E. Fanucci, J. Krzystek, M. W. Meisel, L.-C. Brunel and D. R. Talham, J. Am. Chem. Soc., 1998, 120, 5469 CrossRef CAS .
;
(b) G. Cao, H. G. Hong and T. E. Mallouk, Acc. Chem. Res., 1992, 25, 420 CrossRef CAS ;
(c) G. E. Fanucci, J. Krzystek, M. W. Meisel, L.-C. Brunel and D. R. Talham, J. Am. Chem. Soc., 1998, 120, 5469 CrossRef CAS .
-
(a) J. D. Wang, A. Clearfield and G.-Z. Peng, Mater. Chem. Phys., 1993, 35, 208 CrossRef CAS ;
(b) C. Y. Ortiz-Avila, C. Bhardwaj and A. Clearfield, Inorg. Chem., 1994, 33, 2499 CrossRef CAS .
-
(a) K. Maeda, Y. Kiyozumi and F. Mizukami, J. Phys. Chem. B, 1997, 101, 4402 CrossRef CAS ;
(b) F. Odobel, B. Bujoli and D. Massiot, Chem. Mater., 2001, 13, 163 CrossRef CAS .
-
(a) T. E. Mallouk and J. A. Gavin, Acc. Chem. Res., 1998, 31, 209 CrossRef CAS ;
(b) A. Clearfield, Chem. Mater., 1998, 10, 2801 CrossRef CAS ;
(c) T. Kimura, Chem. Mater., 2003, 15, 3742 CrossRef CAS .
-
G. Alberti and U. Costantino, in Comprehensive Supramolecular Chemistry, ed. J. M. Lehn, Pergamon-Elsevier Science Ltd, London, 1996, p. 1 Search PubMed .
- Z.-M. Sun, J.-G. Mao, Y.-Q. Sun, H.-Y. Zeng and A. Clearfield, New J. Chem., 2003, 27, 1326 RSC .
- H.-H. Song, L.-M. Zheng, Z. Wang, C.-H. Yan and X.-Q. Xin, Inorg. Chem., 2001, 40, 5024 CrossRef CAS .
- J.-L. Song, H.-H. Zhao, J.-G. Mao and K. R. Dunbar, Chem. Mater., 2004, 16, 1884 CrossRef CAS .
- D.-K. Cao, J. Xiao, J.-W. Tong, Y.-Z. Li and L.-M. Zheng, Inorg. Chem., 2007, 46, 428 CrossRef CAS .
- S. Bauer, J. Marrot, T. Devic, G. Férey and N. Stock, Inorg. Chem., 2007, 46, 9998 CrossRef CAS .
- O. R. Evans, H. L. Ngo and W. B. Lin, J. Am. Chem. Soc., 2001, 123, 10395 CrossRef CAS .
- J. A. Groves, P. A. Wright and P. Lightfoot, Dalton Trans., 2005, 2007 RSC .
- J. A. Groves, S. R. Miller, S. J. Warrender, C. M. Draznieks, P. Lightfoot and P. A. Wright, Chem. Commun., 2006, 3305 RSC .
- G. B. Hix, A. Turner, L. Vahter and B. M. Kariuki, Microporous Mesoporous Mater., 2007, 99, 62 CrossRef CAS .
- Z.-Y. Du, A. V. Prosvirin and J.-G. Mao, Inorg. Chem., 2007, 46, 9884 CrossRef CAS .
- Y.-Q. Guo, B.-P. Yang, J.-L. Song and J.-G. Mao, Cryst. Growth Des., 2008, 8, 600 CrossRef CAS .
- P. Yin, S. Gao, L.-M. Zheng and X.-Q. Xin, Chem. Mater., 2003, 15, 3233 CrossRef CAS .
- J.-L. Song and J.-G. Mao, J. Solid State Chem., 2005, 178, 3514 CrossRef CAS .
- N. Stock and T. Bein, J. Mater. Chem., 2005, 15, 1384 RSC .
- D. Kong and A. Clearfield, Cryst. Growth Des., 2005, 5, 1263 CrossRef CAS .
- B.-P. Yang, A. V. Prosvirin, Y.-Q. Guo and J.-G. Mao, Inorg. Chem., 2008, 47, 1453 CrossRef CAS .
-
(a) J. Li, L. Meng, Z.-G. Sun, Z.-M. Liu, Y. Zhao, J. Zhang, Y.-Y. Zhu, X. Lu, L. Liu and N. Zhang, Inorg. Chem. Commun., 2008, 11, 211 CrossRef CAS ;
(b) D.-P. Dong, J. Li, Z.-G. Sun, X.-F. Zheng, H. Chen, L. Meng, Y.-Y. Zhu, Y. Zhao and J. Zhang, Inorg. Chem. Commun., 2007, 10, 1109 CrossRef CAS ;
(c) Z.-G. Sun, L.-Y. Cui, Z.-M. Liu, D.-P. Dong, L. Meng, H. Chen, L.-C. Zhang, Z.-M. Zhu and W.-S. You, Inorg. Chem. Commun., 2006, 9, 1121 CrossRef CAS ;
(d) L.-Y. Cui, Z.-G. Sun, Z.-M. Liu, W.-S. You, Z.-M. Zhu, L. Meng, H. Chen and D.-P. Dong, Inorg. Chem. Commun., 2006, 9, 1232 CrossRef CAS .
- J.-L. Song, C. Lei and J.-G. Mao, Inorg. Chem., 2004, 43, 5630 CrossRef CAS .
- J.-L. Song, J.-G. Mao, Y.-Q. Sun and A. Clearfield, Eur. J. Inorg. Chem., 2003, 4218 CrossRef CAS .
- J. Zhu, X. Bu, P. Feng and G. D. Stucky, J. Am. Chem. Soc., 2000, 122, 11563 CrossRef CAS .
- B.-P. Yang and J.-G. Mao, Inorg. Chem., 2005, 44, 566 CrossRef CAS .
- B. Adair, S. Natarajan and A. K. Cheetham, J. Mater. Chem., 1998, 8, 1477 RSC .
- N. Stock, G. D. Stucky and A. K. Cheetham, Chem. Commun., 2000, 2277 RSC .
- C.-H. Lin and K.-H. Lii, Inorg. Chem., 2004, 43, 6403 CrossRef CAS .
- C.-P. Tsao, C.-Y. Sheu, N. Nguyen and K.-H. Lii, Inorg. Chem., 2006, 45, 6361 CrossRef CAS .
- J.-L. Song and J.-G. Mao, Chem.–Eur. J., 2005, 11, 1417 CrossRef CAS .
- S.-M. Ying and J.-G. Mao, Cryst. Growth Des., 2006, 6, 964 CrossRef CAS .
- Y.-L. Huang, M.-Y. Huang, T.-H. Chan, B.-C. Chang and K.-H. Lii, Chem. Mater., 2007, 19, 3232 CrossRef CAS .
- J.-G. Mao, Coord. Chem. Rev., 2007, 251, 1493 CrossRef CAS .
- A. Cabeza, M. A. G. Aranda and S. Bruque, J. Mater. Chem., 1998, 8, 2479 RSC .
-
(a) A. Cabeza, X. Ouyang, C. V. K. Sharma, M. A. G. Aranda, S. Bruque and A. Clearfield, Inorg. Chem., 2002, 41, 2325 CrossRef CAS ;
(b) Z.-M. Sun, J.-G. Mao, B.-P. Yang and S.-M. Ying, Solid State Sci., 2004, 6, 295 CrossRef CAS .
-
(a) P. Lenaerts, K. Driesen, R. V. Deun and K. Binnemans, Chem. Mater., 2005, 17, 2148 CrossRef CAS ;
(b) Z. He, E.-Q. Gao, Z.-M. Wang, C.-H. Yan and M. Kurmoo, Inorg. Chem., 2005, 44, 862 CrossRef CAS ;
(c) Y. Wang, X. Zheng, W. Zhuang and L. Jin, Eur. J. Inorg. Chem., 2003, 3572 CrossRef CAS .
-
(a) A. de Bettencourt-Dias, Inorg. Chem., 2005, 44, 2737 ;
(b) G. L. Law, K. L. Wong, X. Zhou, W. T. Wong and P. A. Tanner, Inorg. Chem., 2005, 44, 4142 CrossRef CAS .
-
G. M. Sheldrick, SHELXS-97, Program for the solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: Simulated XRD pattern of compound 1 and experimental powder XRD patterns of compounds 1–6. Fig. S2: Experimental powder XRD pattern of compound 1 and of dehydrated samples after calcination at 150 and 180 °C. Table S1: Selected bond lengths (Å) for compounds 1–6. Table S2: Selected bond angles (°) for compounds 1–6. CCDC reference numbers 670477–670481 (1–5) and 686602 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b814400a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.