The plant metallothionein 2 from Cicer arietinum forms a single metal–thiolate cluster†‡
Xiaoqiong
Wan
and
Eva
Freisinger
*
Institute of Inorganic Chemistry, University of Zürich, 8057 Zürich, Switzerland. E-mail: freisinger@aci.uzh.ch; Fax: +41 446356802; Tel: +41 446354621
First published on 18th August 2009
Abstract
The plant metallothionein 2 from Cicer arietinum (chickpea; cicMT2) is a typical member of this subfamily and features two cysteine-rich regions containing eight and six cysteine residues, respectively, separated by a linker region 41 amino acids in length. This metallothionein thus differs significantly from the well-studied vertebrate forms. A synthetic gene encoding cicMT2 was designed, cloned into a suitable vector, and the protein was over-expressed in Escherichia coli. For the first time, an in-depth spectroscopic characterization of cicMT2 in the presence of divalent metal ions is performed showing a binding capacity for five ZnII, CdII, or CoII ions and the typical features of metal–thiolate clusters. Based on proteolytic digestion experiments, the cluster arrangement formed by the divalent metal ions and the cysteine thiolate groups connects the amino-terminal with the carboxy-terminal cysteine-rich region. The cluster formation process, put into effect with the addition of the fourth metal ion to the apo protein, was investigated using the characteristic shift of absorption bands observed in the UV/Vis spectra upon titration with CoII. The pH-dependent ZnII- and CdII-thiolate cluster stability is one of the highest observed for plant MTs so far, but lower than that usually found in vertebrate metallothioneins. The dependence of the pH stability on the ionic strength of the solution is more pronounced for the CdII- than for the ZnII-form of the protein.
Introduction
The typical characteristics shared by all members of the metallothionein (MT) super-family are their low molar mass (Mr < 10 kDa) and their high cysteine (Cys) content. The large number of Cys thiolate groups and their distinctive distribution patterns allow the coordination of metal ions, preferentially with the d10 electronic configuration, in the form of metal–thiolate clusters.1,2 Proposed roles of MTs encompass the storage, transport and regulation of essential metal ions such as ZnII and CuI, as well as the detoxification of excess amounts of essential metal ions and of non-essential heavy metal ions like CdII or HgII.3–7 In addition, MTs have been reported to take part in protection against a variety of stress conditions.8–12 The family of plant MTs is divided into four subfamilies, mainly based on the Cys distribution pattern of the amino- (N-) terminal domain.13 The N- and carboxy- (C-) terminal Cys-rich regions of the p1, p2, and p3 subfamilies are separated by Cys-free linker regions ∼30–45 amino acids long that often, in contrast to the vertebrate forms, contain the aromatic amino acids phenylalanine (Phe) and tyrosine (Tyr) as well as an occasional histidine residue. The fourth subfamily, denoted p4 or pec, is characterized by three Cys-rich regions separated by shorter peptide linkers, which are in addition devoid of Phe and Tyr residues. Plant MTs show differential expression patterns, which vary depending on the organ, the developmental stage and exposure to exogenous factors such as drought, wounding, and heavy metal ion stress.14 The homeostasis of essential transition metal ions includes the tight control of free metal ion concentrations and the requirement for suitable binding partners. While known chaperones for CuI ions include CCH, CCS1, and COX19, no chaperones for ZnII have been identified so far.15 Instead, it has been suggested that ZnII ions are transported in the form of complexes with nicotianamine or organic acids, in analogy to FeII/III ions.15,16 The role of plant MTs in the homeostasis of essential metal ions is still a matter of debate and there is strong demand for further research.A typical plant MT2 from the p2 subfamily consists of ∼80 amino acids (Fig. 1). The distribution pattern of the eight Cys residues in the N-terminal part and the six C-terminal Cys residues is highly conserved. A high degree of conservation is also observed for a part of the amino acids separating the single Cys residues. Glycine (Gly) in particular, as the sterically least demanding amino acid, has a high abundance allowing maximum flexibility for cluster formation. Gene expression studies have shown that mt2gene transcripts are generally more abundant in the aerial parts of a plant, specifically the leaves. However, Arabidopsis thaliana (mouse ear cress) mt2mRNA was also detected in mature roots and flowers, as well as in lower levels in pods,17 and mt2gene transcripts were virtually absent in the leaves of Malus domestica (apple), but abundant in the flowers and the early stages of fruit development.18 Visualization of actual protein expression levels in A. thaliana was possible using the E. coliβ-glucuronidase (GUS) gene reporter system.19 Staining of the translated MT::GUS fusion proteins revealed allocation of MT2B to the vascular tissues of all organs including the roots. MT2A::GUS was mostly found in the mesophyll cells of the leaves as well as in root tips. Abundance of both MT2 forms increases with leaf age reaching maximum values in senescing leaves. Increased concentrations of MT2A::GUS and MT2B::GUS in the trichomes and the roots are observed upon treatment with CuSO4. The influence of exogenous factors was also shown by monitoring the mt2mRNA levels in leaves from Nicotiana glutinosa (tobacco), which became higher upon mechanical wounding, viral infection and exposure to CuSO4 solutions.20 In etiolated epicotyls of C. arietinum, mt2 expression is upregulated and further increases upon polyethylene glycol-induced osmotic stress, while abscisic acid-mediated stress and treatment with CuII-, CdII-, and ZnII-containing solutions had no effect on mt2 expression in this specific tissue.21 However, a general role of the MT2 proteins in metal homeostasis and detoxification was highlighted with yeast complementation studies. A. thaliana MT2A and MT2B are able to re-establish copper tolerance in a mutant deficient in over-expressing the yeast-specific CuI-thionein (CUPI) accompanied by accumulation of copper ions.22 The two MT2 forms also restore a certain amount of ZnII tolerance to a ZnII-hypersensitive yeast mutant lacking two vacuolar ZnII transporters, although being clearly less effective than the A. thaliana MT4, or Ec, proteins and provoking only little ZnII accumulation. Additionally, a remarkable protective effect of MT2A and MT2B against CdII toxicity was observed in this mutant when compared to the wild-type yeast strain. To directly compare A. thaliana MT2a with a known ZnII-thionein, MT2a was expressed in a mutant form of the cyanobacteria Synechococcus PCC 7942 lacking SmtA, a MT with the ability to coordinate four ZnII ions.23,24 MT2a was able to partly complement the ZnIIhypersensitivity of the mutant, although to a lesser extent than SmtA.
 | ||
| Fig. 1 Amino acid sequence alignment of representative members of the plant MT subfamily p2. Cys residues are accentuated with a black, and aromatic amino acids with a gray, background. His residues are framed with a black border. In M. acuminata MT2, one of the otherwise highly conserved Cys residues in the N-terminal domain is mutated to a Ser residue. | ||
At this point, it is essential to be aware of the fact that so far no plant MT has been isolated directly from its natural source with the exception of the wheat MT Ec-1, a seed-specific ZnII binding protein. As a consequence, currently one has to rely on the results of more indirect studies to get a first notion of the nature of the natively-bound metal ions and hence of the metal ion specificity of the respective MT under investigation. The diversity of the results from the experiments described above do not allow to unambiguously decide whether the plant MT2 proteins have a preference for ZnII or CuI ions, or even CdII, in vivo. Hence we decided to first investigate the binding behaviour towards divalent metal ions.
Early experiments showed that recombinant expression of the A. thaliana MT2a glutathion-S-transferase- (GST-) fusion protein in E. coli using ZnII-supplemented media yielded the ZnII form of the protein, although with unknown metal ion content.23 The pH of half dissociation of ZnII ions from the fusion protein was determined to 5.1 suggesting that A. thaliana MT2a might indeed be capable of binding ZnII in the plant. The second member of the p2 subfamily of plant MTs characterized in more detail is the one from Quercus suber (cork oak). Measurements performed on the recombinantly expressed protein with inductively coupled plasma optical emission spectroscopy indicate a ZnII content of 3.5–4.2 and a CdII content of 5.3–6.3 metal ions per protein molecule.25–27ESI-MS studies revealed species containing 3 or 4 ZnII ions and 5–7 CdII ions next to varying amounts of sulfide ions. The titration of the ZnII form of Q. suber MT2 with CdII ions monitored by UV spectroscopy suggested the formation of a Cd4MT2 species.26 Arrangement of the coordinated metal ions in a single metal–thiolate cluster was deduced from comparative studies with the separate N- and C-terminal Cys-rich regions. In the following, we will show that C. arietinum MT2 (cicMT2) forms defined species with ZnII, CdII, and CoII containing five equivalents of metal ions in each case. The divalent metal ions are coordinated in the form of a single clustered structure shaped by the N- and C-terminal Cys-rich stretches based on limited proteolytic digestion experiments combined with mass spectrometry.
Experimental
Chemicals and enzymes
Tritirachium album proteinase K was obtained from Qbiogene (Lucerna Chem AG, Lucerne, Switzerland). All other enzymes were from Promega (Catalys AG, Wallisellen, Switzerland), Roche (Rotkreuz, Switzerland) or New England Biolabs Inc. (Ipswich, MA, USA). LB Broth (Miller), dithiothreitol (DTT), and isopropyl-β-D-thiogalactopyranoside (IPTG) were purchased from Chemie Brunschwig AG (Basel, Switzerland), tris(hydroxymethyl)aminomethane-hydrochloride (Tris-HCl) from Calbiochem (VWR International AG, Lucerne, Switzerland), and Chelex® 100 resin from Bio-Rad (Reinach, Switzerland). All other chemicals were ACS grade from Fluka (Buchs, Switzerland) or Acros organics (Chemie Brunschwig AG, Basel, Switzerland). The deionized water used for the preparation of all solutions was vacuum degassed for approximately 30 min and nitrogen saturated for at least 1 h. Where strictly anaerobic conditions were required, solutions were rendered oxygen-free by three freeze-thaw cycles under vacuum.Cloning, expression and purification of Zn5-cicMT2
The C. arietinum mt2gene (GenBank accession number X95709)28 was optimized for E. colicodon usage (Fig. 2) and constructed from seven commercially obtained oligonucleotides (each 49–53 nt long; Microsynth AG, Balgach, Switzerland) by assembly polymerase chain reaction (PCR ) as described.29 The synthetic gene was cloned into the vector pTYB2 from the IMPACT™-CN system (New England Biolabs Inc.) using the NdeI and XmaI restriction sites, which adds two extra vector-derived amino acids (Pro, Gly) to the C-terminus of the protein. The accuracy of the insert and integrity of the plasmid were confirmed by DNA sequencing. | ||
| Fig. 2 Nucleotide sequences of the native C. arietinum mt2gene (top sequence) and of the synthetic gene designed for optimized expression in E. coli (middle sequence in bold) including restriction sites (horizontal boxes) and 3′- as well as 5′-overhangs (sequences in gray). Codon changes are marked with vertical gray boxes. The encoded amino acid sequence is depicted at the bottom of each row (bold). | ||
Zn5-cicMT2 was over-expressed in the protease-deficient E. colicell line BL21(DE3) in the form of a fusion protein with a vector encoded C-terminal intein tag (protein self-splicing element) and a chitin-binding domain according to procedures described previously.30 After induction of protein synthesis with 1 mM IPTG, growth media were supplemented with 100 μM ZnCl2.
The two-step purification procedure involved affinity chromatography on chitin beads followed by a polishing step using gel filtration. Only the variations to the published procedure are described in the following.30 Cells from an 800 mL bacteria culture were resuspended in 25 mL lysis buffer (20 mM Tris-HCl, pH 8.5, 200 mM NaCl, and 1 mM ZnCl2) before sonication. Tris-HCl buffer was also used during affinity chromatography and the subsequent dialysis steps instead of the considerably more expensive 4-(2-hydroxyethyl)piperazine-1-ethanesulfonate (HEPES-NaOH) as small amounts of the protein–Trisadduct formed during the intein self-splicing process do not interfere with the experiments reported in this work.29 The purity of Zn5-cicMT2 was examined with 12% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) using the Tris-tricine buffer system as described.31Monobromobimane- (mBBr-) modified MT2 was prepared according to the literature but without the addition of EDTA or a reducing agent during the modification procedure.32
Determination of protein concentration and metal ion content
Protein concentrations were assessed by thiol group determination with 2,2′-dithiodipyridine (2-PDS) at pH 4.0 based on the specific absorption of the formed thiopyridinone at 343 nm (ε343 = 7600 M−1 cm−1).33 For the calculation, all 14 Cys residues of the protein were assumed to be present in the reduced state as verified by ESI-MS measurements. For the determination of metal ion concentrations the protein solutions were acidified with 0.2 M HNO3 and analysed with flame atomic absorption spectroscopy (F-AAS) using an AA240FS spectrometer (Varian AG, Zug, Switzerland). The metal-to-protein stoichiometries obtained in this way were confirmed with metal ion substitution reactions and ESI-MS studies (see below).Preparation of apo-cicMT2 and Cd5-cicMT2
Zn5-cicMT2 was incubated with 50 mM DTT for 20 min at room temperature to reduce any disulfide bonds potentially present and subsequently acidified to pH 2.0 with HCl. Apo-cicMT2 and released ZnII ions were separated with size exclusion chromatography (Sephadex G10, pre-equilibrated with 10 mM HCl) at a flow rate of 0.8 mL min−1 in 10 mM HCl. Due to its susceptibility to oxidation and sensitivity to low pH conditions for a prolonged period of time, apo-cicMT2 was used immediately or lyophilized and stored at −20 °C generally not more than 2 days before use.Cd5-cicMT2 was obtained either by substitution of ZnII ions in Zn5-cicMT2 with an excess of CdII or by reconstitution of apo-cicMT2. For the reconstitution, apo-cicMT2 and a surplus of CdII ions were mixed at pH 2.0 followed by a rapid pH increase to 7.5 with 1 M Tris inside a N2-purged glove box. For both procedures, unbound metal ions were removed by incubation with Chelex® 100 resin at 4 °C overnight.
Metal ion titration of apo- and Zn5-cicMT2
14.1 μM freshly prepared apo-cicMT2 in 10 mM HCl was titrated with incremental amounts of CdCl2 (0–8 equiv.) or 33.2 μM apo-cicMT2 for the titration with CoCl2 (0–10 equiv.). Both experiments were performed inside a N2-purged glove box using separate solutions for each titration step. After each metal ion addition the pH of the solution was raised rapidly to pH 7.5 (±0.1) with 1 M Tris and the sample was incubated for 30 min at room temperature before UV/Vis, CD, or MCDspectra were recorded. Final concentrations for titrations with CdCl2 were 10 mM Tris and 12.3 μM cicMT2 and, for titrations with CoCl2, 10 mM Tris and 28.0 μM cicMT2. The titration of 10.0 μM Zn5-cicMT2 with CdII ions was performed in 10 mM Tris (pH 7.5) under a constant flow of nitrogen outside the glove box using the same solution for all titration steps (0–8 equiv.).pH titration of Zn5-cicMT2 and Cd5-cicMT2
600 μL of the respective 10.0 μM protein solution in 1 mM Tris-HCl (pH 7.5) were titrated with increments (typically 0.5–1.0 μL) of diluted HCl solutions under a constant flow of argon. Subsequently to each acid addition, the pH value of the solution was measured and a UV spectrum recorded. Volume increase due to HCl addition and sample evaporation during measurements virtually balanced each other. Curve fitting of experimental data† was performed with Origin 7.0 (OriginLab Corporation, Northampton, MA, USA) with equations derived as described.34Limited proteolytic digestion with Tritirachium album proteinase K
Similar to the method described,30 Cd5.4-cicMT2, obtained from Zn5-cicMT2 by direct metal ion replacement, was incubated for 2 hours with a 30-fold excess of T. album proteinase K in a buffer containing 50 mM Tris-HCl (pH 8.0) and 10 mM CaCl2. Thereafter, the digestion mixture was applied to a Superdex Peptide 10/300 GL size exclusion chromatography column (GE Healthcare, Otelfingen, Switzerland) using 60 mM ammonium acetate (pH 7.5) as elution buffer. The collected fractions were further analyzed by amino acid analysis and MALDI-TOF (matrix assisted laser desorption/ionization) mass spectrometry.UV/Vis, CD, and MCD spectroscopy
UV/Vis absorption spectra were recorded on a Cary 500 scan spectrophotometer (Varian AG) using a scan speed of 600 nm min−1 in the range of 200–800 nm for CoIItitration experiments and of 200–400 nm for all other experiments. CD spectra were acquired with a J-715 spectropolarimeter (JASCO, Japan) over a spectral range of 200–400 nm with a scan speed of 10 nm min−1 and accumulation of four spectra for each measurement. For MCD, a J-810 spectropolarimeter (JASCO, Japan) equipped with a 1.5 T (15 kG) magnet was employed using a scan speed of 20 nm min−1 in the range of 200–400 nm (4 spectra accumulations). All spectra were recorded at room temperature.Recording of ESI-MS and MALDI-TOF spectra
For ESI-MS, samples were diluted with 0.2% formic acid/50% acetonitrile (pH 2.0) or 5 mM ammonium acetate in 50% methanol (pH 7.0–7.5), and mass spectra were recorded in a quadrupole time-of-flight (TOF) Ultima API spectrometer (Waters Corp., UK). Deconvolution of spectra was performed with the MaxEnt1 software. For MALDI-TOF measurements, the sample was diluted threefold with 0.1% trifluoroacetic acid (TFA) in water. Sample preparation was performed on a steel target using a saturated α-cyano-4-hydroxycinnamic acid solution in 50% acetonitrile and 0.1% TFA as matrix. Mass spectra were recorded on an Ultraflex TOF/TOF spectrometer (Bruker Daltonic GmbH, Germany). The ions were accelerated at 25 kV with pulsed ion extraction time of 100 ns in the positive ion linear mode.Results and discussion
Zn5-cicMT2 expression, purity, and metal ion content
After over-expression in E. coli grown in ZnII-supplemented growth media, the cicMT2–intein fusion protein was selectively removed from the cell lysis solution by affinity binding to a chitin column. After washing and DTT-mediated self-cleavage of the C-terminal intein tag, the resulting cicMT2-containing fraction was subjected to a final purification step with size exclusion chromatography, and the ZnII-form of cicMT2 eluted in a single sharp peak. Yields ranged between 1.0 and 1.2 mg of purified protein per L of cell culture. Evaluation of the peak fraction with SDS-PAGE and subsequent silver staining revealed a single band for the ZnII-form of cicMT2 at an apparent molar mass slightly above 27 kDa (Fig. 3, lane 2) according to the molar mass marker run in parallel (lanes 1 and 4). No other protein contaminants are visible. Modification of the Cys thiolate groups with mBBr (see Experimental) causes the release of metal ions from the protein and results in a modified protein species migrating at an apparent molar mass of ∼10 kDa (lane 3). This is in the range of the theoretical molar mass expected for apo-cicMT2 after modification of all 14 Cys residues with mBBr (7969.9 Da + 14 × 190.2 Da = 10632.7 Da). The observed migration behavior agrees well with observations made with Musa acuminata (banana) MT3, a plant MT from the p3 subfamily,34 and has been ascribed to the preservation of the metal–thiolate clusters leaving only the protein surface accessible for SDS binding. This substoichiometrical loading of the protein with SDS leads to a lower charge-to-protein mass ratio compared to the proteins of the molar mass marker, and thus to a reduced migration speed and consequently a higher apparent molar mass. Disruption of the metal–thiolate clusters by mBBr allows complete denaturation of the metallothioneins with SDS and hence results in the theoretically expected molar mass. Also the apparent molar mass of the Triticum aestivum (bread wheat) Zn6Ec-1 plant metallothionein was reported to be higher than expected though the effect is less pronounced.30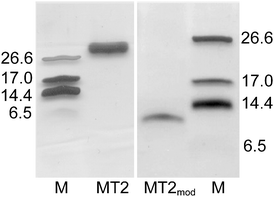 | ||
| Fig. 3 Silver-stained SDS-PAGEgel showing cicMT2 (lane 2), mBBr modified MT2 (lane 3) as well as a protein marker (lanes 1 and 4). The molar masses are given in kDa, the marker band at 6.5 kDa is only faintly visible. | ||
ESI-MS spectra of the ZnII-containing holo-form of cicMT2 obtained directly from recombinant expression in ZnII-supplemented LB media were recorded at acidic as well as neutral pH. At pH 2, the spectrum of apo-cicMT2 is obtained (Fig. 4A) and shows that the N-terminal translation initiator Met is completely removed from the protein, as is common for proteins expressed in E. coli,35–37 giving rise to the signal at 7969.1 Da (calculated average mass 7969.9 Da). The additional signal at 8072.3 Da originates from a C-terminal Trisadduct (calc. 8073.1 Da), formed during the thiol mediated self-splicing of the intein tag by aminolysis of the reactive protein–DTTthioesterintermediate with the Tris buffer as described.29,34 The ESI-MSspectrum of the ZnII form shows Zn5-cicMT2 to be the major species (8285.3 Da, calc. 8286.9 Da) next to a smaller amount of Zn4-cicMT2 (8220.5 Da, calc. 8223.5 Da, Fig. 4B) as well as the corresponding Tris-adducts (Zn4-cicMT2–Tris 8328.2 Da, calc. 8326.7 Da and Zn5-cicMT2–Tris 8388.5 Da, calc. 8390.1 Da). The results from ESI-MS spectrometryagree well with bulk values for the ZnII content of cicMT2, which vary between 4.6 and 5.0 depending on the protein batch. Bulk values are determined by a combination of F-AAS measurements for the concentration of metal ions and 2-PDSassays for the thiol group content and thus the protein concentration. F-AAS measurements also show that no copper ions can be detected in the protein preparations. Similar results are obtained for the CdII form obtained by reconstitution of apo-cicMT2. The ESI-MSspectrum is dominated by Cd5-cicMT2 at 8521.7 Da (calc. 8522.1 Da, Fig. 4C). Again, a small amount of Cd4-cicMT2 (8409.2 Da, calc. 8409.6 Da) next to the Tris-adduct of Cd5-cicMT2 (8625.2 Da, calc. 8625.7 Da) is observed. The peak for Tris-Cd4cicMT2 (calc. 8514.7 Da) is masked by the signal for Cd5-cicMT2. Bulk values for the CdII content are usually slightly larger than for the ZnII content, ranging from 5.0 to 5.4.
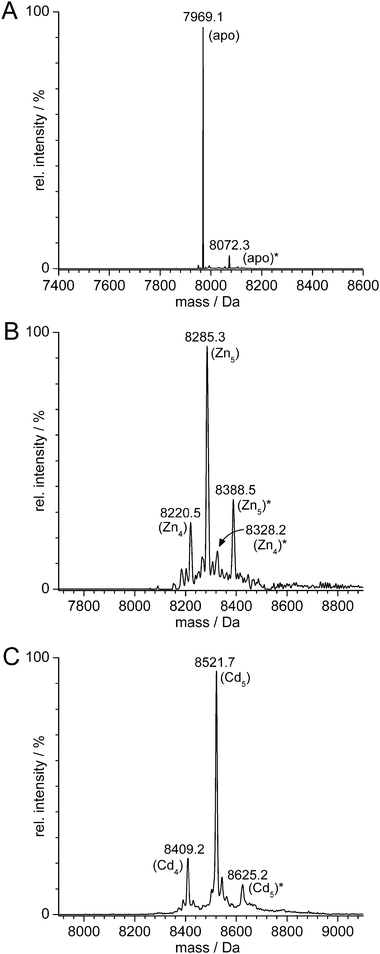 | ||
| Fig. 4 Deconvoluted ESI-MS spectra of (A) apo-cicMT2 recorded at pH 2, (B) ZnII-, and (C) CdII-cicMT2, both measured at pH 7.0–7.5. In all spectra minor signals originating from the C-terminal Tris-adduct are discernable and denoted with *. All species lack the N-terminal Met residue. In (B) as well as (C) both species with five metal ions next to minor amounts with four metal ions are present. See text for more details. | ||
The ZnII content, which was determined with a combination of F-AAS and quantification of Cys residues by the 2-PDSassay and verified with ESI-MS spectra, is significantly higher than that reported for cork oak MT2.26 According to ESI-MS measurements, cork oak MT2 mainly coordinates only four ZnII ions. Additionally, a minor Zn3MT2 species can be detected. The CdII binding ability of cork oak MT2 is higher, revealing coordination of five CdII ions in analogy to cicMT2. However, species with six and seven CdII ions were also found. The increased CdII binding ability can be explained by the concomitant presence of four sulfide ions in the complexes. The origin of the sulfide ions is not finally resolved as both proteins, cork oak as well as cicMT2, were over-expressed in E. coli. However, while the intein system was used for purification in the case of cicMT2, cork oak MT2 was expressed with a glutathione-S-transferase- (GST-) tag.
UV, CD, and MCD spectra
The UV spectra of apo-, Zn5-, and Cd5-cicMT2 are depicted in Fig. 5A. The spectrum of apo-cicMT2 is dominated by the contribution of the peptide bonds leading to a maximum around 190 nm. Additional absorptions in the 190–230 nm region originate from certain protein side chains, namely Asp, Glu, Asn, and Gln. Apart from minor contributions by the three aromatic amino acid residues, i.e. two Phe and one Tyr, the spectrum shows complete transparency above ∼250 nm. The holo-forms of cicMT2 show the characteristic ligand-to-metal charge transfer (LMCT) bands of metal–thiolate clusters visible by an increase in absorption around 230 nm upon ZnII binding and by the formation of a distinct shoulder around 250 nm upon CdII binding.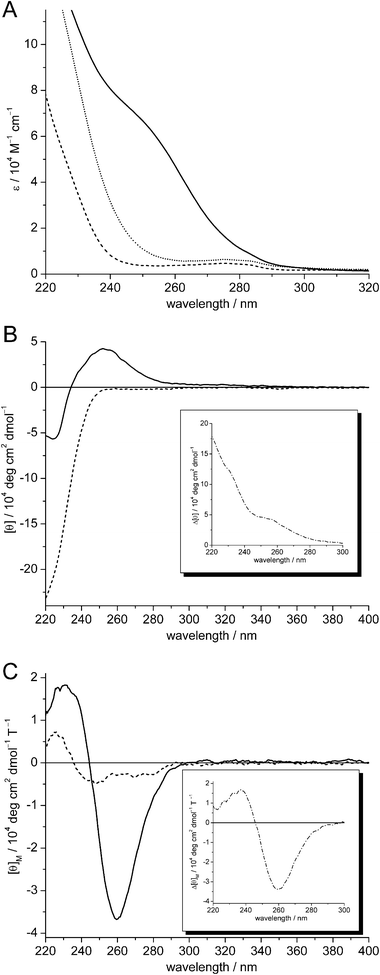 | ||
| Fig. 5 (A) UV, (B) CD, and (C) MCD spectra of apo-cicMT2 (dashed lines), Zn4.9- (dotted line, UV only), and Cd5.2-cicMT2 (solid lines). The insets in (B) and (C) show the difference spectra obtained by subtracting the apo-cicMT2 spectra from the respective Cd5.2-cicMT2 spectra (dash-dotted lines). | ||
To further investigate the coordination geometry of divalent d10 metal ions in cicMT2, the CD and MCD spectra of Cd5-cicMT2 obtained by reconstitution of the apo-form were also examined (Fig. 5B and C). The CD spectrum of Cd5-cicMT2 shows extremities at (+) 252 nm and (−) 224 nm and thus has a similar profile to the one reported for wheat Cd6Ec-1.30 However, when compared to other full-length CdII-MTs,38–42 the two CD bands show shifts towards lower wavelengths, which are even more pronounced than observed for Cd6Ec-1. The notable blue shifts of the two CD bands suggest that the cluster geometry in Cd5-cicMT2 is markedly different from that found in other CdII-MTs. The MCD spectrum of Cd5-cicMT2 is characterized by a biphasic profile with two bands at (−) 259 nm and (+) 231 nm with the inflection point at 245 nm, a feature commonly found in other CdII-form MTs and indicative of tetrahedral tetrathiolate coordination environments.38–39,42,43
Titration of apo- and Zn5-cicMT2 with CdII
Substitution of ZnII ions in tetrahedral tetrathiolate coordination sites by CdII generally results in isomorphous structures.44 To verify the ability of cicMT2 to coordinate up to five ZnII or CdII ions as observed with F-AAS and ESI-MS, Zn5-cicMT2 was titrated with increasing amounts of CdII ions and the increase of the LMCT band of the S → CdII transition at 250 nm observed with UV spectroscopy. The substitution proceeds almost immediately to completion probably aided by the kinetic lability of the clusters in combination with the larger binding constant of CdII in metal–thiolate clusters compared to ZnII. As expected, the absorption at 250 nm increases up to the addition of the fifth equivalent and remains constant thereafter (Fig. 6A). The absorption increase is thereby only linear up to the addition of three CdII ions (ε250: ∼12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 per CdII added), the increase for the fourth (ε250: ∼7800 M−1 cm−1) and the fifth (ε250: ∼4200 M−1 cm−1) equivalent is considerably less pronounced. Again, this behavior is different to that of cork oak MT2. When the ZnII form of cork oak MT2 is titrated with CdII ions only an increase of the LMCT band at 250 nm up to four equivalents occurs. This is especially surprising considering that the amino acid sequences of both proteins are up to 77% identical. If conserved substitutions are also considered, e.g.Tyr → Phe or Thr → Ser, then the similarity increases to 90%. Further experiments are required to recognise the less obvious variations between these two plant MT2 species.
000 M−1 cm−1 per CdII added), the increase for the fourth (ε250: ∼7800 M−1 cm−1) and the fifth (ε250: ∼4200 M−1 cm−1) equivalent is considerably less pronounced. Again, this behavior is different to that of cork oak MT2. When the ZnII form of cork oak MT2 is titrated with CdII ions only an increase of the LMCT band at 250 nm up to four equivalents occurs. This is especially surprising considering that the amino acid sequences of both proteins are up to 77% identical. If conserved substitutions are also considered, e.g.Tyr → Phe or Thr → Ser, then the similarity increases to 90%. Further experiments are required to recognise the less obvious variations between these two plant MT2 species.
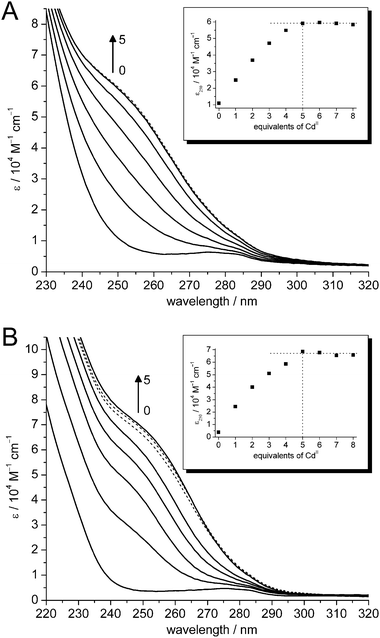 | ||
| Fig. 6 UV spectra of the titration of (A) Zn4.9- and (B) apo-cicMT2 with CdII ions: 0–5 equiv. (solid lines), 6–8 equiv. (dotted lines). Inserts: Plot of molar absorptivity at 250 nm against equiv. of CdII added. | ||
The study of metallothioneins makes it sometimes necessary to reconstitute the metal-free apo form, obtained by size exclusion chromatography at acidic pH, with metal ions. On the one hand, metal ions with lower binding constants than ZnII, such as CoII, cannot be obtained by a substitution reaction. On the other hand, if 100% complete metal exchange is desirable, e.g. with 113CdII for heteronuclear NMR experiments, a too large excess of the precious metal ion would be necessary to completely substitute the originally bound metal ions. To verify retention of the metal ion binding abilities of the apo form, a titration of apo-cicMT2 with CdII ions was performed and the increase of the S → CdII LMCT band at 250 nm again monitored with UV spectroscopy (Fig. 6B). As observed for the experiment with Zn5-cicMT2, the maximum absorptivity is reached with the addition of the fifth CdII ion. A surplus of CdII ions exhibits no further effects. Again, the increase in molar absorptivity upon the addition of four and five equivalents of CdII is smaller. This can be most likely attributed to the formation of thiolate bridges between CdII ions, which leads to a less pronounced increase of the LMCT bands than the coordination by a terminal thiolate group.
pH stability of metal–thiolate clusters
The pH stability of metal–thiolate clusters in MTs can be examined by spectrophotometric pH titrations. The competition between protons and metal ions for the cysteine thiolate groups is reflected in the pH-dependent intensity of the LMCT band at the given wavelength, i.e. at 231 nm for ZnII– and at 250 nm for CdII–thiolate clusters, until complete metal ion release occurs below a certain pH value (Fig. 7A and B).34,45–47 The extent of competition and hence the pH stability of the metal–thiolate clusters can be described by means of apparent pKa values of the Cys thiolate groups in the presence of the respective metal ions. As described,14,30,34,48 such apparent pKa values are obtained by a non-linear curve fit of the plot of molar absorptivity of the LMCT band against the respective pH value (Fig. 7C and D). These absorption plots generally show a single major drop of the absorption at approx. pH 4.5 for the ZnII forms and 3.5 for the CdII forms, and hence curve fitting with an equation considering just a single pKa value usually gives a goodness-of-fit >99%.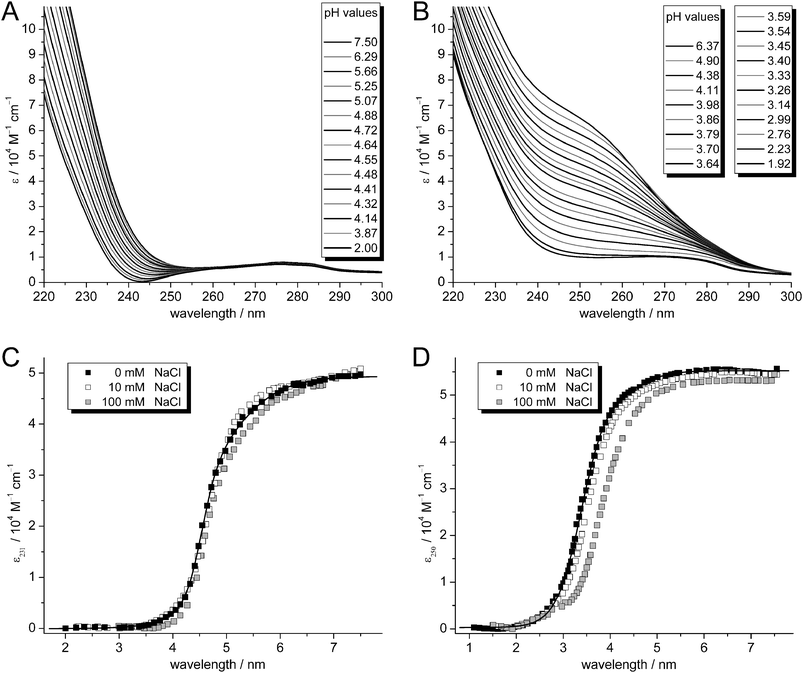 | ||
| Fig. 7 Representative UV spectra of the titration of (A) Zn4.9- and (B) Cd5.4-cicMT2 with increasing amounts of HCl. Plots of molar absorptivity at (C) 231 nm for the ZnII forms and at (D) 250 nm for the CdII forms at different ionic strengths: 0 mM NaCl (black squares), 10 mM NaCl (open squares), and 100 mM NaCl (gray squares). | ||
In this way, a value of 4.68(1) was determined for Zn4.9-cicMT2 and 3.439(7) for Cd5.2-cicMT2, showing that the zinc–thiolate cluster has a lower pH stability than the corresponding cadmium–thiolate cluster as expected. Completely metal-ion-devoid species are obtained at pH values below ∼3.5 for the ZnII form and below ∼2.0 for the CdII form. In addition, the pH stability of the clusters depends to a certain extent on the ion strength of the solution. As shown in Fig. 7C and D as well as in Table 1, the pH stability indeed decreases with increasing salt concentration of the solution. At a salt concentration close to physiological conditions, Cd5.2-cicMT2 shows a pKa value that is 0.4 units higher than the one in a salt-free solution. In contrast, the pH stability of Zn4.9-cicMT2 is only slightly affected by changes in ion strength. This is unexpected as a similar experiment with Zn6Ec-1 showed an increase in the pKa value of 0.2 units when increasing the ionic strength from 10 mM to 100 mM NaCl.14 Generally, differences in the amino acid sequences and variations in the metal-binding microenvironments might explain the different influences of ion strength on different MTs. On a speculative basis, the cluster in Zn4.9-cicMT2 could be more compact than the one in Cd5.2-cicMT2 and/or better shielded from the solvent, minimizing the influence of the ionic strength.
 .
.Having been determined for a considerable number of MTs from different families, such apparent pKa values are currently one of the very few characteristics allowing a basic comparison of the overall stability of the metal–thiolate clusters in MTs across the different phyla of life.14 With respect to other plant MTs, the metal–thiolate clusters of cicMT2 show a high pH stability, i.e. the pKa value of Zn4.9-cicMT2 is 0.2–0.7 units lower and that of Cd5.2-cicMT2 0.6–1.0 units lower than the values observed for the respective ZnII- and CdII-forms of chickpea and garden pea MT1, as well as banana MT3.14,34,48,49 Both cicMT2-forms are, however, less stable by ca. 0.1 pKa units than the respective wheat Ec-1 forms and have a significantly lower pH stability than the mammalian equine and human species.14,30,47,50,51
Titration of apo-cicMT2 with CoII
In MT research, the paramagnetic CoII ion has been used to study the metal–thiolate cluster formation process.30,52 The reaction proceeds without cooperativity, i.e. first all binding sites to terminal thiolate ligands will be saturated by CoII ions before cluster formation and thus the bridging of CoII ions by thiolate ligands takes place. The concomitant antiferromagnetic coupling of CoII nuclei, which is best observed with EPR spectroscopy, is also accompanied by a characteristic shift of absorption bands in the UV/Vis spectra. In the case of cicMT2 this shift can be particularly well observed for the d–d transitions in the visible range. As CoII ions have a lower affinity to thiolate groups than ZnII ions, the CoIItitration experiments have to be performed starting from apo-cicMT2, and therefore strictly anaerobic conditions are required to prevent oxidation to CoIII as well as disulfide bridge formation.The same absorption increase up to five equivalents as observed for the titration with CdII ions can be also seen upon reconstitution of apo-cicMT2 with incremental amounts of CoII (Fig. 8). The spectra of Co1–5-cicMT2 are characterized by minima at 288–300 nm, followed by maxima located at 310–325 nm with pronounced shoulders around 380–400 nm, all originating from S → CoII charge transfer transitions. The d–d transitions in the visible region of the spectra lead to three broad bands at 600–610 nm, 685–694 nm, and roughly 750–743 nm. The splitting into three absorption bands originates from spin–orbit coupling and has been attributed to tetrahedrally coordinated high-spin CoII–tetrathiolate complexes.38,52 The intensity of the bands in the d–d region constitutes approximately 10% of the absorptivity in the LMCT region. During the metal ion titration, the intensities of the bands in the visible range relative to each other change from 1.0 : 1.8 : 1.3 (600 nm : 685 nm : 750 nm) to 1.0 : 1.2 : 1.1 (610 nm : 694 nm : 743 nm). The shift upon cluster formation is most pronounced for the absorption band around 685 nm. Up to the addition of three CoII ions just an increase of absorption is observed. The fourth and also the fifth equivalent additionally give rise to a bathochromic shift of this absorption band (Fig. 8, inset). Thus cluster formation in cicMT2 proceeds as expected based on simple theoretical calculations if binding of metal ions solely by thiolate groups is assumed: The amino acid chain of cicMT2 contains 14 Cys residues and therefore can accommodate three metal ions in independent binding sites with terminal thiolate groups only (3 metal ions × 4 Cys residues ⇒ 12 Cys). For the coordination of the fourth metal ion two terminal Cys residues are still available. Nevertheless, to complete the coordination sphere, two bridging thiolate groups also have to be included, marking the onset of cluster formation. For the coordination of the fifth metal ion, only bridging thiolate ligands are available and therefore another shift of the absorption band is observed.
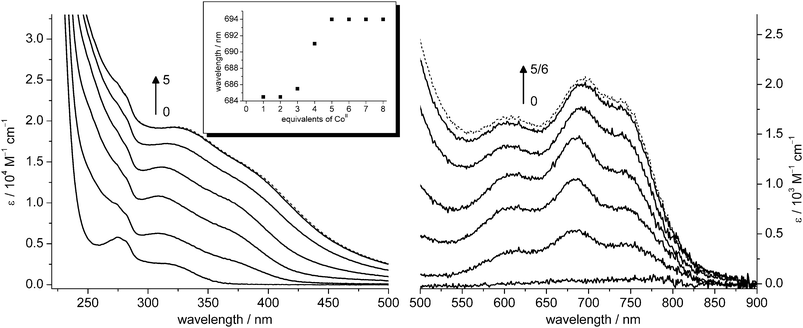 | ||
| Fig. 8 Spectra of the reconstitution of apo-cicMT2 with CoII: charge-transfer region (left), d–d transition region (right). CoII is bound to independent sites up to approximately 3 equiv. and cluster formation occurs from 4 to 5 equiv. No more spectral changes are observed upon addition of more than 5 equiv. of CoII (black dotted line). The shift of absorption bands originating from the process of cluster formation is exemplified in the inset by the red-shift of the local maxima around 690 nm. | ||
When compared with the CoII-substituted forms of other MTs (Table 5), including wheat Co6-Ec-1,30, crab Co6-MT1,38 and rabbit liver Co7-MT1,52,53 the profile of the spectra of Co1–5-cicMT2 is most similar to that of the CoII-substituted α-fragment of rabbit liver MT, which has a stoichiometry of MII4Cys11.53 The molar absorptivity of Co5-cicMT2 is ∼2000 M−1 cm−1, which is higher than that of the CoII-substituted α-fragment of rabbit liver MT1 (∼1600 M−1 cm−1),53 but in the same range as the substoichiometrically substituted rabbit liver Co5–MT1 form (Co4.6MT1 ∼2000 M−1 cm−1, Co5.5MT1 ∼2500 M−1 cm−1).52
Limited proteolytic digestion
To evaluate the coordination mode of divalent d10 metal ions in cicMT2 further, a limited proteolytic digestion of Cd5.4-cicMT2 was performed with T. album proteinase K, a subtilisin-related serine protease with broad specificity. However, earlier experiments showed that the metal–thiolate clusters in MTs usually remain intact even if proteolytic cleavage occurs between amino acids belonging to the same cluster. This method was already used to show that the two Cys rich regions of the garden pea MT1 are connected by CdII ions,54 and also revealed the two-domain structure of the wheat Ec-1 metallothionein.30 The rationale was hence to determine if the five metal ions in cicMT2 are arranged in two separate clusters formed individually by the N- and C-terminal Cys-rich regions of the protein or if the Cys residues of both regions are jointly involved in metal ion coordination. Separation of the digestion mixture was performed with size exclusion chromatography under non-denaturing conditions and gave rise to a single peak with a longer retention time than found for the undigested protein (Fig. 9A). Examination of the metal ion content by F-AAS and of the thiolate group concentration with the 2-PDS assay indicates that the ratio of metal ions to thiolate groups remains the same before and after digestion. The peak fraction was further analysed by MALDI-TOF analysis (Fig. 9B), revealing three major signals, one at 2522.6 Da matching the mass for residues 2–28 (calculated 2523.8 Da) and thus the N-terminal Cys-rich part of the protein, one at 2132.8 Da corresponding to residues 61–81 (calculated 2133.8 Da) and thus the C-terminal Cys-rich part, and the third one at 2003.9 Da corresponding to residues 62–81 (calculated 2004.7 Da). Additional signals originating from the cleaved linker region were not detected. The amino acid analysis of the eluted fraction further corroborates the assignment of the masses to the protein fragments (Table 2).![(A) Gelfiltration profile of undigested (solid lines) and proteinase K digested (dashed lines) Cd5.4-cicMT2 (black lines) and Cd6Ec-1 (gray lines) with assignment of peaks to the molar masses of the proteins and protein fragments. The respective molar masses without metal ions are given in brackets. (B) MALDI-TOF spectrum of the peak fraction from the proteolytic digestion of Cd5.4-cicMT2 revealing signals for the N-terminal (2522.6 kDa, residues 2–28) as well as the C-terminal Cys-rich region of the protein (2003.9/2132.8 kDa, residues 62/61-81). (C) Schematic presentation of the metal ion arrangement, which connects the N- and C-terminal part of cicMT2 (left). Theoretical model of a MII5Cys14 cluster adapted from the solid state structure of [Hg5(SePh)12]2−67 illustrating the feasibility of a connecting metal ion arrangement in the form of a single metal–thiolate cluster (right).](/image/article/2009/MT/b906428a/b906428a-f9.gif) | ||
| Fig. 9 (A) Gel filtration profile of undigested (solid lines) and proteinase K digested (dashed lines) Cd5.4-cicMT2 (black lines) and Cd6Ec-1 (gray lines) with assignment of peaks to the molar masses of the proteins and protein fragments. The respective molar masses without metal ions are given in brackets. (B) MALDI-TOF spectrum of the peak fraction from the proteolytic digestion of Cd5.4-cicMT2 revealing signals for the N-terminal (2522.6 kDa, residues 2–28) as well as the C-terminal Cys-rich region of the protein (2003.9/2132.8 kDa, residues 62/61-81). (C) Schematic presentation of the metal ion arrangement, which connects the N- and C-terminal part of cicMT2 (left). Theoretical model of a MII5Cys14 cluster adapted from the solid state structure of [Hg5(SePh)12]2−67 illustrating the feasibility of a connecting metal ion arrangement in the form of a single metal–thiolate cluster (right). | ||
The question regarding the apparent molar mass of the eluted species still remains. On the one hand, assuming the formation of two separate clusters by the residues of the N- and C-terminal Cys-rich regions, two fragments with masses of roughly 2.0 and 2.5 kDa are expected, which would co-elute in a single peak due to the limited resolution of the column. On the other hand, the joint participation of both regions in metal-ion binding results in an assembly with a molar mass of approximately 4.5 kDa. In both cases only the mass of the protein fragments without metal ions is considered. To determine the apparent molar mass of the species eluting from the Superdex Peptide column, the elution profiles were compared to the profiles obtained from the digestion experiments of wheat Cd6Ec-1 with proteinase K (Fig. 9A).30 Undigested Cd5.4-cicMT2 and Cd6Ec-1 elute in approximately the same volume fractions, which is to be expected considering the similar molar masses of the apo forms (7.9 kDa for Ec-1 compared to 8.0 kDa for cicMT2). Although the variations are small, it is more accurate to compare the molar masses of the apo instead of the holo forms as the largest contribution to the overall size of the protein comes from the amino acid part, provided that the three-dimensional shapes of the holo forms are similar. The contribution of the small metal ions, however, is minor in contrast to their relatively large influence on the molar mass. Comparing the elution profiles of the respective digested proteins, the peak from the cicMT2 sample is closer to the peak of the larger βE-domain of Ec-1 (molar mass of apo-βE-Ec-1 4.5 kDa) than to the peak of the smaller domain (molar mass 2.4 kDa). It can thus be concluded that the molar mass of the digested species of Cd5.4-cicMT2 is around 4.5 kDa, which is exactly the mass of the combined N- and C-terminal Cys-rich regions identified with MALDI-TOF. Thus the formation of two small separate clusters, both smaller than ∼2.5 kDa, can be ruled out. Consequently, the two Cys-rich regions were coeluting from the column at the higher apparent molar mass because they were connected to each other by some means, which was most likely some sort of a metal–thiolate cluster arrangement. On a speculative basis, the connecting metal–thiolate cluster arrangement present in Cd5-cicMT2 could be even just a single cluster as schematically presented in Fig. 9C. This is clearly different from the two separate metal–thiolate clusters found in the wheat Ec-1 MT as well as in the vertebrate, crustacean and echinodermata MTs.30,55–64
The assumption of metal–thiolate cluster connected protein termini is further corroborated considering the results from the SDS-PAGE analysis described above. From the gel (Fig. 3) the apparent molar mass for the unmodified ZnII-cicMT2 species can be estimated to be slightly above 27 kDa, while unmodified wheat Zn6Ec-1 migrates in the SDS gel at approximately 17 kDa.30 As wheat Ec-1 consists of two rather independent metal ion binding domains,30,65,66 its structure is probably less compact than a species with just a single cluster and hence more residues should be exposed to the solvent and thus be accessible for modification with SDS. As a result, Zn6Ec-1 migrates faster in SDS-PAGE than Zn5-cicMT2 and accordingly the presence of a rather compact structure in the latter is corroborated, which is most feasible assuming the formation of a single metal ion binding domain.
But is a single connecting metal–thiolate cluster feasible? To the best of our knowledge, there is no report of a structurally-characterized metal–thiolate cluster containing five divalent metal ions. However, an inorganic metal–selenolate cluster of the form [Hg5(SePh)12]2− has been described.67 If one replaces two of the bridging selenolate ligands with terminal groups a cluster, as depicted in Fig. 9C, with 14 ligands might indeed be realized. In any case, we still await a final structural resolution.
In conclusion, chickpea MT2 has the ability to coordinate up to five divalent metal ions such as ZnII, CdII, and CoIIvia the thiolate groups of its Cys residues. Based on limited proteolytic digestion experiments strong evidence was provided for the presence of a metal ion arrangement or even a single metal–thiolate cluster that connects the N-terminal with the C-terminal Cys-rich region. Additionally, close similarities between the spectroscopic features of cicMT2 and other MTs could be shown with respect to the pH stability of the metal–thiolate clusters and the cluster formation process. In order to achieve more detailed structural information about the cluster(s) formed in cicMT2, additional work is in progress.
Acknowledgements
We thank S. Chesnov and Y. Auchli (FGCZ, Zürich) for recording MS spectra and amino acid analyses, and M. Vašák (Department of Biochemistry, University of Zürich) for access to the MCD spectropolarimeter. Financial support from the Swiss National Science Foundation (SNF grant 200020-113728/1 and SNF Förderungsprofessor PP002-119106/1 to EF) is gratefully acknowledged.References
- J. H. R. Kägi, S. R. Himmelhoch, P. D. Whanger, J. L. Bethune and B. L. Vallee, J. Biol. Chem., 1974, 249, 3537–3542 CAS.
- Y. Kojima, C. Berger, B. L. Vallee and J. H. R. Kägi, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3413–3417 CrossRef CAS.
- M. Brouwer, D. R. Winge and W. R. Gray, J. Inorg. Biochem., 1989, 35, 289–303 CrossRef CAS.
- G. Roesijadi, Comp. Biochem. Physiol. C, 1996, 113, 117–123.
- M. G. Cherian and Y. J. Kang, Exp. Biol. Med., 2006, 231, 138–144 Search PubMed.
- C. Günes, R. Heuchel, O. Georgiev, K. H. Müller, P. Lichtlen, H. Blüthmann, S. Marino, A. Aguzzi and W. Schaffner, EMBO J., 1998, 17, 2846–2854 CrossRef CAS.
- J.-C. Amiard, C. Amiard-Triquet, S. Barka, J. Pellerin and P. S. Rainbow, Aquat. Toxicol., 2006, 76, 160–202 CrossRef CAS.
- W. Maret, J. Nutr., 2000, 130, 1455S–1458S CAS.
- F. Reinecke, O. Levanets, Y. Olivier, R. Louw, B. Semete, A. Grobler, J. Hidalgo, J. Smeitink, A. Olckers and F. H. Van der Westhuizen, Biochem. J., 2006, 395, 405–415 CrossRef CAS.
- S. Suzuki, S. Tohma, N. Futakawa, M. Higashimoto, M. Takiguchi and M. Sato, J. Health Sci., 2005, 51, 533–537 CrossRef CAS.
- W. Feng, F. W. Benz, J. Cai, W. M. Pierce and Y. J. Kang, J. Biol. Chem., 2006, 281, 681–687 CAS.
- M. Sato and I. Bremner, Free Radical Biol. Med., 1993, 14, 325–337 CrossRef CAS.
- P.-A. Binz and J. H. R. Kägi, in Metallothionein IV, ed. C. Klaassen, Birkhäuser Verlag, Basel, edn., 1999, pp. 7–13 Search PubMed.
- E. Freisinger, Dalton Trans., 2008, 6663–6675 RSC.
- C. M. Palmer and M. L. Guerinot, Nat. Chem. Biol., 2009, 5, 333–340 CrossRef CAS.
- C. Curie, G. Cassin, D. Couch, F. Divol, K. Higuchi, M. Le Jean, J. Misson, A. Schikora, P. Czernic and S. Mari, Ann. Bot., 2009, 103, 1–11 CAS.
- J. M. Zhou and P. B. Goldsbrough, MGG, Mol. Gen. Genet., 1995, 248, 318–328 CrossRef CAS.
- S. J. Reid and G. S. Ross, Physiol. Plant., 1997, 100, 183–189 CrossRef CAS.
- W. J. Guo, W. Bundithya and P. B. Goldsbrough, New Phytol., 2003, 159, 369–381 Search PubMed.
- D. Choi, H. M. Kim, H. K. Yun, J.-A. Park, W. T. Kim and S. H. Bok, Plant Physiol., 1996, 112, 353–359 CrossRef CAS.
- F. J. Muñoz, R. V. Ullán, E. Labrador and B. Dopico, Physiol. Plant., 1998, 104, 273–279 CrossRef CAS.
- W.-J. Guo, M. Meetam and P. Goldsbrough, Plant Physiol., 2008, 146, 1697–1706 CrossRef CAS.
- N. J. Robinson, J. R. Wilson and J. S. Turner, Plant Mol. Biol., 1996, 30, 1169–1179 CrossRef CAS.
- C. A. Blindauer, M. D. Harrison, J. A. Parkinson, A. K. Robinson, J. S. Cavet, N. J. Robinson and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9593–9598 CrossRef CAS.
- G. Mir, J. Domènech, G. Huguet, W.-J. Guo, P. Goldsbrough, S. Atrian and M. Molinas, J. Exp. Bot., 2004, 55, 2483–2493 CrossRef CAS.
- J. Domènech, R. Orihuela, G. Mir, M. Molinas, S. Atrian and M. Capdevila, JBIC, J. Biol. Inorg. Chem., 2007, 12, 867–882 CrossRef CAS.
- J. Domènech, G. Mir, G. Huguet, M. Capdevila, M. Molinas and S. Atrian, Biochimie, 2006, 88, 583–593 CrossRef CAS.
- http://www.ncbi.nlm.nih.gov/Genbank/index.html .
- E. A. Peroza and E. Freisinger, Protein Expression Purif., 2008, 57, 217–225 CrossRef CAS.
- E. A. Peroza and E. Freisinger, JBIC, J. Biol. Inorg. Chem., 2007, 12, 377–391 CrossRef CAS.
- H. Schagger, H. Aquila and G. Vonjagow, Anal. Biochem., 1988, 173, 201–205 CAS.
- G. Meloni, M. Knipp and M. Vašák, J. Biochem. Biophys. Methods, 2005, 64, 76–81 CrossRef CAS.
- A. O. Pedersen and J. Jacobsen, Eur. J. Biochem., 1980, 106, 291–295 CAS.
- E. Freisinger, Inorg. Chim. Acta, 2007, 360, 369–380 CrossRef CAS.
- Y. D. Liao, J. C. Jeng, C. F. Wang, S. C. Wang and S. T. Chang, Protein Sci., 2004, 13, 1802–1810 CrossRef CAS.
- K. Ozawa, M. J. Headlam, P. M. Schaeffer, B. R. Henderson, N. E. Dixon and G. Otting, Eur. J. Biochem., 2004, 271, 4084–4093 CrossRef CAS.
- X. Ni and H. K. Schachman, Protein Sci., 2001, 10, 519–527 CrossRef CAS.
- J. Overnell, M. Good and M. Vašák, Eur. J. Biochem., 1988, 172, 171–177 CrossRef CAS.
- H. Willner, M. Vašák and J. H. R. Kägi, Biochemistry, 1987, 26, 6287–6292 CrossRef CAS.
- M. Vašák, D. W. Hasler and P. Faller, J. Inorg. Biochem., 2000, 79, 7–10 CrossRef CAS.
- R. Dallinger, Y. J. Wang, B. Berger, E. A. Mackay and J. H. R. Kägi, Eur. J. Biochem., 2001, 268, 4126–4133 CrossRef CAS.
- D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vašák, Biochemistry, 2000, 39, 14567–14575 CrossRef CAS.
- G. K. Carson, P. A. W. Dean and M. J. Stillman, Inorg. Chim. Acta, 1981, 56, 59–71 CrossRef CAS.
- M. Vašák, Methods Enzymol., 1991, 205, 452–458 CAS.
- J. H. R. Kägi and B. L. Vallee, J. Biol. Chem., 1960, 235, 3460–3465 CAS.
- Y. Wang, E. A. Mackay, M. Kurasakia and J. H. R. Kägi, Eur. J. Biochem., 1994, 225, 449–457 CrossRef CAS.
- J. H. R. Kägi and B. L. Vallee, J. Biol. Chem., 1961, 236, 2435–2442 CAS.
- O. Schicht and E. Freisinger, Inorg. Chim. Acta, 2009, 362, 714–724 CrossRef CAS.
- A. M. Tommey, J. Shi, W. P. Lindsay, P. E. Urwin and N. J. Robinson, FEBS Lett., 1991, 292, 48–52 CrossRef CAS.
- L. J. Jiang, M. Vašák, B. L. Vallee and W. Maret, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2503–2508 CrossRef CAS.
- M. Vašák and J. H. R. Kägi, Met. Ions Biol. Syst., 1983, 15, 213–273 CAS.
- M. Vašák and J. H. R. Kägi, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 6709–6713 CrossRef CAS.
- M. Good and M. Vašák, Biochemistry, 1986, 25, 3328–3334 CrossRef CAS.
- P. Kille, D. R. Winge, J. L. Harwood and J. Kay, FEBS Lett., 1991, 295, 171–175 CrossRef CAS.
- P. Faller, D. W. Hasler, O. Zerbe, S. Klauser, D. R. Winge and M. Vašák, Biochemistry, 1999, 38, 10158–10167 CrossRef CAS.
- A. H. Robbins, D. E. McRee, M. Williamson, S. A. Collett, N. H. Xuong, W. F. Furey, B. C. Wang and C. D. Stout, J. Mol. Biol., 1991, 221, 1269–1293 CAS.
- A. Arseniev, P. Schultze, E. Worgotter, W. Braun, G. Wagner, M. Vašák, J. H. R. Kägi and K. Wüthrich, J. Mol. Biol., 1988, 201, 637–657 CAS.
- B. A. Messerle, A. Schaffer, M. Vašák, J. H. R. Kägi and K. Wüthrich, J. Mol. Biol., 1992, 225, 433–443 CAS.
- D. T. Jiang, S. M. Heald, T. K. Sham and M. J. Stillman, J. Am. Chem. Soc., 1994, 116, 11004–11013 CrossRef CAS.
- K. Zangger, G. Öz, J. D. Otvos and I. M. Armitage, Protein Sci., 1999, 8, 2630–2638 CrossRef CAS.
- S. S. Narula, M. Brouwer, Y. Hua and I. M. Armitage, Biochemistry, 1995, 34, 620–631 CrossRef CAS.
- M. D. Gieselman, Y. T. Zhu, H. Zhou, D. Galonic and W. A. van der Donk, ChemBioChem, 2002, 3, 709–716 CrossRef CAS.
- A. Muñoz, F. H. Forsterling, C. F. Shaw, 3rd and D. H. Petering, JBIC, J. Biol. Inorg. Chem., 2002, 7, 713–724 CrossRef CAS.
- R. Riek, B. Precheur, Y. Wang, E. A. Mackay, G. Wider, P. Guntert, A. Liu, J. H. R. Kägi and K. Wüthrich, J. Mol. Biol., 1999, 291, 417–428 CrossRef CAS.
- E. A. Peroza, A. Al Kaabi, W. Meyer-Klaucke, G. Wellenreuther and E. Freisinger, J. Inorg. Biochem., 2009, 103, 342–353 CrossRef CAS.
- E. A. Peroza, R. Schmucki, P. Güntert, E. Freisinger and O. Zerbe, J. Mol. Biol., 2009, 387, 207–218 CrossRef CAS.
- M. Berardini, T. J. Emge and J. G. Brennan, Inorg. Chem., 1995, 34, 5327–5334 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1: results for curve fitting of pH titration data. See DOI: 10.1039/b906428a |
| ‡ Dedicated with best wishes to Professor Milan Vašák on the occasion of his 65th birthday. |
| This journal is © The Royal Society of Chemistry 2009 |