Denaturing and non-denaturing microsolution isoelectric focussing to mine the metalloproteome†
Barbara
Pioselli‡
ab,
Caroline
Munro‡
a,
Andrea
Raab
c,
Christian L.
Deitrich
c,
Kriangsak
Songsrirote
a,
Jörg
Feldmann
c and
Jane
Thomas-Oates
*a
aDepartment of Chemistry, University of York, York, UK. E-mail: jeto1@york.ac.uk
bDipartimento di Biochimica e Biologia Molecolare, Università degli Studi di Parma, Parma, Italy
cDepartment of Chemistry, Aberdeen University, Aberdeen, UK
First published on 26th August 2009
Abstract
Metals bound to proteins play essential roles in living systems. Elements such as phosphorus, selenium and iodine are commonly covalently linked to proteins while others are non-covalently complexed. Thus, the identification and characterization of the metal–protein complexes require a careful hyphenation of techniques able to separate and detect the intact binding complexes with both high resolution and high sensitivity. This study has investigated for the first time the potential of microsolution isoelectric focussing to separate a mixture of metal-binding protein standards under well-established denaturing conditions and a novel non-denaturing separation protocol has also been developed. SEC-ICP-MS analysis was used to evaluate the ability of the two separation procedures to separate and maintain the integrity of standard metal–protein complexes. Microsolution isoelectric focussing under denaturing conditions separates the metalloprotein mixtures with high resolution, although the stability of the complexes is affected. Microsolution isoelectric focussing under our newly developed non-denaturing conditions shows a lower degree of resolution, although the stability of the metal–protein complexes is preserved. The applicability of the two procedures to a biological metalloproteome has also been evaluated.
Introduction
In living systems, metals are commonly found in their cationic form, and bind and interact with many electron-rich biological molecules such as proteins and DNA. Metal ions bound to proteins are often critical to the protein's function, structure, or stability, with numerous essential biological functions requiring metal ions. Such proteins that contain a metal cofactor are usually described as metalloproteins or metal-containing proteins. In many cases, metals are complexed within proteins via N-, O- or S-ligands contributed by amino acids, (histidine, cysteine, methionine and asparagine) or, in the case of iron, bound in the porphyrin structure. While phosphorus, selenium and iodine are commonly covalently linked to amino acids, other elements are generally non-covalently complexed to the protein. The nature of the chemical link between metal and protein, and the low level at which trace elements are usually present in biological matrices, represent a real challenge to the researcher. The characterisation of metalloproteins in a complex biological system requires optimisation of a multi-step strategy: selective isolation through a high resolution separation technique, detection of the metal bound to the biomolecules through a high sensitivity element-detection technique, and molecular analysis using mass spectrometry to identify the metalloprotein.1A number of different strategies have been used to investigate the speciation of metals bound to proteins. Ion-exchange chromatography (IEC) and size-exclusion chromatography (SEC), followed by detection of metal species with inductively coupled plasma mass spectrometry (ICP-MS) is a standard platform used in bioinorganic speciation,2–4 which allows rapid measurement of a wide range of elements with only limited sample pre-treatment. The ICP source is acknowledged to be the introduction method of choice for element detection, because this high energy ionization source dissociates all molecules into atoms, which are efficiently ionised, giving access to low concentrations of elements even from complex sample matrices. Unfortunately, these liquid chromatography methods do not provide a high enough resolving power to separate all the proteins in a complex biological sample. The resolution of proteins and reproducibility of separation can be greatly improved using polyacrylamide gel electrophoresis (PAGE). The use of denaturing and non-denaturing gel electrophoretic techniques and laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) has been reported. Metal-binding proteins were successfully determined in protein extracts from E. coli, grown in a medium supplemented with cadmium or zinc.5PAGE in combination with LA-ICP-MS has also been reported by Becker’s group to characterize copper- and zinc-binding proteins.6,7 Data were also published showing detection of selenium in proteins separated by both 1D and 2D PAGE after staining with silver.8,9 Although 1D denaturing and non-denaturing PAGE10 and 2D SDS-PAGE used in combination with LA-ICP-MS undoubtedly provide analytical tools for the detection of metal-containing proteins, a number of factors can adversely affect the outcome of such analyses. These include the detection limits of the elements in single gel spots, the pre-preparation of the gel before placement in the ablation chamber and perhaps most importantly, the nature of the interaction between the metals and their proteins, which is likely to be affected by the conditions commonly used for denaturing separations.11,12 Moreover, identification is further complicated as each separate 2D gel spot may only contain very small amounts of protein (around 1 ng for very low abundance proteins detectable using typical silver-staining protocols). Very careful sample preparation is therefore required. Pre-fractionation conducted on intact proteins is a typical strategy for enriching low abundance proteins in biological samples and overcoming the issue of the limited dynamic range of proteome analysis. Multidimensional fractionation has allowed unprecedented proteome coverage.13,14
Preparative solution isoelectric focusing (sIEF) is a method that is gaining use for the pre-fractionation of complex proteome solutions.15 This technique was proposed as a protein pre-fractionation procedure by Bier and colleagues in 1979,16 and extensively reviewed.15,17,18 It is a high resolution method compared to other commonly-used fractionation procedures, such as the widely-used techniques of size exclusion chromatography, differential centrifugation, and selective solubilization, raising the possibility of using it as an orthogonal separation step in a multidimensional separation approach. Preparative sIEF is fully compatible with PAGE analysis, and provides the opportunity to increase sample concentration during focussing and allows retention of very hydrophobic, small and highly basic proteins. Commercial instrumentation for sIEF has been developed primarily for large-scale purifications, but recently, miniaturised instrumentation has been developed to reduce sample size and dilution. The ZOOM® IEF Fractionator (Invitrogen) is one of the commercial versions of the microscale sIEF (microsol IEF) device based on the work of Zuo and Speicher,19 which has multiple chambers with pH-selective partitions to trap proteins at their pI values. This methodology has been based on the basic separation principles described by Righetti and coworkers.20,21 The miniaturization, the elimination of cross-flow recirculation and the increase in gel partition pore size have been recognized as the most significant differences between the two types of multicompartment devices.22,23 Commercial sIEF systems can be classified according to the method of establishing the pH gradient, namely, through carrier ampholytes or through the use of isoelectric membranes, and consequently how proteins are confined within the gradient. In the case of ZOOM® IEF, the pH gradient is formed through both isoelectric membranes and carrier ampholytes . Narrow pI-range protein fractions generated in this way make it possible to detect lower abundance proteins with subsequent 2D PAGE, as it is possible to load the gel only with proteins that will all enter and migrate in the narrow pH range of the first dimension, which, as recognised by Righetti and collaborators, enables much more efficient mining of the proteome.24
Solution IEFfractionation under denaturing conditions has been widely reported. The ability of this approach to significantly reduce biological complexity, absolutely necessary for accurate quantitation in differential proteomic studies, has been recently shown for both prokaryotic and eukaryotic samples.25–28 In contrast, there are very few reports of non-denaturing native sIEF. Shang and colleagues29 successfully performed native IEF on myoglobin and yeast soluble proteins using the Rotofor system and water as the sole solvent. However, the applicability of this approach to the study of the metalloproteome has not been fully investigated and a non-denaturing method has not been established using the ZOOM® IEF fractionator. Ibrahim and colleagues described the extraction and purification of protein which might be associated with silver using IEF and SDS-PAGE,30 but no hyphenation with trace elemental techniques or metal bound to protein detection was attempted. As a proof of concept, our study therefore aimed at the application of microsol IEF under both denaturing and non-denaturing conditions to a comprehensive compositional analysis of a mixed standard and a biological metalloproteome, for its potential use as one step in a multidimensional separation approach aimed at the speciation of metalloproteins. Our scope was to critically evaluate the stability of the different metal–protein complexes under examination, during denaturing and non-denaturing separation. The samples produced in this way were prepared on a scale compatible with subsequent high sensitivity proteomic studies, so that this platform may be suitable for the study of metal–protein binding interactions of species usually present in small amounts in biological samples.
Experimental
Chemicals
Unless otherwise stated all chemicals were of a grade suitable for proteomic analysis. HPLC grade water or ultrapure water (18 MΩ, Elga UK) was used throughout. High-purity solutions of 1000 mg L−1 Cu, Fe and Zn (BDH) were used for the preparation of element standard solutions. Standards of different concentrations containing equimolar amounts of EDTA, Cu and Zn, plus Fe were mixed for quantification of eluting elements. For the isotope exchange experiment copper, zinc and iron metal (CK Gas Products, Hampshire, UK) with a certified isotopic abundance of 63Cu 0.8% and 65Cu 99.2% or 68Zn 99.0%, 64Zn 0.38%, and 66Zn 0.33% or 54Fe 99.88%, 56Fe 0.11%, 57Fe 0.005% and 58Fe 0.005% were used. Solutions of isotopically-enriched Cu, Zn or Fe were made up by dissolving the corresponding metal in concentrated HNO3 and diluting to a working solution of 10 mg L−1.Protein standards
The protein standards used were: superoxide dismutase (EC 1.15.1.1) from bovine liver (SOD), carbonic anhydrase (EC 4.2.1.1) from bovine erythrocytes (CA), cytochrome C (EC 1.1.2.2) from bovine heart (Fluka Biochemika), albumin from bovine serum (BSA), and myoglobin from horse heart (MYO). Stock solutions of proteins were prepared without further purification, dissolving the lyophilized powder in water and storing at −80 °C.Preparation of cytosolic fractions of liver
Cytosolic fractions were prepared from approximately 50 mg of powdered freeze-dried liver from a Blackface male lamb using a ProteoExtract® Subcellular Proteome Extraction Kit from Calbiochem® according to the manufacturer’s instructions. The cytosolic protein fraction was further purified by removing low molecular weight species (below 3000 Da) by centrifugation at 6500 × g for 45 min in a centrifugal filter device (Microgon®). The retentate was removed and stored at −80 °C pending further analysis.Microscale solution isoelectric focusing (microsol IEF)
A ZOOM® IEF fractionator (Invitrogen) was used for solution phase IEF to separate protein standards and cytosolic cell lysates into fractions on the basis of isoelectric point (pI). Separation was performed according to the following protocols.The ZOOM® apparatus was assembled according to the manufacturer’s instructions. Sample chambers were constructed using ZOOM® disks of different pHs placed into the chamber as follows: anode (+), pH 3.0, pH 4.6, pH 5.4, pH 6.2, pH 7.0, pH 10.0 cathode (−). After assembly, samples and buffers were loaded into the chamber. 17.5 mL each of (a) anode buffer containing 42% w/v urea, 15% w/v thiourea and 16.5% v/v Novex® IEF anode buffer (50×) (7 mM phosphoric acid), and (b) cathode buffer containing 42% w/v urea, 15% w/v thiourea and 10% v/v Novex® IEF cathode buffer (10×) (20 mM lysine free base, 20 mM arginine free base) pH 10.1, were placed in the relevant chambers. 670 μL of protein sample solution (containing 0.6 mg mL−1 of total protein) was added to each of the five sample chambers through the fill ports. Fractionation was performed as follows: 100 V, 20 min; 200 V, 80 min; 600 V, 80 min, according to the manufacturer’s instructions for equilibrium conditions. Following fractionation, the samples were removed from each of the chambers and stored at −80 °C pending further analysis.
The ZOOM® apparatus was used as above except that urea and thiourea were omitted from the buffer solutions. 670 μL of protein sample (containing 0.6 mg mL−1 total protein) was added to each of the five sample chambers. Fractionation parameters were optimized on the basis of the denaturing conditions, using a multi-step approach to reach the final voltage. Fractionation was performed as follows: 100 V, 20 min; 200 V, 80 min; the final step was carried out at 600 V for 180 min or was terminated when the current reached an equilibrium value (usually around 0.2 mA for the simple standard mixtures) which indicated that focusing was complete.
HPLC (SEC)-ICP-MS analysis
1D PAGE
Protein assay
Protein fractions from denaturing and non-denaturing experiments were purified using an UPPA™ protein concentration kit (Calbiochem®) to separate the protein components from the chemicals that interfere with the determination of protein concentration using the Coomassie Plus assay . Protein concentrations were measured using the Coomassie Plus assay (Pierce, US). Bovine serum albumin was used to generate standard curves and the microplate procedure was followed according to the manufacturer’s instructions. Absorbance was measured at 570 nm.Isotope exchange experiment
Solutions of enriched 65Cu and 68Zn and 54Fe were added to either the relevant focussing buffer alone or solutions of protein standards in focussing buffer (final solution volume was 4 mL, containing 0 or 2.4 mg total protein and 0.4 μg each of 65Cu, 68Zn and 54Fe). The solutions were subjected to non-denaturing or denaturing microsol IEF. Any binding of the metals with buffer or to proteins, or any exchange with the free metal ions was monitored by measuring the isotope ratios of the added elements in the different ZOOM® fractions. Changes in isotopic ratios of the proteins and the ampholytes were determined by using SEC-ICP-MS.Results and discussion
This study evaluated the potential of sIEF methods as a separation strategy in the investigation of metal–protein complexes, and the applicability of the procedure to the characterisation of a biological metalloproteome where both tightly- and weakly-bound metals can be found. To our knowledge, this is the first reported attempt to run microsol IEF in the absence of chaotropic agents and thus under non-denaturing conditions, and moreover, it is the first reported attempt to develop this approach for combination with a commonly-used metal detection technique. Our study can, thus, open up a new route to hyphenation strategies for metalloproteome analysis.Microsol IEF of standard protein mixtures under denaturing and non-denaturing conditions: evaluating the outcome of the separation protocols
A mixture containing bovine serum albumin (BSA), carbonic anhydrase (CA), superoxide dismutase (SOD), myoglobin (MYO) and cytochrome C (non-denaturing only) was used to investigate IEF separation under both denaturing and non-denaturing conditions. These standard proteins are representative metalloproteins with different metal–protein interactions:31 highly specific co-ordination of zinc and copper for carbonic anhydrase and superoxide dismutase, iron-containing porphyrin group for myoglobin and cytochrome C, and very weak (if any) interaction for bovine serum albumin .Denaturing and reducing IEF conditions fractionated the protein mixture into five pH fractions, ranging between pH 3.0 and pH 10.0. 1D SDS gel analysis (Fig. 1A) was used to visualise the proteins present in the different IEF fractions. Triplicate experiments (data not shown) showed that the procedure consistently and reproducibly separated the proteins: BSA (∼66 kDa) in fraction F3 (pH 5.4–6.2), CA (∼29 kDa) in F4 (6.2–7), SOD (homodimer ∼32 kDa) into F3 (5.4–6.2) and F4 (6.2–7), MYO (∼17 kDa) into F4 (6.2–7) and F5 (7–10). SOD and MYO co-migrated on the SDS gel and were identified in their respective sIEF fractions from 1D gels of ZOOM® IEF fractions of individual proteins (not shown).
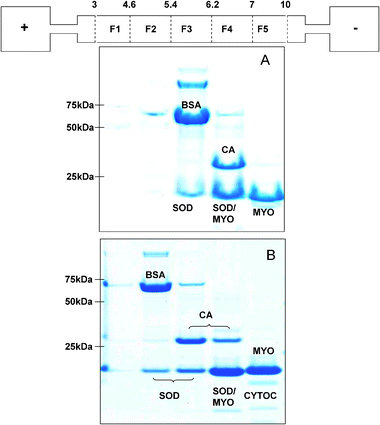 | ||
| Fig. 1 1D SDS gels of standard protein fractions obtained from denaturing solution IEF (panel A) and non-denaturing IEF (panel B) with a schematic of the ZOOM® IEF apparatus. pH fractions are labelled as follows: 3–4.6 (F1), 4.6–5.4 (F2), 5.4–6.2 (F3), 6.2–7 (F4), 7–10 (F5). | ||
The protein mixture was also subjected to sIEF using a modified protocol in which the denaturing and reducing agents were omitted. Focussing conditions were established using time course analyses. Focussing, as assessed by the current equilibrium value (usually around 0.2 mA for the simple standard mixtures) was typically complete within 4.5 hours. BSA (∼66 kDa) reproducibly focused in F2 (pH 4.6–5.4), which is more acidic than under denaturing conditions, and CA (∼29 kDa) into F3 (5.4–6.2) with a minor band in F4 (6.2–7), again a shift to more acidic pIs than under non-denaturing conditions (Fig. 1B). SOD (homodimer ∼32 kDa) was found in F2 (4.6–5.4), F3 (5.4–6.2) and F4 (6.2–7), MYO (∼16 kDa) in F4 (6.2–7) and F5 (7–10) as under denaturing conditions, and cytochrome C in F4 (6.2–7) and F5 (7–10). Monitoring the current and visualising the proteins using 1D SDS PAGE (not shown) showed that focussing for up to 24 hours did not change the results.
We employed a combination of strategies to identify an optimal set of parameters for the non-denaturing sIEF. Water was chosen as the liquid component, as it removes effects from salts and buffer ions, which, although helping to maintain the proteins in solution, could interfere with focusing. Ampholytes (pH 3 to 10) were added at 0.5% v/v to create the pH gradient and to raise the ionic strength to improve protein solubility. To further minimise protein aggregation and precipitation, sucrose (30% w/v) was added, as the stabilizing action of sugars on protein structure has been reported32,33 and sucrose has also been used successfully in microscale, non-denaturing in gel isoelectrofocusing.34 In our study, sucrose did not affect the migration of the protein, but appeared to reduce aggregation and precipitation of the focused proteins, especially when our fractionation procedure was applied to a biological sample (see below).
Different isoelectric migration patterns were thus observed in the denaturing and non-denaturing sIEF fractionations. Particularly, under non-denaturing conditions the proteins’ isoelectric points are shifted towards more acidic pH than under denaturing conditions. Moreover, some proteins reproducibly separate into several pH fractions. This may be explained assuming the existence of different populations of protein molecules having different surface charges. The well-established effects of posttranslational modification in altering the net charge of modified proteins may be responsible for some of this heterogeneity. However, it is also widely accepted that proteins exist in an equilibrium of conformations depending on the environmental conditions.31 This array of conformations will produce a potential range of net charges on the protein surface due to modified tertiary structures, which may be expected to result in different isoelectric migration patterns under denaturing and non-denaturing conditions. The theoretical protein isoelectric point is usually calculated on the basis of the primary structure, which is the linear sequence of the amino acids when the molecule is in a random coil state. Under these conditions, the surface charge of the amphoteric molecule decreases as it moves along a pH gradient, until it reaches its equilibrium position, where the environmental pH matches the protein’s pI, the net charge on the protein is zero, mobility is also zero, and the molecules stop migrating. However, whenever the protein experiences a conformation different from the random coil state, interactions among amino acids may occur which lead to subtle alterations in the experimental pIs; it is reported that reagents such as urea strongly affect a protein’s pI.35 It is also known that the solubility of a protein around its pI may be reduced, especially in the absence of strong solubilizers, such as urea and thiourea. This can become even more pronounced with the increased protein concentrations that are often necessary to reveal minor components. Our results show that sucrose is a relatively mild agent capable of alleviating proteinprecipitation under non-denaturing conditions. Carrier ampholytes were the only background electrolyte used under our non-denaturing conditions. It has been demonstrated33 that during focusing, the local ionic strength of a 1% carrier ampholyte solution can decrease markedly as it focuses. Especially at high ionic strength, protein solubility plotted against pH shows a parabolic relationship with a narrow minimum, while at lower ionic strengths, the minima get wider. Under these low ionic strength conditions, proteinprecipitation may be expected to occur, resulting in broad rather than tight bands of protein, covering at least 0.5 pH units.33 In the pH range 4.6 to 7, the partition discs used in this study differ from one another by 0.8 pH units. Under denaturing conditions, we observed the fractionation to proceed with no proteinprecipitation observable on the immobilized gradient membranes or in the chambers, and the majority of each standard protein focussed into a single pH fraction. However, under non-denaturing conditions, although the fractionation pattern was reproducible, some proteinprecipitation was clearly visible on the partition membranes and within the chambers. As shown in Fig. 1, panel B, a very faint band from cytochrome C was observed under non-denaturing conditions, suggesting a low recovery of this protein in solution. Moreover, different populations of protein molecules for each standard are observed and proteins reproducibly focussed into more than one pH fraction. The presence of sucrose at 30% w/v reduced the amount of proteinprecipitation observed on the discs and the chambers, and has no effect on the separation of a single protein into multiple fractions. This behaviour can, thus, be attributed to the intrinsic lower resolution of the non-denaturing procedure at low ionic strength.
The manufacturer’s instructions suggest that the ZOOM® IEF system is able to reproducibly fractionate 1–2 mg of protein, although up to 10 mg of E. coli lysate have been successfully fractionated under denaturing conditions.26 We have applied our separation approaches to the fractionation of a standard protein mixture containing up to 2.4 mg of total protein. Our rationale for loading a relatively large amount of protein in a single run and evaluating how the two separation protocols functioned with a mixture, was that the analysis of a biological metalloproteome deals with a wide dynamic range of protein concentrations, with the metal-binding proteins usually representing a low abundance fraction of the whole proteome. The sIEF performed under denaturing conditions generally showed better resolution than the non-denaturing approach; as is evident from Fig. 1, panel B, the single protein standards are distributed over at least two fractions, indicating the presence of different populations of protein molecules having a range of different apparent pIs. To determine the total amount of protein recovered in each pH fraction and to correlate this data with the amount of metals, we performed a Coomassie Blue assay on protein pellets obtained after precipitation of the separate sIEF fractions. It should be pointed out that this allows totalprotein determination, and that the protein remains a mixture; it is thus not possible to calculate specific protein recoveries or to correlate the protein amounts with the specific metal recoveries. Precipitation to separate the proteins from the buffer components was necessary because most of the reagents strongly interfere with the Coomassie Blue assay . The UPPA kit was developed as a powerful procedure to quickly and quantitatively precipitate very small protein amounts. Protein recovery after denaturing and non-denaturing sIEF was determined in order to highlight and take into account inevitable protein losses arising for a range of reasons, including our observation that some proteins stick to the membranes. We also wished to evaluate whether significant protein losses occurred due to aggregation at the isoelectric point because of proteins’ decreased solubility at this pH. Fig. 2 shows the percentages of total protein recovered in the different denaturing and non-denaturing fractions after precipitation with the UPPA kit. From a preparative point of view and in line with expectation, after separation and purification by precipitation, protein recovery was not 100% of the protein initially loaded. We calculated the total amount of protein precipitated by summing the amount of proteins purified from each pH fraction. It has been estimated that under denaturing conditions, 1.5% of total protein was recovered after precipitation, whereas under non-denaturing conditions up to 4% of total protein was recovered. However, it should be noted that the main aim of our study was not optimal total protein recovery, but to evaluate the applicability of the newly-developed separation protocols for metalloprotein analysis; subsequent metal determinations needed to be correlated with the amount of metalloproteins effectively recovered in the denaturing or non-denaturing buffer solutions after separation using IEF. Taking into account the potential protein losses, the buffering conditions and purification procedures can be tuned to account for the solubility properties of the samples under examination. Thus, we went on to evaluate the existence of a correlation between the total protein data and the presence of Cu, Zn and Fe as determined by SEC-ICP-MS, to enable us to ascertain the potential to selectively and specifically separate metal–protein complexes under denaturing and non-denaturing conditions by microsol IEF.
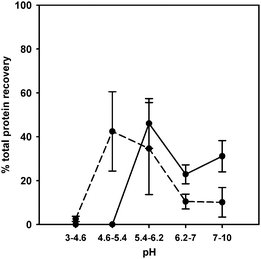 | ||
| Fig. 2 Percentages of total protein recovery for each fraction as determined by Coomassie Blue assay after precipitation of denaturing (circles and solid line) and non-denaturing (circles and short dash line) pH fractions. | ||
Analysis of denaturing and non-denaturing ZOOM® IEF fractionated samples by size exclusion chromatography(SEC)-ICP-MS
Protein fractions produced following separation by sIEF were subjected to SEC-ICP-MS to determine whether metals were still bound to proteins. Based on retention time (tR) (ESI Fig. S1† ) it was possible to distinguish between metal signals eluting around 10–14 min which can be attributed to high molecular weight-bound components, and metal signals eluting around 18–21 min which can be attributed to metals bound to ampholytes (Mr ∼200–900 Da).36,37 Simultaneous recording of UV absorbance at 254 nm allowed us to unequivocally assign the early-eluting ICP signal to the different metals bound to protein.Metal analyses were carried out in order to compare the metals’ and the proteins’ sIEF migration profiles (Fig. 3 and 4, Table S1, S2, S3† ). Under denaturing conditions, the majority of zinc and copper (Fig. 3A and B) was detected in the pH 5.4–6.2 fraction (F3), which contains BSA and SOD (Cu/Zn enzyme), and the pH 6.2–7.0 fraction (F4) containing CA (Zn enzyme) and SOD (Cu/Zn enzyme). It is noteworthy that under denaturing conditions, Cu and Zn are detected in those fractions that we demonstrated contain proteins that specifically associate with these elements (SOD and CA) (Fig. 1A). These results are consistent with migration of intact Cu/Zn–SOD and Zn–CA complexes; the RSD values indicate reliable metal analyses. Fe was consistently detected in F3 and F4 (Fig. 3, panel C). Iron bindingmyoglobin was distributed between F4 and pH 7.0–10.0 fraction (F5) (Fig. 1A). In F3 the most abundant protein was BSA. Under the experimental conditions used, it is possible that BSA weakly interacted with metals. The primary sequence of BSA (UniProtKB/Swiss-Prot database (entry P02769)) has up to 35 cysteine residues, which may represent potential metal-binding sites, available because of the microenvironment that the denaturing IEFbuffer induces. No iron was detected in F5 (Fig. 3C; Table S3† ) where myoglobin was fractionated. This suggests that higher pH possibly causes the loss of the heme group by a population of myoglobin molecules migrating according to a slightly different surface charge. Isolated heme is very hydrophobic and thus not soluble in the aqueous buffers used. Elemental analysis of fractions obtained under non-denaturing conditions showed that Cu and Zn are largely associated with F3 and F4 (Fig. 4A and B), which contain SOD and CA (Fig. 1B). Fe is found in F4 and F5 (Fig. 4C). Fe was largely found in the fractions containing myoglobin and traces of cytochrome C (Fig. 1B). BSA, which has shifted to the pH 4.6–5.4 (F2) fraction (Fig. 1, panel B), does not appear to have a significant amount of metal associated with it under these non-denaturing conditions, which further supports the hypothesis of lower affinity aspecific binding observed under denaturing conditions. These non-denaturing conditions are thus likely to favour separation of proteins adopting globular conformations suitable for specific interactions with metals, although the resolving power of this approach is lower than that under denaturing conditions. Distribution of the protein standards over several pH fractions ultimately led to there being very small amounts of the metals in each separate fraction (less than 1 ng per fraction, Table S1, S2 and S3† ).
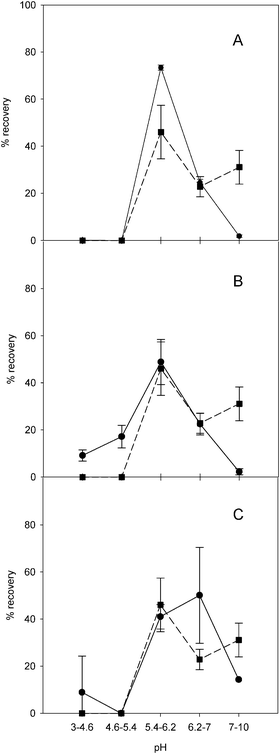 | ||
| Fig. 3 Comparison of average total protein recovery profile (square and short dash line) and the total element recovery profiles (circles and solid line) for each pH fraction under denaturing IEF conditions. Panel A, copper; panel B, zinc; panel C, iron. | ||
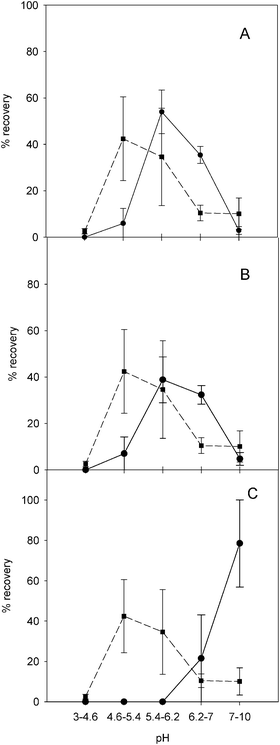 | ||
| Fig. 4 Comparison of average total protein recovery profile (square and short dash line) and the total element recovery profiles (circles and solid line) for each pH fraction under non-denaturing IEF conditions. Panel A, copper; panel B, zinc; panel C, iron. | ||
We performed further experiments with isotopically-enriched element standards at low concentration (65Cu, 68Zn and 54Fe each 0.1 mg L−1) to evaluate isotope exchange phenomena during denaturing and non-denaturing solution ZOOM® IEF separation in the presence and absence of proteins (Fig. 5 and 6). 63Cu/65Cu, 65Zn/68Zn and 54Fe/56Fe ratios were calculated to assess whether there had been any exchange between the spiked isotopically enriched metal ions and the sample solution. Fig. 5 and 6 (panels A and D) show that none of the proteins tested exchanged significant amounts of Cu, although the ampholyte solution can bind additional Cu. Under denaturing conditions Cu shows a slight exchange in F3 (panel D), potentially with BSA. Panels B and E of the same figures show the ratio of 68Zn/66Zn. Some proteins present in the sample can exchange significant amounts of spiked zinc under denaturing and non-denaturing conditions, indicating that (a) zinc–protein complexes are more labile or (b) that Zn-proteins present in the sample are able to bind additional zinc. This exchange is, as expected, especially noticeable in F3 and F4 under both IEF conditions. BSA, CA and SOD migrate in these fractions; the major protein involved in this exchange is probably BSA, which has the least complex stability. Iron, in contrast to these two elements, shows a tendency to bind to high- and low-molecular weight molecules present in the ampholyte mixture (panels C and E of Fig. 5 and 6). As soon as protein is added under denaturing conditions it preferably forms high-molecular weight species, which are only partially recovered in F4 once IEF has been performed, indicating potential losses. Under non-denaturing conditions spiked iron binds to some high molecular weight species present in F2 and F3. The traces for 56Fe and 57Fe in corresponding fractions show that no iron-containing protein is present in these fractions; therefore the iron-binding molecule must be associated with the ampholyte mixture.
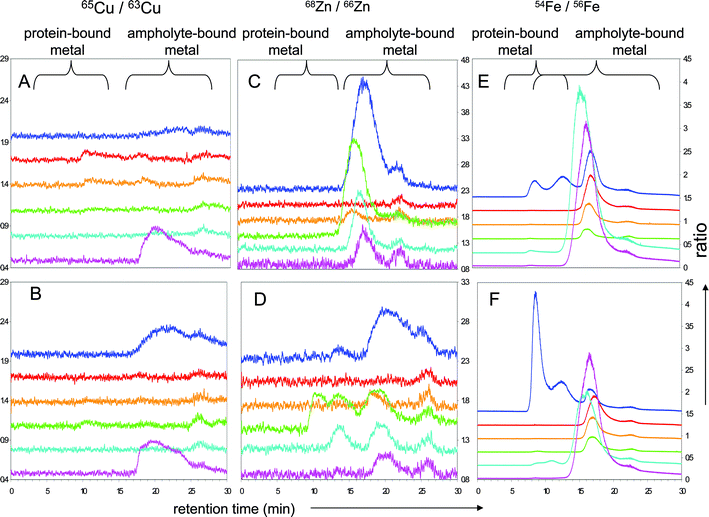 | ||
| Fig. 5 Isotopically-labelled denaturing sIEF fractions separated by SEC-ICP-MS of protein-less (A, C, E) and metalloprotein solutions (B, D, F) spiked with 100 μg L−1 each of isotopically enriched 65Cu, 68Zn and 54Fe; isotopic ratios for 65Cu/63Cu (panels A and B),68Zn/66Zn (panels C and D) and 54Fe/Fe56 (panels E and F), off-set between each fraction is 0.3; retention time windows for protein-bound metal and ampholyte -bound metal are labelled. Traces correspond to: unfractionated mixture (‘neat’): blue trace, F1: red, F2: orange, F3: green, F4: cyan, F5: mauve. Panels A, C and E correspond to protein-less controls, and panels B, D and F to protein-containing experiments. | ||
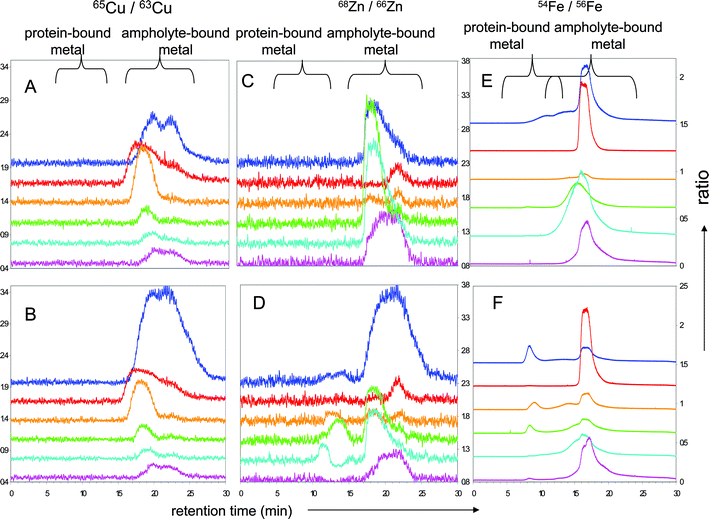 | ||
| Fig. 6 Isotopically-labelled non-denaturing sIEF fractions separated by SEC-ICP-MS of protein-less (A, C, E) and metalloprotein solutions (B, D, F) spiked with 100 μg L−1 each of isotopically enriched 65Cu, 68Zn and 54Fe; isotopic ratios for 65Cu/63Cu (panels A and B), 68Zn/66Zn (panels C and D) and 54Fe/Fe56 (panels E and F), off-set between each fraction is 0.3; retention time windows for protein-bound metal and ampholyte -bound metal are labelled. Traces correspond to: unfractionated mixture (‘neat’): blue trace, F1: red, F2: orange, F3: green, F4: cyan, F5: mauve. | ||
Finally, we have evaluated microsol IEF for fractionation of a biological metalloproteome, using small volumes of protein sample. Both the denaturing and our newly-developed non-denaturing protocols were used to reproducibly (data not shown) fractionate liver cytosolic fractions obtained from sheep liver homogenates. Fig. 7 shows the 1D gel analysis of the resulting fractions; bands were successfully visualised that were not distinguishable in the unfractionated sample (data not shown). As observed with the protein standards, proteins focussed preferentially into more alkaline fractions (F3, F4 and F5) under denaturing conditions, whereas the bands appear more distributed along the pH gradient under non-denaturing conditions. SEC-ICP-MS analysis of the pH fractions allowed us to determine the presence of potentially intact copper, zinc or iron metal–protein complexes with the two procedures (Table S4, S5 and S6, respectively† ). Retention times (tR) compatible with the presence of high molecular weight components have been determined (tR ∼ 9–10 min) for copper and iron under both denaturing and non-denaturing conditions (Table S4 and S6† ). Zinc has been found at longer retention times only under non-denaturing conditions, whereas its association with lower molecular weight components (tR ∼ 18 min) can be hypothesised under denaturing conditions (Table S5† ). This supports the suggestion that zinc binding to protein is weaker than other types of metal–protein interactions in the sample under examination. On the basis of the correlations shown with standard proteins (Fig. 3 and 4), from the metal determinations we can identify both under denaturing and non-denaturing conditions some pH fractions which are likely to contain metal–protein complexes that remain intact after sIEF fractionation, and for which additional characterization using 1D gel LC-MS or 2D gel proteomic analysis38 will provide the necessary characterization of the molecular entities involved.
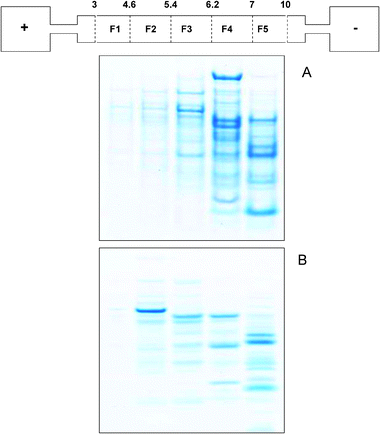 | ||
| Fig. 7 1D SDS gels of liver cytosolprotein fractions obtained from denaturing solution IEF (panel A) and non-denaturing IEF (panel B) with a schematic of the ZOOM® IEF apparatus. pH fractions as follow: 3–4.6 (F1), 4.6–5.4 (F2), 5.4–6.2 (F3), 6.2–7 (F4), 7–10 (F5). | ||
Conclusions
This work is part of a wider project aimed at the characterization of metal–protein interactions through the development of compatible hyphenatable techniques. The specific scope of this research was principally to investigate a separation procedure able to maintain the integrity of different kinds of metalloproteins that would be compatible with subsequent proteomic and trace elemental analysis techniques in order to unequivocally characterize the metalloproteome. Both denaturing and non-denaturing microsol IEF conditions can be applied to separate intact and specific metal–protein complexes. While we anticipated that the metal would be detected in association with metalloproteins following sIEF separation under non-denaturing conditions, our data show that even on denaturing sIEF, specific metal–protein complexes remained intact, giving the researcher an increased scope of separation options. The sIEF fractionation approaches also proved to be successful in the case of a complex biological sample, although further improvements may be required to identify the molecular components and the metals bound to them, especially if a downstream gel electrophoretic approachis to be used, since major effects on the detection of metal–protein complexes using gel electrophoretic separations have to be considered.12 Finally, this study presents a versatile methodology which can be considered a powerful pre-fractionation procedure to enrich the metalloprotein component of biological samples, especially when the non-denaturing approach is carried out, while the denaturing protocol also offers a reliable downstream high-resolution first-dimension step in a multidimensional platform aimed at the characterization of metal–protein complexes.Acknowledgements
The authors acknowledge the UK Engineering and Physical Sciences Research Council for support (EPRSC GRS98689/01 to J. F. and GR/S98696/01 to J. E. T.-O.). K. S. gratefully acknowledges a Royal Thai Scholarship and C. L. D. thanks the College of Physical Sciences at the University of Aberdeen for the award of a PhD studentship. We are grateful to Dr Luca Ronda, Department of Biochemistry and Molecular Biology, University of Parma, for critically reading the manuscript and for helpful comments.References
- J. L. Gómez-Ariza, T. García-Barrera, F. Lorenzo, V. Bernal, M. J. Villegas and V. Oliveira, Anal. Chim. Acta, 2004, 524, 15–22 CrossRef CAS.
- J. Szpunar, Analyst, 2005, 130, 442–465 RSC.
- A. Sanz-Medel, Anal. Bioanal. Chem., 2008, 391, 885–894 CrossRef CAS.
- A. Sanz-Medel, M. Montes-Bayon, M. del Rosario Fernandez de la Campa, J. R. Encinar and J. Bettmer, Anal. Bioanal. Chem., 2008, 390, 3–16 CrossRef CAS.
- M. R. Binet, R. Ma, C. W. McLeod and R. K. Poole, Anal. Biochem., 2003, 318, 30–38 CrossRef CAS.
- J. S. Becker, S. F. Boulyga, J. S. Becker, C. Pickhardt, E. Damoc and M. Przybylski, Int. J. Mass Spectrom., 2003, 228, 985–997 Search PubMed.
- J. S. Becker, M. Zoriy, J. S. Becker, C. Pickhardt, E. Damoc, G. Juhacz, M. Palkovits and M. Przybylski, Anal. Chem., 2005, 77, 5851–5860 CrossRef CAS.
- C. C. Chéry, D. Gunther, R. Cornelis, F. Vanhaecke and L. Moens, Electrophoresis, 2003, 24, 3305–3313 CrossRef.
- H. Chassaigne, C. C. Chéry, G. Bordin, F. Vanhaecke and A. R. Rodriguez, J. Anal. At. Spectrom., 2004, 19, 85–95 RSC.
- C. C. Chéry, K. De Cremer, E. Dumont, R. Cornelis and L. Moens, Electrophoresis, 2002, 23, 3284–3288 CrossRef CAS.
- C. C. Chéry, L. Moens, R. Cornelis and F. Vanhaecke, Pure Appl. Chem., 2006, 78, 91–103 CrossRef CAS.
- A. Raab, B. Pioselli, C. Munro, J. Thomas-Oates and J. Feldmann, Electrophoresis, 2009, 30, 303–314 CrossRef CAS.
- E. Brunner, C. H. Ahrens, S. Mohanty, H. Baetschmann, S. Loevenich, F. Potthast, E. W. Deutsch, C. Panse, U. de Lichtenberg, O. Rinner, H. Lee, P. G. Pedrioli, J. Malmstrom, K. Koehler, S. Schrimpf, J. Krijgsveld, F. Kregenow, A. J. Heck, E. Hafen, R. Schlapbach and R. Aebersold, Nat. Biotechnol., 2007, 25, 576–583 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates 3rd, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- P. G. Righetti, A. Castagna, B. Herbert, F. Reymond and J. S. Rossier, Proteomics, 2003, 3, 1397–1407 CrossRef CAS.
- M. Bier, N. B. Egen, T. T. Allgyer, G. E. Twitty and R. A. Mosher, in Peptides: Structure and Biological Functions, ed. E. Gross and J. Meienhofer, 1979 Search PubMed.
- H. Y. Tang and D. W. Speicher, Expert Rev. Proteomics, 2005, 2, 295–306 Search PubMed.
- A. Thorsell, E. Portelius, K. Blennow and A. Westman-Brinkmalm, Rapid Commun. Mass Spectrom., 2007, 21, 771–778 CrossRef CAS.
- X. Zuo and D. W. Speicher, Anal. Biochem., 2000, 284, 266–278 CrossRef CAS.
- P. G. Righetti, E. Wenisch and M. Faupel, J. Chromatogr., A, 1989, 475, 293–309 CrossRef CAS.
- P. G. Righetti, E. Wenisch, A. Jungbauer, H. Katinger and M. Faupel, J. Chromatogr., A, 1990, 500, 681–696 CrossRef CAS.
- B. Herbert and P. G. Righetti, Electrophoresis, 2000, 21, 3639–3648 CrossRef CAS.
- X. Zuo and D. W. Speicher, Proteomics, 2002, 2, 58–68 CrossRef CAS.
- P. G. Righetti, A. Castagna, P. Antonioli and E. Boschetti, Electrophoresis, 2005, 26, 297–319 CrossRef CAS.
- M. J. Han, M. Herlyn, A. B. Fisher and D. W. Speicher, Electrophoresis, 2008, 29, 695–705 CrossRef CAS.
- M. R. Richardson, S. Liu, H. N. Ringham, V. Chan and F. A. Witzmann, Electrophoresis, 2008, 29, 2637–2644 CrossRef CAS.
- H. Zhong, D. Yun, C. Zhang, P. Yang, H. Fan and F. He, Electrophoresis, 2008, 29, 2372–2380 CrossRef CAS.
- J. C. Tran and A. A. Doucette, J. Proteome Res., 2008, 7, 1761–1766 CrossRef CAS.
- T. Q. Shang, J. M. Ginter, M. V. Johnston, B. S. Larsen and C. N. McEwen, Electrophoresis, 2003, 24, 2359–2368 CrossRef CAS.
- Z. Ibrahim, W. A. Ahmad and A. B. Baba, Braz. Arch. Biol. Technol., 2001, 44, 223–225 Search PubMed.
- F. Arnesano, L. Banci, I. Bertini, F. Capozzi, S. Ciofi-Baffoni, S. Ciurli, C. Luchinat, S. Mangani, A. Rosato, P. Turano and M. S. Viezzoli, Coord. Chem. Rev., 2006, 250, 1419–1450 CrossRef CAS.
- T. Arakawa and S. N. Timasheff, Biochemistry, 1982, 21, 6536–6544 CrossRef CAS.
- M. Conti, M. Galassi, A. Bossi and P. G. Righetti, J. Chromatogr., A, 1997, 757, 237–245 CrossRef CAS.
- Y. Shimazaki, H. Ohnishi, S. Matsuura and T. Manabe, Biochim. Biophys. Acta, Gen. Subj., 2002, 1571, 245–248 Search PubMed.
- P. G. Righetti, J. Chromatogr., A, 2004, 1037, 491–499 CrossRef CAS.
- C. Simó, A. Citterio and P. G. Righetti, Electrophoresis, 2007, 28, 1488–1494 CrossRef CAS.
- C. Simó, M. E. Mendieta, P. Antonioli, R. Sebastiano, A. Citterio, A. Cifuentes and P. G. Righetti, Electrophoresis, 2007, 28, 715–723 CrossRef CAS.
- J. A. Rodrigues, F. J. Lopez-Baena, F. J. Ollero, J. M. Vinardell, R. Espuny Mdel, R. A. Bellogin, J. E. Ruiz-Sainz, J. R. Thomas, D. Sumpton, J. Ault and J. Thomas-Oates, J. Proteome Res., 2007, 6, 1029–1037 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 and additional discussion of it, and six tables of results (Tables S1 to S6) and additional discussion of those results to underpin Fig. 3 and 4 in the main text. See DOI: 10.1039/b903607e |
| ‡ These two authors made equal contributions to the work presented. |
| This journal is © The Royal Society of Chemistry 2009 |