Metal trafficking: from maintaining the metal homeostasis to future drug design
Lalla Aicha
Ba
,
Mandy
Doering
,
Torsten
Burkholz
and
Claus
Jacob
*
Division of Bioorganic Chemistry, School of Pharmacy, Saarland University, PO Box 151150, D-66123 Saarbruecken, Germany. E-mail: c.jacob@mx.uni-saarland.de; Fax: +49 (0)681![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3023464; Tel: +49 (0)6813023129
3023464; Tel: +49 (0)6813023129
First published on 11th June 2009
Abstract
The diverse proteins and enzymes involved in metal trafficking between and inside human cells form numerous transport networks which are highly specific for each essential metal ion and apoprotein. Individual players include voltage-gated ion channels, import and export proteins, intracellular metal-ion sensors, storage proteins and chaperones. In the case of calcium, iron and copper, some of the most apparent trafficking avenues are now well established in eukaryotes, while others are just emerging (e.g. for zinc, manganese and molybdenum). Chemistry provides an important contribution to many issues surrounding these transport pathways, from metal binding-constants and ion specificity to metal-ion exchange kinetics. Ultimately, a better understanding of these processes opens up opportunities for metal-ion-related therapy, which goes beyond traditional chelate-based metal ion detoxification.
 Lalla Aicha Ba Lalla Aicha Ba | Lalla Aicha Ba studied chemistry at the University Paul Verlaine, Metz, France, where she obtained her PhD in October 2007 in the laboratory of Gilbert Kirsch. Afterwards she joined the group of Denyse Bagrel to investigate new synthetic selenium-containing compounds as potential drugs for Alzheimer’s disease. In August 2008, Lalla Aicha became a senior research fellow in the group of Claus Jacob at the University of Saarland. She works on the design and bio-evaluation of new oxygen, sulfur, selenium and tellurium compounds for their potential use as anti-cancer drugs. |
 Mandy Doering Mandy Doering | Mandy Döring studied chemistry at the University of Saarland, where she received her first degree (Diplom) in 2007. She joined Claus Jacob’s group in Bioorganic Chemistry as part of her undergraduate project and started to work on multifunctional, biologically active redox-catalysts. After graduation, Mandy continues to work on the design of multifunctional redox-catalysts with regard to potential applications in medicine and agriculture in Claus Jacob’s research group. |
 Torsten Burkholz Torsten Burkholz | Torsten Burkholz studied Chemistry at the University of Saarland, Saarbruecken, Germany. He obtained his first degree in Chemistry (Diplom) in June 2006. In October 2006, he joined the Division of Bioorganic Chemistry and is currently conducting research as part of his PhD thesis entitled Oxidative stress and electrochemical procedures for surface decontamination in dialysis. This project is supported by Fresenius Medical Care, Bad Homburg, Germany. |
 Claus Jacob Claus Jacob | Claus is a junior professor of bioorganic chemistry at the School of Pharmacy, University of Saarland, Germany. Claus obtained his BSc in Chemistry from the University of Leicester in 1993 and his DPhil from Oxford in 1997 working with Allen Hill FRS. From 1996 to 1999, he spent time as a Postdoc with Bert Valle at Harvard Medical School. In 1998, Claus obtained a Magister degree in philosophy from the University of Hagen. In 1999, he took up the position of lecturer in inorganic chemistry at the University of Exeter, moving to Saarbruecken in 2005. Claus has a particular interest in the chemistry underlying biochemical redox events, most notably reactive sulfur species (RSS). |
1. Introduction
Metal trafficking between and inside living cells is one of the most important and, at the same time, fascinating areas of Metallomics. We nowadays know that the journey of (most) metal ions between and within cells is tightly controlled at virtually every step of this journey: ions such as calcium, copper, iron or zinc do not simply enter and leave cells by chance—they are imported and exported in a highly controlled manner. Here, each individual essential metal is able to rely on its own transport system(s). Voltage-gated ion channels, pumps, import and export proteins (often called ‘transporters’), endo- and exocytosis all play an important part in these processes. The complexity of metal ion import into and export from cells is mirrored by systems responsible for the maintenance of an appropriate metal ion homeostasis inside the cell and between different cellular compartments. There are proteins able to sense metal ion concentrations, to store and transport as well as to release metal ions into the cytosol. Ultimately, the intracellular concentration of ‘free’ (i.e. readily available) essential metal ions, such as copper, manganese and zinc, is incredibly small. The inclusion of metal ions into de novo synthesized proteins and enzymes therefore does not occur spontaneously or by chance: it represents a highly regulated process and often involves proteins able to escort (‘chaperone’) metal ions to the respective sites where they are required by specific apoproteins.Not surprisingly, the task of regulating the various metal ions inside the cell is far from trivial and requires the interplay of numerous, often quite sophisticated, proteins and enzymes (see Table 1). We must remember that there are many different metal ions present within a human cell, among them various (redox active) transition metal ions. These ions occupy a multitude of pre-destined sites in peptides , proteins and enzymes—and some of them also attach to membranes, DNA and smaller molecules (such as to adenosine triphosphate, ATP). To traffic so many different metal ions to different sites is a truly Herculean task, and there is little room for error. If the incorrect metal ion ends up at the ‘active’ site of an enzyme, the latter is likely be inactive. This has been demonstrated in the past in vitro, for instance for Cd2+ inhibited zinc enzymes. In contrast, metal ion promiscuity of an (apo-)protein which may result in misincorporation of the wrong metal ion seems to be rare in vivo. One example is iron-loaded mitochondrial superoxide dismutase 2(SOD2) in higher organisms. Unlike the ‘normal’ manganese enzyme (Mn-SOD2) found in these organisms, Fe-SOD2 is inactive. This misincorporation event may serve a regulatory purpose in response to elevated iron levels, although the precise role and extent of this misincorporation, if any, especially in eukaryotes including humans is largely unknown.1
| Trafficking component | Function | Example |
|---|---|---|
| a Please note that certain proteins possess chaperone and insertase activities, while others only escort, but do not insert the metal ion into the target apoprotein. | ||
| Transporter | Metal ion transport across membranes | CTR1 (copper) |
| DCT1 (divalent cations) | ||
| ZIP (zinc) | ||
| Channel | Metal ion transport across membranes | Voltage gated Ca2+ channel (allows entry of calcium into the cell) |
| Exchanger | Formal exchange of two metal ions across a membrane | Na+/Zn2+ exchanger (exchanges sodium and zinc across outer membrane, may import and expel zinc from cells) |
| Endocytosis | Import of metal ion transportprotein into cell | Transferrin (import of protein-bound iron into cell) |
| Chaperonea | Metal ion escorting inside cell, often targeted towards intracellular compartments | CCS (escorts and inserts copper into apo-SOD1) |
| Frataxin (escorts iron to ferrochelatase) | ||
| Metalloinsertasea | Metal ion insertion into apoproteins | CCS (escorts and inserts copper into apo-SOD1) |
| Sco1/Sco2 (insert copper into cytochrome c oxidase) | ||
| Chelatase | Metal ion insertion into small molecules, co-factors | Ferrochelatase (inserts iron into protoporphyrin IX) |
| Pump | Metal ion transport across membranes | Ca2+-ATPase pump (calmodulin regulated, exports Ca2+ out of the cell) |
Misincorporation appears to be more widespread, however, in the realm of metal transport systems, where toxic metals enter the cell by molecular mimicry, i.e. by using transport systems designed to traffic essential metal ions. The divalent cation transporter DCT1, for instance, may transport iron, zinc, manganese and copper, but also cobalt, cadmium, nickel and lead. Similarly, the phosphate transporter may be used by structurally similar vanadate and arsenate to gain entry into the cell, whilst the sulfate transporter is prone to being ‘abused’ by chromate, selenate and molybdate.2
Apart from misincorporation, ‘adventitious’ metal binding of metal ions to proteins at inhibitory sites or sites not designed for metal binding at all, such as active sites containing catalytic cysteine and/or histidine residues, is of major concern. There are various examples of ‘inhibitory’ or ‘adventitious’ metal binding and reactivity, ranging from Zn2+ binding to an inhibitory site in carboxypeptidase A (CPA)3,4 and to cysteine residues in hydrolytic or redox active enzymes, to ‘free’ iron and copper ions participating in Fenton-type reactions.5 It is therefore important that the amount of readily available, ‘free’ metal ions in the cell is controlled tightly—and in some instances, such as copper and zinc, is approximately zero.6 This matter will be discussed in more detail in sections 2.4 and 2.5.
Not surprisingly, disturbed metal trafficking has been implicated in a range of human diseases, such as Wilson and Menkes disease, some of which will be discussed later on.7 It is therefore essential for us to understand how such a complex regulatory network of cellular metal ion maintenance may function. During the last ten-to-fifteen years, considerable progress has been made in this area of research, in many cases fuelled by genomics , which has provided insight into the various metal transporters and their presence in pro- and eukaryotes. At the same time, new and refined analytical techniques have become available to follow metal ions within the cell, and to identify and characterize proteins involved in metal trafficking.
While most of the groundbreaking studies have been performed in bacteria and in yeast (Saccharomyces cerevisiae) as a model system for eukaryotes, evidence of metal trafficking systems in humans is increasing rapidly. We will therefore focus on metal trafficking in humans, and refer to yeast, bacteria or plants in cases where the human systems are not yet fully understood. This will allow us to briefly consider some of the issues surrounding the cellular trafficking of calcium, iron, copper, manganese, zinc and molybdenum. Several of these individual trafficking systems have recently been reviewed, yet a comprehensive overview of the various metal ions and their respective systems is largely missing.
Interestingly, intracellular metal trafficking—and our knowledge thereof—has already led to some rather promising metal (trafficking) related therapeutic approaches. We will demonstrate the pharmaceutical potential residing therein by presenting a few selected highlights, such as chelate-assisted photodynamic therapy (PDT). Throughout, we will focus on examples only. Hence, this review should not be considered as a comprehensive or even complete treatise on metal ion transport in human cells.
2. Components of metal ion trafficking
2.1 Metal binding constants
Metal trafficking in vivo is, in the true meaning of the word, a highly ‘complex’ matter. It may be dissected and considered at various layers of complexity, starting from basic metal–ligand interactions, to complicated apoprotein–chaperone interactions. Without attempting to reduce such intricate interactions to the level of basic inorganic chemistry, it is worthwhile to consider the chemistry behind metal–protein interactions first.Interestingly, peptides , proteins and enzymes do not differ per se from small inorganic metal complexes. Whilst being considerably larger in size and molecular weight, the major concepts of (inorganic) complex chemistry, such as the hard and soft acid and base concept (HSAB), coordination numbers for metal ions and ligand field theory, still apply. In fact, metal binding constants provide an excellent tool to rationalize some of the basic findings of metal trafficking.† For instance, they do, at least in part, explain why certain proteins bind the ‘correct’ metal ions, such as copper and zinc, whilst at the same time avoiding others, such as calcium and magnesium. This specificity is hardly obvious—numerous inorganic complexes, such as EDTA complexes, feature binding constants which are rather similar for Cu2+, Zn2+, Ca2+ and Mg2+, and the use of the correct medley of hard oxygen, intermediate nitrogen and soft sulfur (and even selenium) ligands is key to selectivity.
Such metal–protein binding constants play a key role in several important biological processes, in particular in pathways responsible for sensing and controlling intracellular levels of ‘free’ metal ions. Here, comparably weak metal ion binding with the option of reversible association and dissociation is important (see section 3).
In the case of the calcium sensor calmodulin, for instance, synergistic binding of two to four labile, ‘free’ Ca2+ ions to the four EF-hands of the sensor triggers a change in the calmodulin structure from the inactive to the active form. The latter then binds to various proteins and enzymes and activates Ca2+-ATPase pumps, which in turn lower intracellular Ca2+ levels. Once these levels have fallen below 10−7 to 10−8 M, Ca2+ ions begin to dissociate from the calcium–calmodulin complex. This returns calmodulin to its metal-free, inactive form, and the proteins and enzymes activated by calmodulin switch-off. The Ca2+-ATPase pumps, in particular, are also turned off, which ensures that intracellular Ca2+ levels do not fall below a critical level required by the cell (around 1.0–5.0 × 10−8 M).8 If Ca2+ levels rise again, binding to calmodulin occurs and the regulatory feedback loop is triggered once more.
The binding of Ca2+ ions to calmodulin with binding constants9 (for the four individual Ca2+ ions) of 48 × 109 M−1 to 403 × 109 M−1 is a crucial factor in this process. It determines the intracellular Ca2+ concentration at which the calcium-loaded, activated form of calmodulin is formed and Ca2+ export is initiated. If this binding constant would be too high, considerably lower intracellular levels of labile Ca2+ would result—if the binding constant would be too low, a Ca2+ overload inside the cell may occur. While the calcium–calmodulin interaction does not fully describe the intracellular calcium homeostasis—there are several additional players involved, such as the calsequestrin calcium storage proteins—the calcium–calmodulin system with its ‘fine-tuned’ binding constants forms a crucial part of it.
2.2 Pre-formed metal binding sites in proteins and enzymes
In addition to metal binding constants, proteins and enzymes often contain pre-formed metal binding sites, which may be formed by the action of special molecular ‘protein folding’ chaperones. Because of size and coordination geometry, such sites may only hold the ‘correct’ metal ion. Nonetheless, there are significant exceptions, such as the above-mentioned transporter DCT1, SOD2 and the anion transporters for phosphate and sulfate. In fact, metal poisoning by toxic metals such as lead and aluminium is the result of binding the ‘wrong’ metal, of misincorporation due to an override in binding site specificity.The example of aluminium toxicity illustrates this rather nicely. Al3+ ions are usually not present in significant amounts in humans and their diet. If, in exceptional cases, Al3+ ions enter the body, they replace (primarily) Fe3+ ions from the iron transporter transferrin (see section 3.3). This (and related) misincorporation events allow Al3+ to travel within the human body, cross the blood–brain barrier and to accumulate in glial and neuronal cells.10
In the case of some proteins, such as the metallothioneins (MTs), promiscuity is actually beneficial. These proteins contain metal binding sites which can bind a variety of different metal ions (e.g. Zn2+, Cd2+) and hence are able to sequester toxic metal ions and exchange them for zinc (see section 4). While MTs contain kinetically labile metal ions prone to rapid exchange, most metal ions in proteins and enzymes appear to be bound kinetically stable, i.e. adventitious metal ion exchange, even if favoured thermodynamically, is very slow.
Finally, it should be pointed out that many proteins and enzymes contain metal ions bound to small organic molecules, such as porphyrins and pterins. These binding sites differ somewhat from the sites formed by the amino acid residues of the peptide chain. Here, several distinct processes need to be considered, including the biosynthesis of such small molecules, insertion of a metal ion into the small molecule and finally insertion of the small (metal ion bearing) molecule into the target protein. These processes are fairly complex and involve several ‘assisting’ proteins and enzymes, such as chelatases and (molecular) chaperones. Section 3.5 will discuss aspects of molybdenum insertion into pterins while section 5.3 will discuss some of these issues related to porphyrin synthesis.
2.3 Chaperones
Binding constants and coordination geometries are not enough, however, to avoid erroneous metal ion incorporation.“A naïve expectation was that each protein would pluck the correct element from mixed-metal cell solutions and exclude all others solely based on its metal affinities. However, the full suite of protein–ligand chemistries and coordination geometries is inadequate for such perfect metal partitioning in one step”.11
Although there are exceptions, aluminium toxicity, Fe-SOD2 and similar examples illustrate the importance of this statement rather nicely. It is therefore important to add one more aspect to metal trafficking, namely the metallochaperone and ion-inserting proteins, which are able to escort the ‘correct’ metal ion to the site of incorporation and subsequently insert it into the appropriate apoprotein. Although this process—or processes—are rather complicated, still little understood and often highly diverse depending on the metal ion and even the individual apoproteins in question, they form an unifying link in the metal trafficking chain which, although long-missing, appears to explain some of the mysteries behind metal ion transport, homeostasis and insertion into proteins. Such chaperones and ‘metalloinsertases’ are known or emerging for copper, manganese, iron, zinc and molybdenum proteins and will be discussed in section 3.‡
Interestingly, the reliance of apoproteins on specific chaperones and metal inserting proteins implies that ‘free’ metal ions are either not available directly for activation, or, as has been discussed in detail for SOD enzymes, that ‘free’ metal ions are not able to simply insert themselves into certain apoproteins.6,7
2.4 The labile metal ion pool
As already mentioned, most metal ions, including essential ones, become toxic at higher concentrations and therefore cannot roam freely inside the cell. There are too many small molecules, proteins and enzymes, which may be affected by uncontrolled, adventitious metal ion interactions. This problem is particularly apparent for ions with (second) messenger functions (such as Ca2+, Mg2+), ions able to inhibit proteins and enzymes (e.g. Zn2+, which interferes with cysteineproteins), and redox active ions (e.g. Cu+/2+, Fe2+/3+). Intracellular concentrations of such metal ions, especially in their ‘free’ state, need to be controlled tightly, and, as already mentioned, there are some indications that certain metal ions, such as copper or zinc, do not occur in a ‘free’ state at all.6,12In order to specify the concept of ‘free’ metal ions, we can roughly distinguish between three different forms of metal ions inside the cell: (a) metal ions bond to proteins and enzymes; (b) metal ions bound to small molecules; and (c) metal ions in the form of aquo-complexes. Metal ions in proteins and enzymes are tightly bound with binding constants easily exceeding 1010 M−1. A classic example, carbonic anhydrase, binds Zn2+ with a binding constant of 1012 M−1.13 Only (b) and (c) may therefore count as readily available, labile or ‘free’ metal ions (see section 3).
During the last decade, various attempts have been made to estimate the extent of the pool of labile metal ions inside cells, either by direct measurements or by calculations based on metal–protein binding constants. Table 2 provides a summary of estimated intracellular concentrations of metal ions. Although these values are often estimates only, they indicate that the extent of labile metal ion pools varies widely (even if accounting for different biological species and cell types), and that in the case of copper and zinc in particular, there appear to be few ‘free’ metal ions inside the eukaryotic cell.§ It should be emphasized that Table 2 contains values for labilemetal ion concentrations. The corresponding total metal ion concentrations, which include protein- and enzyme-bound metal ions, are generally orders of magnitude higher. For instance, Finney and O’Halloran have provided an estimate of total metal ion concentrations for Escherichia coli which is more than 10−2 M for potassium and magnesium, around 10−4 M for iron, calcium and zinc and approximately 10−5 M for copper, manganese, molybdenum and selenium.14
| Metal ion | Labile ion concentration | Organism/cell type | References |
|---|---|---|---|
| a The total concentration of labile metal ions are provided regardless of oxidation state. b Please note that such concentrations may on average correspond formally to less than one free copper ion per cell. | |||
| Potassium | 10−1 M | 123 | |
| Sodium | 10−2 M | 123 | |
| Magnesium | 10−3 M | 123 | |
| Irona | 5 × 10−6 M | Rat hepatocyte (cytosol) | 18 |
| 11.8 × 10−6 M | Rat hepatocyte (nucleus) | ||
| 9.8 × 10−6 M | Rat hepatocyte (mitochondria) | ||
| Manganese | 10−7 M | 123 | |
| Calcium | 1–5 × 10−8 M | Eukaryote (cytosol) | 8 |
| Zinc | 10−11 M | 48, 123, 124 | |
| 10−12 M | Eukaryotic cells | ||
| <10−9 M (i.e. none) | E. coli (bacteria) | ||
| Coppera | 10−15 M | 6, 123 | |
| <10−18 Mb | Yeast | ||
Surprisingly, the rather damaging iron ion, which may become engaged in Fenton-type radical formation chemistry, seems to be present rather readily in its ‘free’ form, albeit in low micromolar concentrations only. Indeed, the labile iron pool (LIP) inside living cells is large enough to be measured, for instance by using (fairly) specific fluorescent dyes, such as calcein and phen-green, and in combination with laser scanning microscopy.15–18 For instance, (rat) hepatocytes contain an average of 5.0 μM of chelatable iron, with 11.8 μM in the hepatocyte nucleus and 9.8 μM in the mitochondria.18¶ These findings confirm the existence of a rather significant amount of labile iron in the mammalian cell even under normal conditions (when compared to other metal ions). They also point towards a certain compartmentalization of the LIP, which is of particular interest when considering possible (mis-)incorporation of iron into proteins (see sections 3.2 and 3.3). One should also note that the LIP is not constant but, for instance, increases significantly under conditions of oxidative stress (OS).
2.5 Metal ion release during oxidative stress
Inside the living cell, metal binding and redox events enter into an exciting, and highly complicated interplay, often resulting in extensive cellular signalling pathways and control networks.19,20 These interactions involve redox control of metal binding as well as various forms of metal-controlled redox activity. The biochemical condition of OS exemplifies these interactions. It also provides insight into changes of intracellular pools of labile metal ions, which in turn form the basis for the trafficking and rescuing systems described in sections 3 and 4.OS is characterized by a significant increase in intracellular oxidants (in particular reactive oxygen species, ROS), which is often accompanied by a loss in antioxidant defence. One of the key events linked to this kind of disturbed intracellular redox balance is the activation of oxidant-induced cellular signalling pathways.|| OS is found in many human diseases, including various neurodegenerative and autoinflammatory diseases, rheumatoid arthritis, diabetes, glaucoma and several types of cancer. It is also present at sites of inflammation, accompanies bacterial and viral infections and increases as part of the normal ageing process. An increase of ROS above normal levels changes the intracellular redox state. This affects in particular redox sensitive proteins and enzymes, many of which contain metal ions.
One of the most devastating events caused by ROS is the oxidation of ligands and subsequent metal ion release. Cysteine, methionine and inorganicsulfide (S2−) ligands in proteins and enzymes are oxidized, resulting in disulfides and higher sulfur oxidation states.** Metals released during OS include zinc, but also the redox active iron and copper ions. The released metal ions initially end up in the respective labile ion pools, until the cell is able to mount an antioxidant defence, which often also includes a metal sequestering component (see sections 3 and 4).
Importantly, the redox behaviour of metal ions depends critically on their coordination state and accessibility inside a metal-complex. Iron and copper ions tightly bound to proteins and enzymes exhibit neither the correct redox potentials nor are they accessible for ‘dangerous’ reactions, such as the ones of Fe2+ and Cu+ ions with hydrogen peroxide. This type of reaction is known as the Fenton reaction and results in the formation of highly damaging hydroxyl radicals (HO˙), as illustrated for iron in eqn (1). Interestingly, labile iron and copper ions appear to participate readily in Fenton type reactions. These processes may turn catalytic in form of a combined Fenton and Haber–Weiss reaction (eqn (3)).
| Fe2+ + H2O2 → Fe3+ + HO− + HO˙ | (1) |
| Fe3+ + O2˙− → Fe2+ + O2 | (2) |
| Overall: O2˙− + H2O2 → O2 + HO− + HO˙ | (3) |
Uncontrolled metal ion release during OS is not limited to iron and copper. Increased concentrations of labile ions such as zinc and calcium may also, at least temporarily, cause inconveniences, for instance by binding to active site cysteine residues in enzymes or by interfering with cellular signalling pathways.
The question of whether OS triggers adventitious metal ion release, or if accidentally released metal ions are a cause of OS, is often a ‘chicken and egg’ question, which is rather difficult to answer. UVA radiation, for instance, is known to attack a range of proteins, including iron-containing proteins such as ferritin, which are damaged and release iron ions.23 While UVA radiation may therefore be seen as an initial ‘trigger’ of events, the subsequent increase in the labile iron pool is responsible for a sharp increase in ROS formation, especially of HO˙ radicals.
3. Towards comprehensive metal trafficking networks
As already mentioned in the Introduction, metal trafficking networks are highly complex and, in most cases, not yet fully understood. Several examples have, however, recently been emerging, and it is now possible to map out some of the key elements of such trafficking networks. In most cases, the maintenance of the intracellularmetal ion homeostasis is combined with specific metallochaperones. Copper, for instance, enters the cell via a set of (more or less specific) transporters, which pass on the ion to a series of proteins, some of which escort the metal to its site and apoprotein of destination, where copper is then inserted into the active site of the destined protein (for details see section 3.1). Copper ion insertion is an active process, which often includes direct interactions between the chaperone and the apoprotein. In some cases, complex formation between chaperone and apoprotein and even transient covalent bonds between the two have been observed.6Metal ion insertion may also be accompanied by additional processes, such as redox events. Most importantly, there is no ‘free’ copper involved in any of these processes.The three key steps, i.e. import, escorting and insertion of metal ions are commonly found for several tightly regulated metal ions (copper, zinc, iron, manganese), although individual details differ widely. In order to function properly, such trafficking pathways need to be backed up by sensors of intracellularmetal ions, storage proteins and export systems.
3.1 Transport and insertion of copper ions
The trafficking of copper ions represents perhaps the best-studied system to date, and copper trafficking has coined some of the key terminology in this area of metallomics, such as the name ‘copper chaperone’.24 Basic components of this trafficking system are sketched in Fig. 1. Copper enters the cell via a high-affinity transport system, provided by the cell surface transporters (CTRs), which have been studied extensively in yeast. This system is backed up by low-affinity transporters, such as FET4 (in yeast) and DCT1, which are less specific and may also transport other metal ions. | ||
| Fig. 1 Schematic overview of copper trafficking in the human cell. Copper enters the cell via the high-affinity CTR transporters or the low-affinity DCT1 (the latter may also be misused by toxic metal ions in order to gain entry into the human cell). Once inside the cell, copper is passed on to one of the chaperones, which escort the ion either to the Golgi (ATOX1), apo-SOD1 (CCS) or to the mitochondria (Cox17, Cox19). Upon reaching its destination, copper is either imported into the Golgi and released (using a P-type ATPase denoted as cP, including the Wilson and Menkes disease proteins), inserted into apo-SOD1 or incorporated into cytochrome c oxidase located in the mitochondria (using proteins such as Sco1, Sco2 and Cox11). Most of the copper trafficked appears to be Cu+, yet redox processes and Cu2+ may also play an important part in copper trafficking. Please note that additional or alternative copper transport, escort and insertion pathways may also exist. | ||
Once inside the cell, the destination of the copper ion depends on its trafficking protein. For instance, copper is transported to the Golgi using the ATOX1 protein (also known as HAH1) in humans, an analogue of the ATX1 protein in yeast. It is passed on to copper-transporting P-type ATPase molecules, among them the Wilson and Menkes disease proteins, which enable passage of copper (as Cu+) into the lumen of the Golgi. Once inside the Golgi, copper is released and may bind to its target proteins which form part of the secretory pathway, including extracellular SOD (EC-SOD).1,7,25 It is still vastly unknown if insertion of copper ions into EC-SOD requires the assistance of a ‘metalloinsertase’ protein, as has been shown for cytosolic SOD (SOD1, see below), or if this process occurs spontaneously.
A mutation and subsequent dysfunction of either Wilson or Menkes disease proteins (both have 56% overall identity) has dire consequences. The Wilson disease protein is mostly present in the liver, where it normally assists excretion of copper by transporting copper into the trans-Golgi network. This excretion pathway is impaired in Wilson disease and results in accumulation of copper in many tissues, including the liver, brain, kidneys and in the cornea.2 In Menkes disease, the relevant transport protein is found in tissues other than liver, where it pumps copper into endosomal/lysosomal compartments. Dysfunction in these tissues results in systemic copper deficiency.
Interestingly, copper transfer between proteins, such as ATOX1 and its acceptor (called CCC2 in yeast), appears to occur via a direct interaction of donor and acceptor protein ligands, involving an interconversion of the metal ion’s coordination sphere with 2- and 3-coordinant copper–thiol binding intermediates.7,24
Not all chaperones employ such ‘simple’ transfer of the metal ion by ligand exchange (which does not involve any ‘free’ metal ion). Copper destined for Cu,Zn-SOD (SOD1) is loaded onto the ‘copper chaperone for SOD1’, CCS, a protein which escorts the metal ion to apo-SOD1 (Fig. 1). The transfer of copper between CCS and apo-SOD1 is rather intricate. In brief, CCS consists of three domains which fulfil different functions. Domain I, which is similar to the ATOX1 transporter and contains a MXCXXC copper binding site, binds the copper ion during transport. Domain II bears striking similarities to the SOD1 monomer itself (a mutation of aspartic acid in position 200 to histidine actually endows CCS with SOD-like activity!26). Domain II is responsible for tight binding of CCS to SOD1. During metal ion transfer, CCS replaces one SOD1 monomer from the SOD1 dimeric complex.7 Domain III contains a CXC motif for copper binding and ultimately facilitates the metal insertion into SOD1. The underlying insertion process is rather complicated. It is accompanied by disulfide formation between Cys229 of CCS and Cys55 of SOD1 in an oxygen-dependent reaction and subsequent copper transfer. The CCS/SOD1 heterodimer is cleaved by thiol/disulfide exchange resulting in the Cys55–Cys144 disulfide of SOD1 which is essential for the enzyme’s function.
The interplay between the disulfide redox chemistry on the one hand, and copper loading and transfer on the other, is still not yet fully understood, but may include redox processes involving cysteine residues as well as the copper ion itself.6 It is also unclear when, where and how Zn2+ is loaded onto SOD1. Interestingly, a CCS-independent activation of SOD1 has also been observed, which strongly depends on the presence of reduced glutathione (GSH).1
Yet another system is used to transport copper to its target proteins in mitochondria, such as cytochrome c oxidase (Fig. 1). In this case, the membrane transporter CTR1 passes its copper on to chaperones Cox17 or Cox19. Cox17 binds copper in Cys-ligated polynuclear clusters. Upon reaching the mitochondria, copper for the CuA centre of cytochrome c oxidase is passed on to the apo-enzyme via the Sco1 or Sco2 proteins, which bind copper with a His residue and a CXXXC motif. The CuB site is also supplied by the Cox protein(s), yet probably via a membrane bound Cox11 and Sco1.27,28
The intracellular trafficking network for copper illustrates the complexity of such networks. It also shows that the same metal ion may be trafficked by quite different proteins, depending on the final destination, i.e. the site and apoprotein in question. Furthermore, it should also be noted that there are various backup systems, for instance low-affinity transporters for metal ion import, and GSH as a ‘substitute’ for certain copper chaperones and/or metalloinsertases. It is likely that a similarly diverse situation also exists for several other essential metal ions.
3.2 Manganese
Compared to copper, considerably less is known about the transport of manganese into the cell and trafficking within. Manganese SOD (Mn-SOD, SOD2) is an eukaryotic protein found in the mitochondria of various organisms, including humans, where it serves as a major antioxidant enzyme sequestering the superoxide radical anions generated by the respiratory process. In contrast to copper-containing SOD1, SOD2 does not seem to require a chaperone for metal ion uptake. This is especially surprising, since eukaryotic SOD2 has a strong homology with bacterial SOD—and the latter is an iron enzyme. Nonetheless, iron incorporation into eukaryotic SOD2 is normally not a major issue in vivo (see section 1), and this has been explained with the fact that iron is normally less ‘bioavailable’ in mitochondria when compared to manganese. Only a minor fraction of SOD2 may therefore be associated with iron in eukaryotic cells, as has been discussed for yeast.1This raises the question how manganese is transported to the mitochondria. While specific manganese transporters have been identified in yeast—such as SMF-1, but also PHO84, MTM1 (for SOD2), and a manganese excretion system targeting the Golgi and based on the P-type ATPase PMR1—the case is more complicated and less-understood in humans.25 Manganese may enter the human cell as Mn2+via the DCT1 ( Fig. 2). Its mode(s) of transport inside the cell and its import into the mitochondria are barely understood, however, but may occur via human analogues of the yeast system.
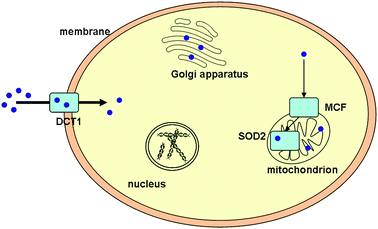 | ||
| Fig. 2 Basic features of manganese trafficking in human cells. Although the cellular pathways of this important metal ion are only just emerging, it appears that manganese enters the cell via DCT1 and relies on MCF to cross the mitochondrial membrane, where it binds to SOD2. | ||
Interestingly, manganese is also required in the Golgi apparatus to activate enzymes such as mannosetransferase and to facilitate proteinglycosylation. Here, a human PMR1 analogue called hSPCA1 has been identified which functions in human manganese and calcium homeostasis. Mutations of hSPCA1 result in defects in proteinglycosylation and lead to a dermatological disorder known as Hailey–Hailey disease.25,29–31
Since such impairments of the manganese trafficking system—or parts thereof—have been associated with serious human disorders, a better understanding of manganese import, export, transport and intracellular compartmentalization is urgently required. Such knowledge will not only shed light on the various transport and escort proteins involved, but may also pave the way to new therapies able to ‘fix’ or bypass such disrupted manganese trafficking pathways. At present, Hailey–Hailey disease is treated by Botulinum toxin type A31 and by photodynamic therapy (PDT, see section 5.3).30
3.3 Iron
As mentioned in section 2.5, free copper and iron ions are frequently associated with the chemistry of OS, in particular with hydroxyl radical (HO˙) generation via the Fenton and Haber–Weiss reactions. It is therefore surprising that the level of labile iron inside the cell is considerably higher than the one for copper—and indeed the one for redox—inactive zinc. As already mentioned, the intracellular labile iron pool can be estimated by using dyes such as calcein and phen-green in combination with laser scanning microscopy—and under normal conditions appears to be in the micromolar range.17The various iron trafficking pathways have been studied extensively during the last decades (Fig. 3). In brief, plasma iron present as Fe2+ becomes bound to its transport protein transferrin upon oxidation to Fe3+, a process assisted by the multifunctional plasma copper proteinceruloplasmin. Transferrin is then imported as a whole into most human cells by endocytosis, a process which may appear to be somewhat cumbersome when compared to other metal transport proteins, yet ensures that iron is not released or available for redox processes during cross-membrane transport. Transferrin contains around 99% of iron in plasma, and the cell provides specific transferrin receptors to control its import. Once inside the cell, transferrin is shipped to acidic compartments where iron is released and apo-transferrin generated. The latter is exported out of the cell viaexocytosis, which allows reuse of the transport protein in the plasma. Alternative transport mechanisms for iron into the cell also exist, such as the DCT1. In the case of hepatocytes, iron may also be transported into the cell by internalization of ferritin. Export of iron (as Fe2+) is facilitated by a separate set of proteins, including the iron-regulatory protein-1 (IREG-1, also called ‘ferroportin’) and the hephaestin iron export complex. Iron inside the cell is either stored in ferritin as Fe3+ or is ‘sequestered’ by apoproteins.2
 | ||
| Fig. 3 Iron trafficking in the human cell. Iron enters the human cell as part of the iron-transport protein transferrin, which is taken up by endocytosis. Inside the cell, iron is either stored in ferritin or escorted to appropriate apoproteins. The chaperone frataxin is particularly important. It provides iron for the assembly of iron/sulfur clusters in proteins and also for the protoporphyrin IX cofactor. The latter receives its iron from ferrochelatase, which in turn is supplied by frataxin. Disruption of the frataxin chaperone results in Friedreich’s ataxia, a serious cardiovascular and neurodegenerative human disorder. Excess of iron is removed from the cell by a set of proteins, including IREG-1 and the hephaestin iron export complex. | ||
Importantly, there are also some examples of directed iron escorts similar to the copper chaperones, such as frataxin (Fig. 3). The latter forms part of a specific iron trafficking pathway responsible for Fe2+ insertion into iron/sulfur clusters and porphyrins. Within this context, it should be emphasized that porphyrins, such as protoporphyrin IX, do not simply ‘pick up’ their metal ion(s) spontaneously. Indeed, iron is not the only metal ion ultimately found in porphyrins, and there appear to be specific proteins with the task of inserting either iron, cobalt or magnesium into this particular macrocycle (see below).
Frataxin is a highly conserved multifunctional mitochondrial protein found in prokaryotes and eukaryotes where it fulfils an essential task in maintaining the intracellular iron homeostasis.32 Deficiency of this protein results in severe cardiovascular and neurodegenerative disorders, such as Friedreich’s ataxia. Symptoms of this disorder are often treated with antioxidants (such as high doses of vitamin E, coenzyme Q10, idebenone, N-acetyl cysteine and selenium), which are able to prevent some of the damage caused by out-of-control, redox active iron in the cell.33,34 Frataxin is a non-haem protein which binds high spin Fe2+ in a six-coordinate environment employing acidic amino acid residues (e.g.Asp and Glu) as ligands. The dissociation constant Kd for iron is around 10 μM.35 Frataxin has been considered as an iron escort (chaperone), delivering iron to proteins involved in iron/sulfur cluster synthesis as well as porphyrin synthesis.35 In the case of iron–porphyrins, the iron chaperone frataxin passes its iron on to the ‘iron metalloinsertase’ ferrochelatase.
Ferrochelatase is an iron–sulfur protein containing a [2Fe–2S] cluster. The process of iron transfer between frataxin and ferrochelatase has been studied in some detail employing methods of (protein) structural determination and docking simulations.36 In brief, the monomeric form of iron-loaded frataxin docks to the ferrochelatase dimer. Rearrangement of (mostly acid) ligands of frataxin and ferrochelatase facilitates the ‘passing on’ of the metal ion to ferrochelatase, which subsequently channels the received Fe2+ ion to its iron/sulfur loading site. Importantly, iron transfer between the two proteins does not involve ‘free’ Fe2+ ions. Throughout this ligand exchange and rearrangement process, the metal ion remains bound to up to six donor groups.
Iron bound to ferrochelatase is subsequently inserted into protoporphyrin IX to form protohaem IX. Ferrochelatase and the iron transfer resulting in iron insertion into protoporphyrins have been studied in great detail 37–39 and most of this work has been reviewed recently.40 Although some questions regarding the precise mechanism of iron transfer/insertion still remain,41 it seems that protoporphyrin enters and binds to the ‘active site’ of ferrochelatase, where the iron ion is transferred to a geometrically distorted protoporphyrinmacrocycle amenable to Fe2+ insertion.
It must be emphasized, however, that iron insertion into protoporphyrin IX is not directly comparable with metal ion insertion into an apoprotein. Protoporphyrin IX is considerably smaller in size when compared to an apoprotein. While the former ‘simply’ fits into the active site of the chelatase, transfer of a metal ion to an apoprotein requires more eloquent docking procedures between the proteins involved in the metal ion transfer.
Iron transfer between ferrochelatase and protoporphyrin IX is a key biochemical process. Inhibition of ferrochelatase by metal ions such as Cu2+ and Zn2+ results in a decrease in intracellularhaem and an increase in protoporphyrin IX.41 It has been proposed that inhibition of ferrochelatase by metal ions such as lead (or manganese) may serve a therapeutic purpose as part of PDT (see section 5.3).
The synthesis of iron/sulfur clusters may also involve the frataxin iron delivery system, although spontaneous assembly of such clusters has been observed upon addition of high concentrations of Fe2+ and S2−.6 In addition, chelatases similar to ferrochelatase have been identified for cobalt and magnesium. A cobalt chelatase in bacteria delivers cobalt (as Co2+) to cobalamin (vitamin B12).42 Similarly, a specific magnesium chelatase loads Mg2+ onto protoporphyrin IX to form Mg-protoporphyrin IX which is then incorporated into chlorophyll α.43
3.4 Zinc
Although Zn2+ cannot participate in (uncontrolled) redox events, adventitious zinc binding to proteins and enzymes with subsequent loss of protein function/enzyme activity poses a serious risk to the cell and requires a tight control of intracellular zinc against a 500-fold Zn2+transmembrane gradient at the outer membrane. The various mechanisms of cellular zinc transport have recently been reviewed by Sekler and colleagues44 and are summarized in Fig. 4. As far as we know today, the mammalian cell provides several points of entry and exit for zinc ions. Import may occur via diffusion or the ZIP proteins (Zrt and Irt-like proteins).45,46 Export seems to be facilitated by Na+/Zn2+ exchangers and zinc transporters (known as ZnT-1 to ZnT-10), while a possible role of Zn2+ pumps remains to be confirmed. Members of the ZnT protein family are also responsible for zinc transport within the cell and across intracellular membranes. | ||
| Fig. 4 Zinc trafficking and homeostasis in the human cell. Zn2+ enters the cell via the ZIP transporters and in part also by diffusion (there is a 500-fold excess of Zn2+ outside the cell) and possibly by the (apparently bi-directionally active) Na+/Zn2+ exchangers. Once inside the cell, Zn2+ is taken up by apoproteins or is sequestered by the thioneins, the apo-form of the metallothionein (MT) proteins. Either directly or via the MT proteins, Zn2+ is passed on to the zinc transport (ZnT) proteins which traffic zinc within the cell and also expel it from the cell. Zinc may also leave the cell via the Na+/Zn2+ exchangers, although this is still a matter of ongoing investigations. Intracellular concentrations of labile Zn2+ are extraordinarily low. They are regulated by a complex feedback loop which, among other components, involves the Zn2+ sensing transcription factor MTF-1, the MRE of DNA and de novo synthesis of thionein and ZnT. It is still vastly unknown how apoproteins acquire Zn2+. This may be a spontaneous process, or—more likely—may involve zinc escorts and metalloinsertases, possibly including the MT proteins. | ||
Here, the ZnT proteins provide part of a sophisticated intracellular feedback loop, which maintains the intracellular zinc homeostasis and involves a range of proteins, including the metal responsive element-binding transcription factor-1 (MTF-1) and the metallothionein proteins (MTs). The ‘active’ zinc form of MTF-1 exhibits an apparent zinc binding constant Ka of all six domains collectively of around 32 × 109 M−1.47 It is formed when levels of labile zinc rise above critical, picomolar concentrations.†† Active MTF-1 then binds to metal-responsive elements (MREs) of the DNA and causes the expression of various proteins, including the ZnT proteins and also the thioneins (T), i.e. the apo-forms of metallothioneins (MT-1 and MT-2).‡‡ Together, the ZnT and T proteins are able to efficiently lower the intracellular concentration of labile Zn2+. While the ZnT transporters can distribute and export zinc, the thioneins effectively sequester free Zn2+ ions (Fig. 4). For this purpose, the thionein proteins consist of around 60 amino acids, 20 of which are cysteine residues, which are able to bind simultaneously seven Zn2+ ions in a Zn4Cys11 and a Zn3Cys9 cluster (α- and β-cluster, respectively). Apparent zinc binding constants Ka for Zn7MT have been estimated to be around 3.2 × 1013 M−1 at pH 7.4, which makes this protein an excellent reservoir for ‘locked up’ zinc.50–52 So far, it is unknown if there is a direct interaction between the ZnTs and the MTs.44 It is possible that in a first step the thioneins ‘mop up’ labile Zn2+ to form the folded MTs, which in a second step interact with the ZnTs to pass on the sequestered Zn2+ ions. In a final step, excess Zn2+ loaded onto ZnT is transported out of the cell.44
Although purely hypothetically at this time, there is some indirect experimental support for this pathway. First of all, the MT proteins contain eight highly conserved lysine residues.53 These positively charged residues may not only counter-balance the negative charges of the zinc/sulfur clusters but also provide a docking site or even motif for interactions with ZnT (or similar zinc export proteins). Interestingly, this lysine surface is only generated upon MT formation, i.e. thionein would not interact in the same way. Secondly, MT proteins also fulfil the requirement of thermodynamically stable, yet kinetically labile zinc binding and have already been shown to transfer one Zn2+ ion per MT molecule to apoproteins.51,54,55 The two clusters even provide access for docking and metal ion transfer, for instance via ligand rearrangements. Thirdly, experiments with zinc/sulfur models have confirmed the possibility of rapid Zn2+ transfer between peptides via a mechanism which involves docking of the two peptides , and exchange/rearrangement of ligands “in the virtual absence of free zinc ions”.56 Considered together, these aspects reflect the ‘classical’ interactions between metal trafficking proteins, such as the ones described above for copper and iron transport. It should be noted that MT is also redox sensitive and may release labile Zn2+ upon oxidation, an event which may endow MT proteins with ROS and OS sensing properties (Fig. 5).51,54
 | ||
| Fig. 5 The various emerging roles of the metallothionein (MT) proteins in zinc trafficking. Apart from their well-known function as zinc sequestering and zinc storage proteins (magenta arrows), the metallothioneins are known to donate (one) zinc ion to apoproteins (light green arrow). Upon oxidation, MTs are able to release all seven bound metal ions, which has also led to the notion of MTs as intracellular zinc reservoirs and as reactive oxygen (ROS) ‘sensors’. Furthermore, MTs are able to exchange metal ions, such as Cd2+ (which is readily taken up) for Zn2+ (which is released). This pro-active ‘detoxification’ of heavy metal ions is not limited to free toxic metal ions (dark green arrows). A series of experiments has indicated that MTs may also act as ‘rescue’ proteins able to repair Cd2+ poisoned proteinsvia an effective reciprocal metal ion exchange. MTs may also pass on Zn2+ to ZnT for excretion (blue arrow). Finally, thionein is an excellent cellular antioxidant reducing agent (pink arrows). The image of MT has been obtained from the Protein Data Bank (identifier 4MT2).125 The image of the carbonic anhydrase used as part of the graphical abstract is PDB entry 1CAB.126 | ||
3.5 Molybdenum and others
Molybdenum plays an important part in pro- and eukaryotic proteins and enzymes in form of the molybdenum cofactor (Moco), which is based on a molybdopterin structure (Fig. 6a and b). Moco is found in around 50 prokaryotic enzymes, such as various hydroxylases, reductases and nitrogenases.6,57 In humans, Moco is present in three important enzymes, i.e. in sulfite oxidase (SO), xanthine oxidase (XO) and aldehyde oxidase. In plants, Moco is additionally found in nitrate reductase. Furthermore, molybdenum forms part of the iron–molybdenum cofactor FeMoco, which only occurs in the bacterial enzyme nitrogenase and is structurally unrelated to Moco. Molybdenum is redox-active under biological conditions with oxidation states of +4, +5 and +6. As part of Moco, it is—almost exclusively—found in redox proteins where it provides an excellent ‘relay’ to connect electron transfer processes involving different numbers of electrons. To avoid any misunderstandings, it must be emphasized that molybdenum—in contrast to the other metal ions discussed here—does not occur in form of ‘naked’ molybdenum ions (such as Mo4+/5+/6+) in biological systems. | ||
| Fig. 6 (a) Elements of molybdenum trafficking. Unlike the other metal ions discussed as part of this review, molybdenum is trafficked not as a cation but as molybdate (MoO42−) anion. It almost exclusively ends up in the molybdenum cofactor (Moco), which exhibits a trafficking system on its own. Please note that the area of molybdenum trafficking is still highly speculative. The Figure shown therefore combines known elements of molybdenum (and Moco) trafficking from various sources, including plants (algae) and bacteria. It is likely that the emerging molybdenum trafficking networks in human cells ultimately will be similar, but not identical to the illustration provided here. Tungsten appears to follow similar trafficking avenues, yet several tungsten-specific transporters (TupA, WtpA) have been identified recently, which do not deal with molybdenum. (b) Synthesis of the molybdenum cofactor (Moco). Details are discussed in the text. | ||
The synthesis of the pterin scaffold from guanosine triphosphate (GTP) via precursor Z with subsequent metal insertion are complicated processes which are still not fully understood. Nonetheless, several transport and chaperone proteins involved in molybdenum and tungsten metabolism have recently been discovered, albeit mostly in plants.57–60
It appears that molybdenum enters the (plant) cell as molybdate (MoO42−), either via a specific, high affinity uptake system based on an ABC-type transporter or via a phosphate and/or sulfate uptake system (Fig. 6a).59 Present knowledge of such systems is still limited and keeps the door open for new discoveries. Only recently, a distinct molybdate uptake system has been identified in the green algae Chlamydomonas rheinhardtii,61 while molybdate storage proteins have been found in some bacteria.62 At the same time, two distinct ABC transporter systems selectively transporting tungsten (rather than molybdenum and tungsten) have been identified in bacteria and archaea, and have been named TupA and WtpA (T and W in these abbreviations denote ‘tungsten’ and stand for tungsten uptake, and tungsten transport protein, respectively).63 Together, these discoveries point towards molybdenum and tungsten trafficking systems, which are more sophisticated than previously thought.
In accordance with this view, processing and insertion of molybdenum into the preformed pterin structure also appears to be surprisingly well-regulated (Fig. 6b).57 Although speculative at this time, it seems that—at least in plants—the pterin (MPT) precursor, once synthesized, binds a copper ion which protects its sulfur ligands from oxidation. MPT then binds tightly to the C-terminal domain of the Cnx1 protein, which facilitates a series of events leading to the fully functional Moco molecule. First, MPT is adenylated to form MPT-AMP. MPT-AMP is then transferred to the N-terminal domain of Cnx1, where the adenylate moiety is cleaved in a molybdate-dependent reaction. At the same time, copper is released and molybdenum inserted (formally as a ‘MoO2’ unit).
Since Moco is highly sensitive (e.g. to oxidation), it is thought that it is stored in particular Moco storage proteins and chaperoned to target proteins where it is inserted by specific Moco trafficking proteins. Indeed, a Moco carrier protein (MCP) able to bind four Moco molecules has been identified in C. rheinhardtii, which protects the cofactor against oxidation.64,65 Furthermore, Moco insertion into nitrate reductase from E. coli may require a specific chaperone (called NarJ), similar to xanthine dehydrogenase from Rhodobacter capsulatus, which requires the XDHC protein for Moco insertion.66,67 In contrast, human apo-SO appears to incorporate Moco spontaneously, i.e. in the absence of a specific chaperone protein.68
In some enzymes, such as xanthine oxidase and aldehyde oxidase, further activation of Moco occurs when terminal inorganic sulfur is added to the molybdenum atom (Fig. 6b).57 Such activation is not random but, as Mendel and colleagues have shown, involves a sulfurase enzyme (e.g. ABA3, a Moco sulfurase).69
Related pathways exist in bacteria, such as in E. coli. Here, proteins such as MoaA, MoaD, MoaE, MoeA, MoeB and MogA are involved in the pterin synthesis and facilitate the incorporation of molybdenum into the metal free pterin (the metal insertion step is catalyzed by MoeA and enhanced by MogA).70 It is possible that similar proteins are also involved in molybdenum insertion in human Mo-enzymes, such as SO.71
Finally, it should be mentioned that apart from the essential metal ions, some of which have been discussed here, certain non-essential and even toxic metal ions, such as cadmium and lead, are also able to enter and travel within the human cell. Those metal ions employ molecular mimicry to ‘hijack’ the trafficking systems of essential metal ions or certain anions (e.g.phosphate, sulfate). The DCT1, in particular, is prone to this kind of misuse. Trafficking of toxic metal ions poses an additional biochemical complication and requires specific medical detoxification strategies (see section 5).
4. Metal exchange between proteins and enzymes
4.1 A repair service for proteins containing the incorrect metal ion?
The previous section has considered at some length aspects of metal ion escorting and insertion into proteins and enzymes. Although the processes governing the insertion of the ‘correct’ metal ion into a given protein or enzyme appear to be highly regulated and hence reliable, there is still the possibility that something may go wrong. What is the fate of a protein which has received the incorrect metal ion—either accidentally or as a result of an adventitious, spontaneous metal ion exchange? For instance, it may be possible that certain SOD2 molecules are loaded with iron instead of manganese if the mitochondrial concentration of labile iron is unusually high. Similarly, zinc enzymes may exchange Zn2+ for Cd2+—and hence become adversely affected—in the presence of elevated intracellular concentrations of cadmium, as has been discussed for zinc-dependent proteins involved in maintaining genomic stability.72In this case, one may foresee several possible consequences. Most obviously, the protein incorporating the incorrect metal ion may be degraded. From a perhaps more-naïve perspective, one may also envisage that the correct metal ion ‘appears’ and simply replaces the incorrect one spontaneously, for instance due to its higher affinity for the given protein. As we have seen, however, such ‘spontaneous’ processes are surprisingly rare in metal trafficking, and hence are also unlikely to take place in this particular situation. Alternatively, the incorrect metal ion may be selectively removed from the protein and replaced by the correct one. This possibility is particularly attractive. It would allow the ‘rescue’ of the protein, avoid de novo synthesis and circumvent the need for any ‘free’ metal ions.
To date, such rescue systems have hardly been explored. It is likely that such systems exist for certain metal ions and/or metalloproteins, similar to the protein disulfide isomerases, which rescue proteins with incorrectly formed disulfide bonds. In any case, if such rescue proteins do exist, they are likely to differ from the chaperones and metalloinsertases discussed above. We will briefly illustrate the potential workings of such a rescue system using the example of the metallothioneins. It should be emphasized from the outset that this discussion is highly speculative—and that to date there is no reliable evidence in vivo which would unambiguously identify MTs as rescue proteins.
4.2 Metallothioneins as metal ion exchange proteins
Despite their high zinc binding constant, MTs are not a ‘grave’ for zinc ions. In the past, several studies have shown that cadmium and probably copper binding constants of MT (3.2 × 1017 M−1 at pH 7.4 for Cd2+ and estimated at >1019 M−1 for Cu+) by far exceed the binding constant for zinc (3.2 × 1013 M−1 at pH 7.4).50,51,73 In essence, the relative magnitude of these constants implies that Zn7MT will release Zn2+ ions in the presence of ‘free’ Cd2+ and Cu+ ions to form the corresponding cadmium and copper complexes (i.e. Cd7MT and a less-well-defined, highly redox-sensitive Cu+ complex). In turn, Cd7MT will release Cd2+ ions in the presence of Cu+. Indeed, metal exchange between ‘free’ Cd2+ and Zn7MT has been observed and occurs almost instantaneously. This finding has led to the notion that zinc (and cadmium) MT is thermodynamically stable yet kinetically labile.Interestingly, these exchange reactions are unidirectional, i.e. Zn2+ is normally not able to release Cd2+ or Cu+ from the corresponding cadmium and copper MT, while Cd2+ cannot release Cu+ from copper MT.§§ Since the ranking of zinc, cadmium and copper binding-constants of MT is reflected by the toxicity of the corresponding metal ions, MT is able to sequester the ‘free’ toxic ions (Cd2+, Cu+), and exchange them for the beneficial Zn2+ ions. Once released, Zn2+ ions are then able to initiate an antioxidant and detoxification cascade, for instance by binding to MTF-1 (see section 3.4) which results in the formation of more thionein (and ZnT). This rather ‘intelligent’ detoxification and response system against Cd2+ (and Cu+) ions is still not fully understood, yet appears to take advantage of appropriate metal binding constants. It should also be noted that some detoxification and antioxidant response pathways appear to be triggered exclusively by Zn2+, but not by Cd2+.74 The exchange of Cd2+ and Zn2+ at MT would therefore form a crucial step in the cell’s response towards cadmium. Not surprisingly, several attempts are underway to ‘mimic’ these exchange properties of MT for therapeutic purposes.
4.3 Metallothioneins as metal exchanging rescue proteins
The intracellular concentrations of Cd2+ and Cu+ are usually rather low and significant increases in these concentrations, for instance due to cadmium intoxication or oxidative release of copper ions from proteins, are rare under normal conditions. Nonetheless, conditions such as OS may result in uncontrolled release of metal ions (see section 2.5). In these situations, incorporation of the incorrect metal ion into proteins and subsequent rescuing may play an important role. We will therefore briefly consider the ability of Zn7MT to repair the inactive cadmium ‘poisoned’ forms of human enzymes by reciprocal metal ion transfer.75 This kind of rescue (or ‘repair’¶¶) has been demonstrated in vitro for a range of human proteins, including several enzymes, zinc finger proteins and actin.The underlying mechanism is illustrated by the rescue of the cadmium form of the human enzyme carbonic anhydrase (Cd–CA). This enzyme actually prefers zinc over cadmium (Ka = 1012 M−1 for Zn2+ and 1011.1 M−1 for Cd2+) and therefore can be re-activated with ‘free’ Zn2+ as well as with Zn7MT.77 Nonetheless, cadmium/zinc exchange between Zn7MT and Cd–CA, which results in enzymatically active Zn–CA and a cadmium form of MT, is considerably more efficient when compared to the reaction of ‘free’ Zn2+ ions with Cd–CA. While the free Zn2+ ions restore the zinc form of CA with a rate constant of 0.021 M−1 s−1, the Zn2+ bound to MT exchanges with a rate constant of 2.3 M−1 s−1, i.e. about one hundred times faster.77 This rather surprising finding, i.e. that the bound form of Zn2+ (in Zn7MT) is actually more reactive than free Zn2+, may be explained with the high affinity of (apo-)MT for Cd2+, which provides a second driving force in addition to the binding affinity of CA for Zn2+.|||| In fact, a similar metal exchange between Zn7MT and a cadmium protein may even be possible if the protein itself would not prefer Zn2+ over Cd2+, as long as MT drives the exchange due to its own preference for Cd2+. This possibility is lacking as far as ‘free’ Zn2+ is concerned.
Apart from restoring the activity of CA fairly rapidly, the exchange with Zn7MT also has the advantage that Cd2+ is not released, but rather sequestered within MT. This type of reciprocal metal ion exchange is particularly interesting since it avoids the presence of labile metal ions (Zn2+, Cd2+), in line with the notion of metal chaperone proteins (see section 3). Nonetheless, appropriate metal binding constants are crucial for such a reciprocal metal ion exchange to occur, and this may generally limit the number of possible reciprocal metal ion exchange reactions between proteins.
Mechanistically, this kind of metal ion exchange process, which appears to resemble a ‘rochade/castling’ of two metal ions between two proteins, is not yet fully understood. It may involve a transient MT/CA complex similar to the one described for the CCS/SOD complex or the frataxin/ferrochelatase complex. Indeed, protein complex formation would allow a direct metal ion transfer between the proteins. It must be pointed out, however, that the reciprocal zinc/cadmium exchange is mechanistically more complicated when compared to metal transfer between a metal-containing chaperone protein and an apoprotein. In the case of the apoprotein, the chaperone is able to ‘donate’ the metal ion to an empty binding site as part of a one-way transfer. In contrast, the rescue protein must remove one erroneous metal ion from the inhibited site of the target proteinand simultaneously donate another one, i.e. the correct one, as part of a two-way exchange. Apart from such a metal ion rochade, it is therefore possible that, alternatively, small amounts of metal ions may be temporarily set free which would facilitate the transfer as part of several intertwined equilibrium reactions.
How, and to which extent such metal exchange reactions occur in vivo, and which role MT and similar proteins play as part of such a ‘rescue’, is still a matter for future investigations. Apart from Cd–CA, the Cd2+ forms of carboxypeptidase A (Cd-CPA), the zinc finger transcription factor TFIIIA, the Tramtrack zinc finger protein and actin, as well as nickel-containing Sp1-3 zinc finger peptide have all been rescued/reactivated by Zn7MT.75,76,78–80 Interestingly, synthetic compounds able to restore the activity of proteins incorporating the incorrect metal ion may be of considerable pharmaceutical interest as combined detoxification/supplementation agents. This issue will be discussed further as part of the next section.
5. From simple chelate therapy to protein mimics and rational drug design
5.1 Chelate therapy in the context of metal poisoning and OS
The medical use of metal binding agents against acute metal intoxication has a long tradition. Agents such as EDTA, desferrioxamine (DFO), 2,3-dithiopropanol (British anti-Lewisite, BAL) and N-acetyl-D-penicillamine (NAPA) have been used in the context of lead, cadmium, mercury and arsenic poisoning since the beginning of the 20th century (Fig. 7).81–84***These agents bind to the toxic metal ions, hence preventing their random interactions with biomolecules, such as proteins and enzymes, counteract their deposition in organs and ultimately aid their excretion from the body. The diverse applications of metal binding agents in acute or chronic exposure to metal ions have been the subject of many books and reviews. We will therefore move on by briefly mentioning the use of chelators in the context of a disturbed metal metabolism. Here, several human diseases associated with a malfunctioning metal ion uptake and excretion are known, among them Wilson disease (see section 3.1). | ||
| Fig. 7 Overview of selected current drug development programmes related to metal trafficking. Apart from the ‘classical’ chelate therapies for metal detoxification or reduction of metal ion overload, more sophisticated chelators have been developed which are selectively activated, for instance by oxidation or UVA radiation. Other approaches include apo-pro-drugs which are activated by ‘picking up’ or by exchanging certain metal ions and chelators controlling the level of protoporphyrin IX (PP-IX), which is a key component in photodynamic therapy (PDT). | ||
While chelate therapy in Wilson disease acts against copper overload at the level of the whole cell and organism (Table 3), most irregularities in metal trafficking (including in Wilson disease) are due to the malfunctioning of just one trafficking protein. Such disorders may be addressed by specifically targeting the impaired metal ion transport system. The following sections will provide a glimpse of current investigations in this area. One should point out from the outset that most of these approaches are still in the developmental phase, and are therefore speculative at this time. Furthermore, only a few selected examples may be provided here in order to stake out the general area of interest. This summary is neither comprehensive nor complete as the area of chelate therapy is highly diverse and also rapidly expanding.
| Metal ion | Related disease | Manifestation | Associated protein | Current treatment |
|---|---|---|---|---|
| Copper | Wilson disease | Copper overload in various tissues | Wilson disease protein (copper transporter) | Low copper diet, chelators (e.g. penicillamine, BAL), Zn2+ (to stimulate MT expression) |
| Menkes disease | Copper deficiency | Menkes disease protein (copper transporter) | Copper supplements | |
| Manganese | Hailey–Hailey disease | Skin disorder associated with defects in proteinglycosylation | hSPCA1 pumps | Botulinum toxin, PDT |
| Iron | Friedreich’s ataxia | Cardiovascular and neuro-degenerative disorders | Frataxin (iron chaperone) | Antioxidants |
5.2 Strategies to lower the labile ion pools
As mentioned in sections 2.4 and 2.5, the intracellular pools of labile metal ions may vary and increase to ‘unhealthy’ levels due to external and internal stresses, such as UVA radiation and OS. Not surprisingly, various strategies have been developed during the last two decades to re-adjust such unhealthy increases in the labile metal ion pools, in particular in the case of iron. For this purpose, the therapeutic potential of various chelators has been evaluated, for instance, as part of the CIBA oral chelator programme between 1992 and 1996.Interestingly, many of these ‘early’ chelators were based on siderophore-like structures. Siderophores themselves are iron-chelating agents produced by a variety of microorganisms.85,86 They usually feature carboxylate, hydroxamate and catecholate ligands, which effectively sequester iron ions, mostly in form of octahedral complexes. Some of the siderophores are chemically rather ‘simple’ structures, while others, such as enterobactin, are chemically very complex (Fig. 8).87,88 The biological role of individual siderophores differs, yet is usually related to the microorganism’s iron uptake and/or homeostasis. Due to their paramount role in iron metabolism (of lower organisms), siderophores and siderophore mimics have also been considered for therapeutic applications. Such compounds may, for instance, be useful in the treatment of metal poisoning (e.g. Al3+) and in the regulation of the labile iron pool (e.g. DFO). Furthermore, there is evidence that siderophore mimics may also be used to deprive microbes of iron ions essential for microbial growth and hence act as antimicrobial agents.89 Similar considerations apply to cancer cells, where compounds such as DFO appear to retard cancer cell growth by interacting with copper and iron ions.90 In contrast, other siderophore mimics resemble the ‘natural’ function of siderophores and hence promote bacterial growth by delivering iron ions to these organisms.91 Jean-Louis Pierre and colleagues have recently reviewed aspects of synthetic siderophore mimics and their potential therapeutic uses.92,93
 | ||
| Fig. 8 Chemical structure of the siderophoreenterobactin, which is found in organisms such as E. coli and Salmonella typhimurium. Based on three combined catecholate moieties, this natural chelator exhibits an extraordinarily high binding constant for iron ions (Ka = 1052 M−1).127 It is employed by its parent bacteria to mobilize and traffic iron ions required for their growth. | ||
The lead structures for iron chelators identified at the time, further developments since then and the design criteria for such oral chelators have recently been reviewed by Robert Hider and colleagues (Fig. 7).94,95 Relevant design criteria involve the redox potential of the iron–chelator complex (which indicates if redox active iron has indeed been ‘disarmed’ by chelation), binding constants (to ensure only the LIP is affected while iron-containing proteins remain largely intact) and lipophilicity (to ensure a good pharmacokinetic profile). Apart from the various compounds described in these reviews, the weak iron binding agents derived from the amino acidserine and vitamin B6 are also worth mentioning.96
In addition, several rather intriguing strategies have been developed during the past decade, all of which employ pro-drugs selectively activated by cells under stress (Fig. 7). In their inactive form, such agents are supposed to bind iron rather weakly, i.e. they do not remove the metal from iron-proteins and hence are on the whole non-toxic. Once activated, however, these agents turn into strong chelators able to sequester and hence detoxify labile iron ions. Within this context, N,N′-bisdibenzylethylenediaminediacetic acid (DBED) can be activated in the presence of H2O2 by hydroxylation to form N-2-hydroxybenzyl-N′-benzylethylenediaminediacetic acid (HBBED), with an increase in iron binding constant from less than 1015 M−1 to 1028 M−1.97 Researchers at the L’Oréal Basic Research Center have since developed a range of additional oxidatively activated chelators.98–100 Such compounds respond to elevated levels of OS, which in turn go hand in hand with increased levels of labile metal ions (see section 2.5).
Apart from these compounds, which are activated by hydroxylation, several rather interesting boronate-masked (pro-)chelators have been developed by Katherine Franz and colleagues (Fig. 9).101–103 In essence, these compounds are based on a boron-containing precursor of a phenol. The phenol is formed upon oxidation of the pro-chelator by H2O2 and subsequently takes part in effective iron ion binding. Importantly, while the prochelators are non-toxic and do not sequester iron ions, the ‘unmasked’ chelators are able to effectively, yet selectively, lower (toxic) levels of labile iron ions at sites of OS. Recently, the protective effects exerted by such boron-based pro-chelators against H2O2 have been demonstrated in cultured retinalpigmentepithelial cells.101
![Redox-activation of the pro-chelator isonicotinic acid [2-(4,4,5,5,-tetramethyl-[1,2,3]dioxaborolan-2-yl)benzylidene]hydrazide (BSIH) in the presence of elevated levels of H2O2. Once activated, salicylaldehyde isonicotinyl hydrazone (SIH) efficiently sequesters iron ions in form of a [Fe(SIH)2]+complex, hence preventing iron from reacting with H2O2 to form the highly damaging HO˙ radical (Fenton reaction). Importantly, since BSIH is only activated at the ‘point of need’, and the resulting iron complex itself does not undergo Fenton-type chemistry, the pro-chelator strategy provides both, efficiency and selectivity against iron-mediated OS.](/image/article/2009/MT/b904533c/b904533c-f9.gif) | ||
| Fig. 9 Redox-activation of the pro-chelator isonicotinic acid [2-(4,4,5,5,-tetramethyl-[1,2,3]dioxaborolan-2-yl)benzylidene]hydrazide (BSIH) in the presence of elevated levels of H2O2. Once activated, salicylaldehyde isonicotinyl hydrazone (SIH) efficiently sequesters iron ions in form of a [Fe(SIH)2]+complex, hence preventing iron from reacting with H2O2 to form the highly damaging HO˙ radical (Fenton reaction). Importantly, since BSIH is only activated at the ‘point of need’, and the resulting iron complex itself does not undergo Fenton-type chemistry, the pro-chelator strategy provides both, efficiency and selectivity against iron-mediated OS. | ||
As part of a similar approach, the research groups of Charareh Pourzand and James Dowden have developed so-called caged-iron chelators, which are activated by UVA radiation.104 These chelators are particularly useful in the protection against UVA radiation (see section 2.5), for instance against prolonged exposure to sunlight (Fig. 7). In contrast to conventional chelators, the caged pro-drugs are inactive in the absence of the external UVA stressor and hence do not damage skin cells by removing iron from proteins and enzymes, or by causing widespread iron deficiency. These substances are activated by UVA radiation, which as the same time is the stressor actually responsible for increases in the LIP. In doing so, UVA radiation itself initiates the protective mechanism against the damage it causes. Together with oxidatively activated metal binding agents, caged-iron chelators provide a promising new avenue to modulate the intracellularmetal ion homeostasis without causing metal ion deficiency. It must be emphasized that these approaches are still in the developmental phase.
5.3 Chelate assisted photodynamic therapy
Chelate assisted PDT has recently been developed as a treatment for various tumours that can be exposed to light. It is particularly effective, but not limited to, tumours of the skin (e.g. low risk basal cell carcinoma) and other, non-cancerous skin conditions, such as Hailey–Hailey disease (see section 3.2).105 In short, PDT employs a photosensitizer in combination with (usually visible) light to generate singlet oxygen (1O2) and a host of related ROS. ROS formation occurs selectively at sites which contain the photosensitizer and are exposed to light.The basics of PDT have been known since the beginning of the 20th century and the method itself has been developed for anticancer treatment since the 1970s. Originally, photosensitizers such as the ‘hematoporphyrin derivative’ (HpD), a chemically rather ill-defined mixture of porphyrins, have been employed.106 This has gradually given way to more intricate methods. Some of them rely on a ‘photosensitizer’ which occurs naturally in human cells, i.e.protoporphyrin IX. As already discussed in section 3.3, the latter is a precursor of metal porphyrins, and as such present in human cells as a normal metabolite. The fact that it may also serve as a photosensitizer is, of course, more or less coincidental, yet provides a fertile ground for a range of interesting photodynamic therapeutic approaches. It is possible, for instance, to stimulate excess production of protoporphyrin IX in cells by administering its precursor, 5-aminolevulinic acid (ALA).107 The latter increases the amount of protoporphyrin IX and hence the photosensitivity of tissue. Since tumours enrich ALA, they are especially sensitized after its administration.
Under normal conditions, protoporphyrin IX is converted rapidly to haem, a process which involves the uptake of Fe2+ and results in loss of photosensitizing properties. Iron uptake of protoporphyrin IX is controlled by ferrochelatase (see section 3.3). This process provides two points for therapeutic intervention that would both result in an increase of protoporphyrin IX: inhibition of ferrochelatase and depletion of the pool of available Fe2+. Since ferrochelataseinhibitors available today are not specific and include toxic metals such as lead108 and manganese,109 this approach results in various undesired side effects. The main focus has therefore been on depleting the pool of available iron ions by employing chelating agents. For instance, DFO, an established iron chelator, has been evaluated alongside novel iron chelators, such as 1,2-diethyl-3-hydroxypyridin-4-one hydrochloride, which is also known as CP94 (Fig. 7).110,111
This approach has already shown promise in clinical studies.112 Interestingly, one of the key criteria for success seems to be the ‘correct’ iron binding constant of the chelator. Agents which bind iron ions too loosely will be ineffective, and CP94 exhibits an iron binding constant of 1037.2 M−1.113 In contrast, agents designed to ‘merely’ lower the LIP exhibit lower metal binding constants in order not to disrupt iron bound to iron-proteins.113 It should be mentioned that other promising avenues in PDT involve the control of protoporphyrin IX levels via nitric oxide (˙NO).114
5.4 Metal uptake and exchange as basis for pro-drug activation
The use of chelators to ‘lock away’ or even remove metal ions from cells should not diminish the beneficial role metal ions may play as part of therapy. Here, we are not addressing the vast area of trace element research. Rather, we are considering the usage of metal ions already available inside cells for specific therapeutic purposes. For instance, various copper- and iron-containing porphyrin derivatives exhibit a significant superoxide dismutase (SOD)-like catalytic activity. Such SOD-mimics have been considered as antioxidants and in anticancer therapy.21,115–119 Interestingly, elevated levels of labile copper and iron ions in cells suffering from OS or in cancer cells therefore provide the basis for the use of (metal-free) porphyrins as therapeutic agents. The latter may be activated selectively in cells with high concentrations of labile copper and iron ions, while cells with ‘normal’ levels of labile copper or iron ions would be less affected. The labile metal ion present as part of the disorder (copper or iron) would therefore not only stake out the sick cell for therapeutic action, but would also become an active part of it. Again, the correct metal binding constants would be pivotal for both, selectivity and activity.One argument against such an approach is, of course, the presence of other divalent metal ions (such as zinc), which could bind to such porphyrins and similar compounds well before such a compound may reach its cellular destination. In this case, the concept of metal binding constants and metal exchange reactions comes into play. It has recently been shown that zinc-containing porphyrins are rather stable, yet do exchange zinc ions for iron in the presence of (reasonable amounts of) labile iron ions.120 Such an exchange system, which is governed by the respective zinc and iron porphyrin binding constants, also allows activation of the catalytically active SOD mimic in cells with an elevated LIP. Interestingly, such systems not only generate the SOD mimic in target cells in situ (viametal ion exchange), but also sequester toxic iron ions and release beneficial zinc ions.
The combination of these three beneficial, antioxidant effects should be considerably higher when compared to simply administering an antioxidant (such as vitamin C or a particular SOD mimic) or employing an antioxidantchelator (such as DFO). Since such metal ion exchange systems reflect some of the properties of the MTs, the design and development of MT mimics, or, indeed, combined MT/SOD mimics (with exchange-based activation) provide ample opportunities for future investigations.
6. Conclusions
In summary, we have shown that metal trafficking between and inside human cells is a highly complex issue. Inorganic chemistry can contribute a range of valuable concepts, such as metal binding constants and parameters governing metal exchange reactions. Based on these concepts, we can rationalize some of the more intricate aspects of inter- and intracellular metal trafficking, such as selectivity for a particular metal ion, the ‘passing on’ of a metal ion from weaker to stronger binding sites, metal ion escorting and insertion and metal ion exchange between proteins. While the systems trafficking copper, iron and zinc are rather well understood today, other metal ions are likely to follow in the near future.Many aspects of metal trafficking have been addressed by biochemists and cell biologists with a rather limited input of (inorganic) chemistry. This has been rather unfortunate, yet may be understood if one considers the minute concentrations of trafficking proteins involved. Future research in this area will therefore rely on refined analytical techniques as well as on close interactions between coordination chemists and biologists.121 Metal trafficking in plants and lower organisms in particular provides exciting opportunities for the development of ‘green’ metal detoxifying systems.
At the same time, the notion of metal trafficking between cells, but also inside cells—between bound and labile metal ion pools and between cellular compartments—opens up many promising avenues of future drug design.122 Rather than simply locking away or removing metal ions from the cell, more sophisticated approaches suddenly emerge, such as chelate assisted PDT, ‘intelligent’ metal exchange reactions and metal/redox activated pro-drugs. Most of these avenues are still hardly explored today, and the development of such therapeutic molecules is in its very early stages. Nonetheless, the first sensible designs are now possible, and this area of investigation is particularly important in the field of cancer research, where metal ions play a role in cancer cell formation (via OS), and also in the development of novel anticancer therapies.
| ALA | 5-Aminolevulinic acid |
| AMP | Adenosine monophosphate |
| ATOX1 | Human metallochaperone antioxidant-1 |
| ATP | Adenosine triphosphate |
| ATPase | Adenosine triphosphatase |
| ATX1 | Yeast metallochaperone antioxidant-1 |
| BAL | British anti-Lewisite |
| CA | Carbonic anhydrase |
| CCS | Copper chaperone for SOD1 |
| Cnx | Cofactor for nitrate reductase and xanthine dehydrogenase |
| Cox | Copper chaperone for cytochrome c oxidase |
| CPA | Carboxypeptidase |
| CTRs | Cell surface transporters |
| DBED | N,N′-Bisdibenzylethylenediaminediacetic acid |
| DCT1 | Divalent cation transporter 1 |
| DFO | Desferrioxamine |
| DNA | Deoxyribonucleic acid |
| DPA | Pyridine-2,6-dicarboxylic acid |
| EC-SOD | Extracellular SOD |
| EDTA | Ethylenediaminetetraacetic acid |
| GSH | Glutathione |
| GTP | Guanosyl triphosphate |
| HBBED | N-2-Hydroxybenzyl-N′-benzylethylenediaminediacetic acid |
| HpD | Hematoporphyrin derivative |
| HSAB | Hard and soft acid and base concept |
| hSPCA1 | Human secretory pathway calcium ATPase |
| IREG-1 | Iron regulatory protein 1 |
| K a | Association constant |
| K d | Dissociation constant |
| LIP | Labile iron pool |
| Moco | Molybdenum cofactor |
| MPT | Molybdopterin |
| MREs | Metal-responsive elements |
| MTF-1 | Metal responsive element-binding transcription factor-1 |
| MTM1 | Manganese transporter for the mitochondrion |
| MTs | Metallothioneins |
| NAPA | N-Acetyl-D-penicillamine |
| ˙NO | Nitric oxide |
| OS | Oxidative stress |
| PDT | Photodynamic therapy |
| PPi | Pyrophosphate |
| PP-IX | Protoporphyrin IX |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SO | Sulfite oxidase |
| SOD | Super oxide dismutase |
| Sp1-3 | Third zinc finger of the transcription factor Sp1 |
| TF IIIA | Transcription factor IIIA |
| XO | Xanthine oxidase |
| ZIP | Zinc-iron related transport protein |
| ZnT | Zinc transporter |
Acknowledgements
The authors would like to thank the University of Saarland, the Ministry of Economics and Science of Saarland and the Deutsche Forschungsgemeinschaft (DFG; grant number Be 1540/10-1 to K. B.) for financial support. We also acknowledge the support of the European Union in form of the RedCat Initial Training Network (215009) and the Corena Interreg IV programme.References
- V. C. Culotta, M. Yang and T. V. O’Halloran, Biochim. Biophys. Acta: Mol. Cell Res., 2006, 1763, 747–758 CrossRef CAS.
- N. Ballatori, Environ. Health Perspect., 2002, 110, 689–694 CAS.
- K. S. Larsen and D. S. Auld, Biochemistry, 1989, 28, 9620–9625 CrossRef CAS.
- K. S. Larsen and D. S. Auld, Biochemistry, 1991, 30, 2613–2618 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, Oxford, 2007 Search PubMed.
- J. Kuchar and R. P. Hausinger, Chem. Rev., 2004, 104, 509–525 CrossRef CAS.
- L. S. Field, E. Luk and V. C. Culotta, J. Bioenerg. Biomembr., 2002, 34, 373–379 CrossRef CAS.
- E. A. Permyakov and R. H. Kretsinger, J. Inorg. Biochem., 2009, 103, 77–86 CrossRef CAS.
- O. Minowa and K. Yagi, J. Biochem., 1984, 96, 1175–1182 CAS.
- S. Verstraeten, L. Aimo and P. Oteiza, Arch. Toxicol., 2008, 82, 789–802 CrossRef CAS.
- S. Tottey, D. R. Harvie and N. J. Robinson, Acc. Chem. Res., 2005, 38, 775–783 CrossRef CAS.
- T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O’Halloran, Science, 1999, 284, 805–808 CrossRef CAS.
- K. A. McCall and C. A. Fierke, Biochemistry, 2004, 43, 3979–3986 CrossRef CAS.
- L. A. Finney and T. V. O’Halloran, Science, 2003, 300, 931–936 CrossRef CAS.
- N. C. Andrews, AJP Cell Physiol., 2004, 287, C1537–C1538 Search PubMed.
- F. Petrat, H. de Groot and U. Rauen, Biochem. J., 2001, 356, 61–69 CrossRef CAS.
- F. Petrat, H. de Groot, R. Sustmann and U. Rauen, Biol. Chem., 2002, 383, 489–502 CAS.
- M. Kruszewski, Acta Biochim. Pol., 2004, 51, 471–480 CAS.
- C. Jacob and P. G. Winyard, Redox Signaling and Regulation in Biology and Medicine, Wiley-VCH, 2009 Search PubMed.
- N. M. Giles, A. B. Watts, G. I. Giles, F. H. Fry, J. A. Littlechild and C. Jacob, Chem. Biol., 2003, 10, 677–693 CrossRef CAS.
- F. H. Fry and C. Jacob, Curr. Pharm. Des., 2006, 12, 4479–4499 CrossRef CAS.
- C. Jacob and A. Anwar, Physiol. Plant., 2008, 133, 469–480 CrossRef CAS.
- C. Pourzand, R. D. Watkin, J. E. Brown and R. M. Tyrrell, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6751–6756 CrossRef CAS.
- R. A. Pufahl, C. P. Singer, K. L. Peariso, S. J. Lin, P. J. Schmidt, C. J. Fahrni, V. C. Culotta, J. E. PennerHahn and T. V. OHalloran, Science, 1997, 278, 853–856 CrossRef CAS.
- E. Luk, L. T. Jensen and V. C. Culotta, JBIC, J. Biol. Inorg. Chem., 2003, 8, 803–809 CrossRef CAS.
- P. J. Schmidt, M. Ramos-Gomez and V. C. Culotta, J. Biol. Chem., 1999, 274, 36952–36956 CrossRef CAS.
- A. C. Rosenzweig, Acc. Chem. Res., 2001, 34, 119–128 CrossRef CAS.
- A. C. Rosenzweig, Chem. Biol., 2002, 9, 673–677 CrossRef CAS.
- L. Missiaen, L. Raeymaekers, L. Dode, J. Vanoevelen, K. Van Baelen, J. B. Parys, G. Callewaert, H. De Smedt, S. Segaert and F. Wuytack, Biochem. Biophys. Res. Commun., 2004, 322, 1204–1213 CrossRef CAS.
- M. F. Guarino, A. M. Ryan, A. Harto, B. Perez-Garcia, J. M. Arrazola and P. Jaen, J. Dermatol. Treatment, 2008, 19, 288–290 Search PubMed.
- W. J. Koeyers, S. Van der Geer and G. Krekels, J. Dermatol. Treatment, 2008, 19, 251–254 Search PubMed.
- K. Z. Bencze, K. C. Kondapalli, J. D. Cook, S. McMahon, C. Millan-Pacheco, N. Pastor and T. L. Stemmler, Crit. Rev. Biochem. Mol. Biol., 2006, 41, 269–291 CrossRef CAS.
- J. M. Cooper and A. H. V. Schapira, Biofactors, 2003, 18, 163–171 Search PubMed.
- R. Lodi, C. Tonon, V. Calabrese and A. H. V. Schapira, Antioxid. Redox Signaling, 2006, 8, 438–443 Search PubMed.
- T. Yoon and J. A. Cowan, J. Am. Chem. Soc., 2003, 125, 6078–6084 CrossRef CAS.
- K. Z. Bencze, T. Yoon, C. Millan-Pacheco, P. B. Bradley, N. Pastor, J. A. Cowan and T. L. Stemmler, Chem. Commun., 2007, 1798–1800 RSC.
- H. A. Dailey, C. K. Wu, P. Horanyi, A. E. Medlock, W. Najahi-Missaoui, A. E. Burden, T. A. Dailey and J. Rose, Biochemistry, 2007, 46, 7973–7979 CrossRef CAS.
- M. Hoggins, H. A. Dailey, C. N. Hunter and J. D. Reid, Biochemistry, 2007, 46, 8121–8127 CrossRef CAS.
- T. Karlberg, M. D. Hansson, R. K. Yengo, R. Johansson, H. O. Thorvaldsen, G. C. Ferreira, M. Hansson and S. Al-Karadaghi, J. Mol. Biol., 2008, 378, 1074–1083 CrossRef CAS.
- S. Al-Karadaghi, R. Franco, M. Hansson, J. A. Shelnutt, G. Isaya and G. C. Ferreira, Trends Biochem. Sci., 2006, 31, 135–142 CrossRef CAS.
- G. A. Hunter, M. P. Sampson and G. C. Ferreira, J. Biol. Chem., 2008, 283, 23685–23691 CrossRef CAS.
- E. Raux, H. L. Schubert and M. J. Warren, Cell. Mol. Life Sci., 2000, 57, 1880–1893 CAS.
- C. J. Walker and R. D. Willows, Biochem. J., 1997, 327, 321–333 CAS.
- I. Sekler, S. L. Sensi, M. Hershfinkel and W. F. Silverman, Mol. Med., 2007, 13, 337–343 CAS.
- M. L. Guerinot, Biochim. Biophys. Acta, Biomembr., 2000, 1465, 190–198 CrossRef CAS.
- J. P. Liuzzi and R. J. Cousins, Annu. Rev. Nutr., 2004, 24, 151–172 CrossRef CAS.
- A. L. Guerrerio and J. M. Berg, Biochemistry, 2004, 43, 5437–5444 CrossRef CAS.
- R. A. Bozym, R. B. Thompson, A. K. Stoddard and C. A. Fierke, Chem. Biol., 2006, 1, 103–111 CAS.
- Z. C. Ding, Q. Zheng, B. Cai, F. Y. Ni, W. H. Yu, X. C. Teng, Y. Gao, F. Liu, D. Chen, Y. Wang, H. M. Wu, H. Z. Sun, M. J. Zhang, X. S. Tan and Z. X. Huang, J. Inorg. Biochem., 2008, 102, 1965–1972 CrossRef CAS.
- J. D. Otvos, D. H. Petering and C. F. Shaw, Comments Inorg. Chem., 1989, 9, 1–35 CAS.
- C. Jacob, W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3489–3494 CrossRef CAS.
- A. Krezel and W. Maret, J. Am. Chem. Soc., 2007, 129, 10911–10921 CrossRef CAS.
- S. G. Bell and B. L. Vallee, ChemBioChem, 2009, 10, 55–62 CrossRef CAS.
- W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3478–3482 CrossRef CAS.
- A. Z. Mason, R. Moeller, K. A. Thrippleton and D. Lloyd, Anal. Biochem., 2007, 369, 87–104 CrossRef CAS.
- U. Heinz, M. Kiefer, A. Tholey and H. W. Adolph, J. Biol. Chem., 2005, 280, 3197–3207 CAS.
- R. R. Mendel, Dalton Trans., 2005, 3404–3409 RSC.
- G. Schwarz and R. R. Mendel, Annu. Rev. Plant Biol., 2006, 57, 623–647 CrossRef CAS.
- R. R. Mendel, J. Exp. Bot., 2007, 58, 2289–2296 CrossRef CAS.
- R. R. Mendel, A. G. Smith, A. Marquet and M. J. Warren, Nat. Prod. Rep., 2007, 24, 963–971 RSC.
- A. Llamas, K. L. Kalakoutskii and E. Fernandez, Plant, Cell Environ., 2000, 23, 1247–1255 CrossRef CAS.
- R. N. Pau and D. M. Lawson, Molybdenum and Tungsten: Their Roles in Biological Processes vol. 39, 2002, pp. 31–74 Search PubMed.
- J. R. Andreesen and K. Makdessi, Ann. N. Y. Acad. Sci., 2008, 1125, 215–229 CAS.
- M. Aguilar, K. Kalakoutskii, J. Cardenas and E. Fernandez, FEBS Lett., 1992, 307, 162–163 CrossRef CAS.
- F. S. Ataya, C. P. Witte, A. Galvan, M. I. Igeno and E. Fernandez, J. Biol. Chem., 2003, 278, 10885–10890 CrossRef CAS.
- F. Blasco, J. P. Dos Santos, A. Magalon, C. Frixon, B. Guigliarelli, C. L. Santini and G. Giordano, Mol. Microbiol., 1998, 28, 435–447 CrossRef CAS.
- S. Leimkuhler and W. Klipp, J. Bacteriol., 1999, 181, 2745–2751 CAS.
- S. Leimkuhler and K. V. Rajagopalan, J. Biol. Chem., 2001, 276, 1837–1844 CrossRef CAS.
- F. Bittner, M. Oreb and R. R. Mendel, J. Biol. Chem., 2001, 276, 40381–40384 CrossRef CAS.
- J. Nichols and K. V. Rajagopalan, J. Biol. Chem., 2002, 277, 24995–25000 CrossRef CAS.
- J. D. Nichols and K. V. Rajagopalan, J. Biol. Chem., 2005, 280, 7817–7822 CAS.
- A. Hartwig, M. Asmuss, H. Blessing, S. Hoffmann, G. Jahnke, S. Khandelwal, A. Pelzer and A. Burkle, Food Chem. Toxicol., 2002, 40, 1179–1184 CrossRef CAS.
- J. D. Otvos, X. L. H. Li, G. Shen and M. Basti, in Metallothionein III. Biological Roles and Medical Implications, eds. K. T. Suzuki, N. Imura and M. Kimura, Birkhäuser Verlag, Basel, 1993, pp. 57–74 Search PubMed.
- B. Zhang, O. Georgiev, M. Hagmann, C. Gunes, M. Cramer, P. Faller, M. Vasak and W. Schaffner, Mol. Cell. Biol., 2003, 23, 8471–8485 CrossRef CAS.
- G. Roesijadi, Cell. Mol. Biol., 2000, 46, 393–405 Search PubMed.
- G. Roesijadi, R. Bogumil, M. Vasak and J. H. R. Kagi, J. Biol. Chem., 1998, 273, 17425–17432 CrossRef CAS.
- J. Ejnik, A. Munoz, T. Gan, C. F. Shaw and D. H. Petering, JBIC, J. Biol. Inorg. Chem., 1999, 4, 784–790 CrossRef CAS.
- P. C. Huang, in Metallothionein III. Biological Roles and Medical Implications, ed. K. T. Suzuki, N. Imura and M. Kimura, Birkhäuser Verlag, Basel, 1993, pp. 407–426 Search PubMed.
- M. C. Posewitz and D. E. Wilcox, Chem. Res. Toxicol., 1995, 8, 1020–1028 CrossRef CAS.
- M. Huang, C. F. Shaw and D. H. Petering, J. Inorg. Biochem., 2004, 98, 639–648 CrossRef CAS.
- C. Kelley, D. E. Sargent and J. K. Uno, Curr. Pharm. Des., 1999, 5, 229–240 CAS.
- M. Blanusa, V. M. Varnai, M. Piasek and K. Kostial, Curr. Med. Chem., 2005, 12, 2771–2794 CrossRef CAS.
- K. Kalia and S. J. S. Flora, J. Occup. Health, 2005, 47, 1–21 Search PubMed.
- C. Gourlaouen and O. Parisel, Int. J. Quantum Chem., 2008, 108, 1888–1897 CrossRef CAS.
- H. Haas, Appl. Microbiol. Biotechnol., 2003, 62, 316–330 CrossRef CAS.
- L. Johnson, Mycol. Res., 2008, 112, 170–183 Search PubMed.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584–3588 CrossRef CAS.
- R. J. Abergel, M. C. Clifton, J. C. Pizarro, J. A. Warner, D. K. Shuh, R. K. Strong and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11524–11534 CrossRef CAS.
- K. L. Stirrett, J. A. Ferreras, V. Jayaprakash, B. N. Sinha, T. Ren and L. E. N. Quadri, Bioorg. Med. Chem. Lett., 2008, 18, 2662–2668 CrossRef CAS.
- J. L. Buss, B. T. Greene, J. Turner, F. M. Torti and S. V. Torti, Curr. Top. Med. Chem., 2004, 4, 1623–1635 CrossRef CAS.
- K. A. Mies, P. Gebhardt, U. Mollmann and A. L. Crumbliss, J. Inorg. Biochem., 2008, 102, 850–861 CrossRef CAS.
- J. L. Pierre, P. Baret and G. Serratrice, Curr. Med. Chem., 2003, 10, 1077–1084 CrossRef CAS.
- A. D. d’Hardemare, S. Torelli, G. Serratrice and J. L. Pierre, BioMetals, 2006, 19, 349–366 Search PubMed.
- Z. D. Liu and R. C. Hider, Med. Res. Rev., 2002, 22, 26–64 CrossRef CAS.
- R. C. Hider and T. Zhou, Cooley’s Anemia Eighth Symposium, 2005, vol. 1054, pp. 141–154 Search PubMed.
- M. Kitazawa, Y. Ishitsuka, M. Kobayashi, T. Nakano, K. Iwasaki, K. Sakamoto, K. Arakane, T. Suzuki and L. H. Kligman, Photochem. Photobiol., 2005, 81, 970–974 CrossRef CAS.
- J. B. Galey, J. Dumats, I. Beck, B. Fernandez and M. Hocquaux, Free Radical Res., 1995, 22, 67–86 CrossRef CAS.
- J. B. Galey, O. Destree, J. Dumats, P. Pichaud, J. Marche, S. Genard, G. Bracciolli, L. Le Capitaine, H. Plessix, L. Brambilla and O. Cantoni, Free Radical Biol. Med., 1998, 25, 881–890 CrossRef CAS.
- J. B. Galey, O. Destree, J. Dumats, S. Genard and P. Tachon, J. Med. Chem., 2000, 43, 1418–1421 CrossRef CAS.
- S. Seite, E. Popovic, M. P. Verdier, R. Roguet, P. Portes, C. Cohen, A. Fourtanier and J. B. Galey, Photodermatol. Photoimmunol. Photomed., 2004, 20, 47–52 CrossRef CAS.
- L. K. Charkoudian, T. Dentchev, N. Lukinova, N. Wolkow, J. L. Dunaief and K. J. Franz, J. Inorg. Biochem., 2008, 102, 2130–2135 CrossRef CAS.
- L. K. Charkoudian, D. M. Pham and K. J. Franz, J. Am. Chem. Soc., 2006, 128, 12424–12425 CrossRef CAS.
- L. K. Charkoudian, D. M. Pham, A. M. Kwon, A. D. Vangeloff and K. J. Franz, Dalton Trans., 2007, 5031–5042 RSC.
- A. Yiakouvaki, J. Savovic, A. Al-Qenaei, J. Dowden and C. Pourzand, J. Invest. Dermatol., 2006, 126, 2287–2295 CrossRef CAS.
- A. Pye, S. Campbell and A. Curnow, J. Cancer Res. Clin. Oncol., 2008, 134, 841–849 CrossRef CAS.
- T. J. Dougherty, J. E. Kaufman, A. Goldfarb, K. R. Weishaupt, D. Boyle and A. Mittleman, Cancer Res., 1978, 38, 2628–2635 CAS.
- S. H. Battah, C. E. Chee, H. Nakanishi, S. Gerscher, A. J. MacRobert and C. Edwards, Bioconjugate Chem., 2001, 12, 980–988 CrossRef CAS.
- G. Bhasin, H. Kausar and M. Athar, Cancer Lett., 2002, 187, 9–16 CrossRef CAS.
- Y. Okimura, H. Fujita, T. Ogino, K. Inoue, T. Shuin, H. Yano, T. Yasuda, M. Inoue, K. Utsumi and J. Sasaki, Physiol. Chem. Phys. Med. Nucl. Magn. Reson., 2007, 39, 69–82 Search PubMed.
- A. Curnow, B. W. McIlroy, M. J. Postle-Hacon, J. B. Porter, A. J. MacRobert and S. G. Bown, Br. J. Cancer, 1998, 78, 1278–1282 CAS.
- A. Curnow and A. Pye, J. Environ. Pathol. Toxicol. Oncol., 2007, 26, 89–103 Search PubMed.
- S. M. Campbell, C. A. Morton, R. Alyahya, S. Horton, A. Pye and A. Curnow, Br. J. Dermatol., 2008, 159, 387–393 CrossRef CAS.
- Z. D. Liu and R. C. Hider, Coord. Chem. Rev., 2002, 232, 151–171 CrossRef CAS.
- F. Yamamoto, Y. Ohgari, N. Yamaki, S. Kitajima, O. Shimokawa, H. Matsui and S. Taketani, Biochem. Biophys. Res. Commun., 2007, 353, 541–546 CrossRef CAS.
- A. Laurent, C. Nicco, J. T. Van Nhieu, D. Borderie, C. Chereau, F. Conti, P. Jaffray, O. Soubrane, Y. Calmus, B. Weill and F. Batteux, Hepatology, 2004, 39, 1277–1285 CrossRef CAS.
- A. Laurent, C. Nicco, C. Chereau, C. Goulvestre, J. Alexandre, A. Alves, E. Levy, F. Goldwasser, Y. Panis, O. Soubrane, B. Weill and F. Batteux, Cancer Res., 2005, 65, 948–956 CAS.
- C. Nicco, A. Laurent, C. Chereau, B. Weill and F. Batteux, Biomed. Pharmacother., 2005, 59, 169–174 CrossRef CAS.
- J. Alexandre, F. Batteux, C. Nicco, C. Chereau, A. Laurent, L. Guillevin, B. Weill and F. Goldwasser, Int. J. Cancer, 2006, 119, 41–48 CrossRef CAS.
- J. Alexandre, C. Nicco, C. Chereau, A. Laurent, B. Weill, F. Goldwasser and F. Batteux, J. Natl. Cancer Inst., 2006, 98, 236–244 CrossRef CAS.
- S. Mecklenburg, C. A. Collins, M. Doring, T. Burkholz, M. Abbas, F. H. Fry, C. Pourzand and C. Jacob, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 863–888 Search PubMed.
- D. Hare, B. Reedy, R. Grimm, S. Wilkins, I. Volitakis, J. L. George, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. Doble, Metallomics, 2009, 1, 53–58 RSC.
- H. Kodama and C. Fujisawa, Metallomics, 2009, 1, 42–52 RSC.
- J. J. R. F. D. Silva and R. J. P. Williams, The Biological Chemistry of the Elements: The Inorganic Chemistry of Life, Oxford University Press, Oxford, 2001 Search PubMed.
- C. E. Outten and T. V. O’Halloran, Science, 2001, 292, 2488–2492 CrossRef CAS.
- W. Braun, M. Vasak, A. H. Robbins, C. D. Stout, G. Wagner, J. H. Kagi and K. Wuthrich, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10124–10128 CAS.
- K. K. Kannan, M. Ramanadham and T. A. Jones, Ann. N. Y. Acad. Sci., 1984, 429, 49–60 CAS.
- C. T. Walsh, J. Liu, F. Rusnak and M. Sakaitani, Chem. Rev., 1990, 90, 1105–1129 CrossRef CAS.
Footnotes |
| † The numbers are generally provided as association constants Ka, which are sometimes measured in vitro or in vivo as Km values, or as dissociation constants Kd. |
| ‡ Proteins escorting and inserting metal ions to and into apoproteins are often considered together as ‘chaperones’. One should notice, however, that the two functions are rather separate and, as we will see, are sometimes—but not always—carried out by quite different proteins. The escorting proteins deliver the metal ion to the site of the apoprotein, while the metalloinsertases insert the metal ion into the active site of the apoprotein (see also Table 1). We will emphasize this distinction and use the term ‘metalloinsertase’ as a working hypothesis in order to distinguish them from proteins which function solely as escorts. ‘Metalloinsertases’ are presently known individually under different names, including ‘chelatase’ (e.g.ferrochelatase). |
| § In yeast, it has been postulated that just one free copper or zinc ion may be found per cell. Assuming a cellular volume of around 10−14 l, this would imply concentrations in the range of around 10−10 M.6 Similar calculations exist for other cell types. |
| ¶ Since disruptive procedures are frequently used before or during the measurement of the LIP, such numbers may vary significantly.17 The numbers provided here appear to be reasonable, yet should still be considered with some caution. |
| || Apart from OS, there are also other imbalances of the intracellular redox homeostasis. Some tumours, for instance, exhibit an unusually reducing intracellular environment related to hypoxia. Such reducing conditions enable the deployment of so-called ‘bioreductive’ drugs, which have been the subject of various recent reviews and will not be discussed here any further.21 |
| ** ‘Inorganic’ sulfide ions (S2−) are found primarily in iron/sulfur clusters. Recent studies on cellular signalling have also revealed an important role of H2S/HS− as a cellular signalling molecule (for more information on this topic see a recent review by Jacob and Anwar22). |
| †† The exact level of labile Zn2+ inside the (human) cell is still a matter of investigation (Table 2). In any case, the literature available to date points towards very low, picomolar concentrations of labile Zn2+ in the cytosol.48 |
| ‡‡ Apart from human MT-1 and MT-2, which are primarily found in the liver and kidneys, there are also other metallothioneins, such as MT-3, which is found in the human brain. The latter exhibits a rather complicated (bio-)chemistry on its own.49 We will refer here to MT-1 and MT-2 only. |
| §§ We are dealing here with similar concentrations of protein and ‘free’ metal ions. Of course, if a massive excess of ‘free’ Zn2+ would be used, some of the Cd2+ would be replaced by Zn2+ as part of the thermodynamic equilibrium. |
| ¶¶ We will use the term ‘rescue’ rather than ‘repair’, since the latter is usually associated with rescue via covalent, ‘organic’ modifications in DNA and/or proteins. Both expressions have been used in the metal trafficking literature.76 |
| |||| Indeed, chelators such as EDTA and pyridine-2,6-dicarboxylic acid (DPA) foster the rescuing process by removing Cd2+ from Cd–CA with rate constants of 1.9 × 10−6 s−1 and 3.4 × 10−6 s−1, respectively.77 |
| *** The origins of this area of research are associated with an early collaboration between two founding fathers of modern chemistry and medicine, namely Alfred Werner at the ETH Zurich and Paul Ehrlich at Frankfurt. These and related aspects of the history of chelate therapy have recently been reviewed.83 |
| This journal is © The Royal Society of Chemistry 2009 |