Inhibition of transcription by platinum antitumor compounds
Ryan C.
Todd
and
Stephen J.
Lippard
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: lippard@mit.edu
First published on 26th May 2009
Abstract
Cisplatin, carboplatin, and oxaliplatin are three FDA-approved members of the platinum anticancer drug family. These compounds induce apoptosis in tumor cells by binding to nuclear DNA, forming a variety of structural adducts and triggering cellular responses, one of which is the inhibition of transcription. In this report we present (i) a detailed review of the structural investigations of various Pt–DNA adducts and the effects of these lesions on global DNA geometry; (ii) research detailing inhibition of cellular transcription by Pt–DNA adducts; and (iii) a mechanistic analysis of how DNA structural distortions induced by platinum damage may inhibit RNA synthesis in vivo. A thorough understanding of the molecular mechanism of action of platinum antitumor agents will aid in the development of new compounds in the family.
 Ryan C. Todd and Stephen J. Lippard | Ryan C. Todd received his bachelor’s degree in chemistry from Johns Hopkins University in 2003, where he was an undergraduate research assistant under Professor David P. Goldberg. He then worked for two years as an analytical chemist at Merck & Co., Inc., before beginning his graduate studies at the Massachusetts Institute of Technology in 2005. His research in the laboratory of Professor Stephen J. Lippard focuses on the effects of platinum binding on the structure and functions of DNA and the nucleosome. Stephen J. Lippard is the Arthur Amos Noyes Professor of Chemistry at MIT. His research spans the fields of inorganic chemistry, biochemistry, and neurochemistry, with a focus on synthetic and mechanistic studies of platinum anticancer drugs, the preparation of dimetallic complexes as models for non-heme iron metalloenzymes, structural and mechanistic investigations of methane monooxygenase and related bacterial non-heme diiron multicomponent monooxygenases, and inorganic neurotransmitters, especially nitric oxide and zinc. |
Introduction
One of the great success stories in the field of cancer chemotherapy is that of cisplatin (cis-diamminedichloroplatinum(II), or cis-DDP) a curative treatment for testicular tumors.1 Approved by the Food and Drug Administration in 1978, cisplatin is also administered for several other forms of cancer, including ovarian, cervical, head and neck, esophageal, and non-small-cell lung cancers.1–3 Only in testicular cancer, however, does the drug reach greater than 90% cure rates, approaching 100% in early stage cases.4,5 Treatment can be limited by toxic side effects, including nephrotoxicity, emetogenesis, and neurotoxicity.1 Resistance to the drug, either acquired or inherent, is also common.6 Two other members of the platinum antitumor drug family, carboplatin and oxaliplatin (Fig. 1), have subsequently been approved for use in the United States. Whereas carboplatin and cisplatin are cross-resistant,7oxaliplatin has a different spectrum of activity and has become a first-line therapy for colorectal cancer.8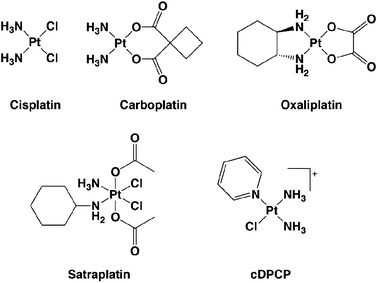 | ||
| Fig. 1 Chemical structures of platinum anticancer agents. Cisplatin, carboplatin, and oxaliplatin are FDA-approved for chemotherapy use in the United States. Satraplatin was the first Pt(IV) complex to reach Phase III clinical trials as an orally available platinum compound. cDPCP is a non-classical, monofunctional platinum complex with antitumor activity in colorectal cancer cells that inhibits transcription in vitro. | ||
Since the serendipitous discovery of its antineoplastic activity,9,10 many research groups have focused on revealing the molecular details of the mechanism of action of cisplatin and related compounds. The early steps of triggering cell death by platinum(II) compounds involve four stages. They are (1) cellular accumulation by both passive and active uptake; (2) activation of the platinum(II) complex; (3) binding to nucleic acids to form a variety of Pt–DNA adducts; and (4) the cellular response to DNA damage.11,12 For years it was thought that cisplatin entered cells primarily through passive diffusion, owing to data that showed platinum uptake was neither saturable nor inhibited by structural analogues.13–15 However, a growing body of evidence suggests a role for active uptake by membrane proteins, such as the copper transporter CTR1, in cisplatin accumulation.16,17Cisplatin activation involves replacement of the chloride ligands with water molecules in consecutive first order processes, driven by a drop in chloride ion concentration as the compound crosses the cell membrane.18 The aquated forms of cisplatin bind DNA at the N7 position of purine bases to form primarily 1,2-intrastrand adducts between adjacent guanosine residues.19 A smaller number of interstrand and monofunctional Pt–DNA adducts also form. The DNA damage leads to disruption of several cellular processes including transcription and replication. After cell cycle arrest occurs, the Pt lesions are either removed by nucleotide excision repair or apoptosis is triggered.
Early mechanistic studies led to the formation of several classical structure–activity relationships.20 In particular, it was hypothesized that an active platinum antitumor complex should have square-planar geometry, contain two labile leaving groups in a cis conformation, be neutral to facilitate passive diffusion across cell membranes, and contain inert amine ligands in the non-leaving-group positions. Since that time, however, many non-classical “rule-breakers,” including polynuclear platinum compounds,21 platinum(IV) complexes,22 monofunctional platinum(II) complexes,23 and compounds with trans sterochemistry,24,25 have been discovered with significant ability to destroy cancer cells.
The present article focuses on the DNA binding and cellular processing aspects of the mechanism of action of platinum anticancer complexes. We first review structural studies of various DNA adducts that arise from binding different members of the platinum antitumor drug family. Cisplatin and related complexes bound to DNA have been thoroughly studied by both X-ray crystallography and NMR spectroscopy, yielding abundant information about platinum modification of DNA structure. We next describe studies demonstrating that cisplatin blocks transcription and discuss data that implicate transcription inhibition as a major pathway involved in cancer cell death. Finally, we discuss current hypotheses detailing how platinum–DNA adducts block transcription by RNA polymerases and how this disruption can promote apoptosis through p53-dependent and -independent pathways.
DNA adducts formed by platinum antitumor agents
Cisplatin/carboplatin
Cisplatin forms a spectrum of intra- and interstrand DNA cross-links, which have been identified both in vitro and in vivo.26–29 The major adduct, comprising ∼65% of total products, is a cis 1,2-{Pt(NH3)2}2+-d(GpG) intrastrand cross-link. Other minor products include 1,2-d(ApG) (∼25%) and 1,3-d(GpNpG) (5–10%) intrastrand adducts, as well as a smaller number of interstrand cross-links (ICL) and monodentate adducts. Surprisingly, the 1,2-d(GpA) lesion is not observed either in vitro or in vivo.30 Although carboplatin forms the same type of adducts as cisplatin, the product profile is markedly different in cells.31 The major carboplatin adduct identified was cis-[Pt(NH3)2(dG)2] (36%), which could arise from 1,3-d(GpNpG) intrastrand cross-links. Minor products included 1,2-d(GpG) (30%), 1,2-d(ApG) (16%), as well as a small number of interstrand (3–4%) cross-links and monofunctional adducts. Because trans-diamminedichloroplatinum(II) is incapable of forming 1,2-intrastrand cross-links on DNA,32,33 and because this complex has insignificant antitumor activity in cells,34 intrastrand adducts are more likely to be responsible for the cytotoxicity of cisplatin in cancer cells. Further investigations of platinum interstrand cross-links revealed no correlation between the frequency of ICL formation and cytotoxicity, providing additional evidence that intrastrand cross-links are essential to tumor cell death.35X-Ray structural investigation of Pt–DNA adducts initially focused on platinated di- or trinucleotides.36–38 However, it was not until a platinated DNA dodecamer duplex containing a site-specific 1,2-{Pt(NH3)2}2+-d(GpG) intrastrand cross-link was solved by X-ray crystallography that fine details of the structure of Pt-damaged DNA began to emerge (see Fig. 2a).39,40 The X-ray crystal structure revealed that the Pt adduct induces a global bend in the DNA duplex by 35–40° and unwinds the double helix by ∼25°. The major groove is compacted and the minor groove widened and flattened. The DNA takes on A-form properties to the 5′ side of the Pt cross-link and a B-form structure on the 3′ side of the 1,2-d(GpG) adduct. The roll angle between platinated guanine bases is 26°. This relatively shallow roll angle results in considerable strain being placed on the Pt–N7 bonds, displacing the Pt atom out of the guanine ring planes by approximately 1 Å each. Subsequent NMR spectroscopic studies41,42 revealed differences between the solid state and solution structures, which could be traced to crystal packing interactions in the former. The solution structures showed bend angles of 60–70° and an exaggerated roll of 49° at the 1,2-{Pt(NH3)2}2+ d(GpG) cross-link. In addition, the NMR structures contained exclusively B-form DNA.
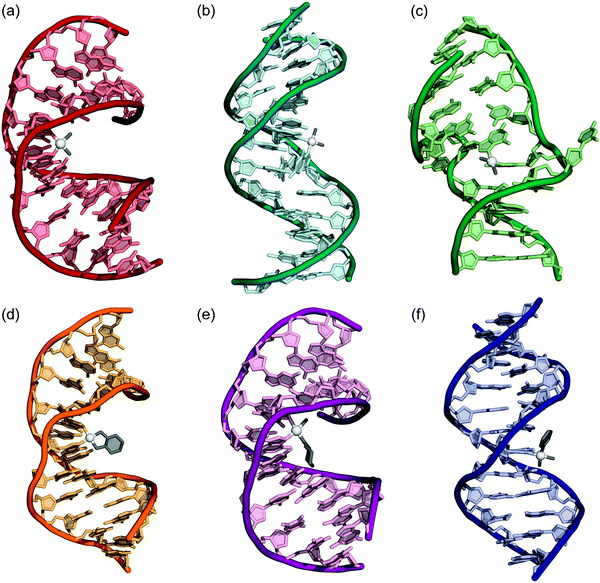 | ||
| Fig. 2 X-Ray crystal and NMR structures of double stranded DNA containing adducts of various platinum anticancer agents. (a) Cisplatin 1,2-d(GpG) intrastrand cross-link (1AIO). (b) Cisplatin 1,3-d(GpTpG) intrastrand cross-link (1DA4). (c) Cisplatin interstrand cross-link (1A2E). (d) Oxaliplatin 1,2-d(GpG) intrastrand cross-link (1PG9). (e) Satraplatin 1,2-d(GpG) intrastrand cross-link (1LU5). (f) cDPCP monofunctional adduct (3CO3). PDB accession codes are given in parentheses. | ||
Examination of the NMR structures of duplex DNAs containing a 1,2-{Pt(NH3)2}2+-d(GpG) cross-link revealed significant distortion of the DNAbase pair step to the 5′ side of the adduct.43,44 This conformational change is marked by unusually large and positive shift and slide values, indicating that the platinated base is significantly displaced toward the major groove. As will be discussed in more detail later, this feature is also present in structures of platinated DNA containing bound proteins and is believed to be a key recognition element for proteins that interact with platinated DNA.
In addition to the structure of the 1,2-intrastrand cross-link, that of the 1,3-intrastrand cross-link on duplex DNA has also been solved by NMR spectroscopy.45,46 This lesion, a likely major adduct of carboplatin–DNA binding, distorts double-stranded DNA in a different manner than the 1,2-d(GpG) cross-link (Fig. 2b). In this structure the duplex is bent by ∼30° and the double helix displays local unwinding and widening of the minor groove, similarly to features of the structure of the 1,2-d(GpG) cross-link. The 1,3-d(GpTpG) adduct differs, however, in that base pairing of the 5′ G*-C, where the asterisk denotes a platination site, is disrupted and the internal thymidine of the adduct is extruded from the minor groove. Although the area of the duplex in the immediate vicinity of the 1,3-d(GpTpG) adduct is more severely distorted than in the 1,2-d(GpG) counterpart, the global effects of the 1,3-cross-link on the DNA duplex are more subtle than for the 1,2-lesion, with a less dramatic bend angle.
The structure of a DNA molecule containing a site-specific interstrand cisplatin cross-link was solved both by X-ray crystallography47 (Fig. 2c) and by NMR spectroscopy.48 This Pt–DNA adduct is structurally unique in many ways compared to the intrastrand cross-links. In the ICL, the {Pt(NH3)2}2+ moiety binds in the minor groove and bends the helix by 47° in that direction. The double helix is severely unwound, by 110°, with the result that the two cytosine bases opposite the bound guanosines are pointed outward, away from the duplex. As in the intrastrand cross-links, the Pt–N7 bonds are strained, with the Pt atom being displaced from the guanine ring planes by 0.3–0.6 Å. Platinum ICLs on DNA also adopt a unique head-to-tail binding conformation whereby the ligating guanine bases are oriented in opposite directions.49 Head-to-head binding is observed in all intrastrand cross-links of cisplatin on double-stranded DNA.
Oxaliplatin
Oxaliplatin produces a similar type of DNA adduct spectrum to cisplatin and carboplatin, with the difference being that oxaliplatin–DNA lesions contain a {Pt(DACH)}2+ (DACH = trans-R,R-diaminocyclohexane) rather than a {Pt(NH3)2}2+ group.50,51DNA duplexes containing site-specific 1,2-{Pt(DACH)}2+-d(GpG) adducts have been studied both by X-ray crystallography52 and by NMR spectroscopy.53 The X-ray crystal structure is very similar to that of the analogous cisplatin-damaged DNA, with the duplex bent toward the major groove and the double helix taking on an A/B-form hybrid structure. The NMR solution structure (Fig. 2d) revealed the oxaliplatin-damaged DNA to be mostly B-form, further emphasizing the effect of crystal packing on the X-ray structure. The solution structure was overall very similar to that of DNA bearing a cisplatin 1,2-d(GpG) cross-link, but the global bend angle was only 31°, compared to 80° for the cisplatin adduct.Despite the similarity of the oxaliplatin-1,2-d(GpG) structure to that of the cisplatin lesion, several conformational differences were observed.54,55 The cisplatin cross-link preferentially forms hydrogen bonding interactions on the 5′ side of the adduct and causes more structural distortion to the base pair step at the 5′ end. Conversely, oxaliplatin forms hydrogen bonds more readily to the 3′ side of the intrastrand cross-link. Particularly pronounced is an interaction between a hydrogen atom of the NH2 group of the DACH ligand and the O6 oxygen atom of the 3′ guanine base.52 This interaction can form only with the biologically active R,R-isomer of oxaliplatin and not the inactive S,S-isomer. It has been postulated that these conformational differences between oxaliplatin– and cisplatin–DNA adducts may be responsible for differences in protein recognition and cellular processing of the two platinum antitumor compounds.54–56
Non-classical platinum compounds
In addition to cisplatin/carboplatin and oxaliplatin, the DNA adducts of several additional cytotoxic platinum compounds have been structurally characterized. The first of these complexes to be investigated was satraplatin, c,c,t-ammine(cyclohexylamine)dichlorodiacetatoplatinum(IV), a platinum(IV) complex that reached Phase III clinical trials for treatment of hormone-refractory prostate cancer.57,58 The axial acetate ligands are released as the platinum complex is reduced in the bloodstream, and the resulting platinum(II) complex binds DNA in a manner analogous to that of cisplatin. Two orientational isomers form, in which the cyclohexylamine ligand is pointed either toward the 3′ or the 5′ end of the platinated DNA strand.59 These adducts appear in approximately a 2 : 1 ratio in favor of the 3′-orientational isomer. The structure of the major isomer of this asymmetric bifunctional 1,2-d(GpG) adduct was characterized crystallographically on a dodecamer duplex. cis-Ammine(cyclohexylamine)platinum(II)–DNA adducts derived from satraplatin cause the same conformational changes to the double helix as other platinum 1,2-d(GpG) cross-links (Fig. 2e).60Recently a cationic platinum(II) complex containing three inert amine ligands and only one labile leaving group, cis-diammine(pyridine)chloroplatinum(II), or cDPCP, was crystallized bound to a single guanosine residue of a DNA duplex.61 The resulting X-ray crystal structure provided the first geometric information about an antitumor active monofunctional platinum–DNA adduct (Fig. 2f).23,62 This complex inhibits transcription at a level comparable to that of cisplatin as revealed by in vitro studies. Like oxaliplatin, it is taken up by cells bearing organic cation transporters (OCTs).61,63 This property presents an opportunity for selective delivery to colorectal tumor cells that express OCT membrane proteins in high abundance.63,64 The global structure of cDPCP-damaged DNA is quite different from that of DNA containing a platinum intrastrand d(GpG) cross-link. The latter platinated duplex is bent by ∼40° towards the major groove at the site of the cross-link, yet the monofunctional platinum–dG lesion causes no significant distortion of the double helix. Like the cisplatin intrastrand cross-link, however, the monofunctional adduct creates a distorted base pair step to the 5′ side of the platinum site that may be correlated to antitumor activity.61
Effects of platinum binding on nucleosome structure
In eukaryotic organisms, ∼80% of genomic DNA is wrapped in nucleosomes, which consist of 146 base pairs of DNA wrapped in a left-handed superhelix around a core of eight histone proteins.65,66 It is therefore necessary to consider this component of the cellular environment when studying the interactions of platinum compounds with their biological target. Our lab has used chemical methods to study the effects of cisplatin binding on nucleosome structure and positioning.67–69Double-stranded DNAs containing a centralized site-specific 1,2-d(GpG) or 1,3-d(GpTpG) cross-link of cisplatin were reconstituted into nucleosome core particles and analyzed by hydroxyl radical and exonuclease footprinting. These investigations revealed that cisplatin intrastrand cross-links direct nucleosome positioning to a preferred rotational and translational setting, with the {Pt(NH3)2}2+ moiety directed inwards toward the histone octamer protein core. This preferred position overrides that of strong native DNA positioning sequences and occurs in nucleosomes prepared from both native, containing a variety of post-translational modifications, and recombinant histones . Studies from another group demonstrated that cisplatin or oxaliplatin adducts inhibit ATP-independent nucleosome mobility in samples of nucleosome core particles treated with either drug.70 These data demonstrate that platinum complexes influence not only the structure of the DNA double helix, but also that of nucleosomes.Protein binding to platinum–DNA adducts
A number of proteins have been identified that bind to Pt–DNA adducts with specificity over unmodified DNA, including those associated with DNA repair, HMG-domain proteins, transcription factors, and others.11 Within the scope of this review, only proteins that play a role in eukaryotic transcription will be discussed. Transcription factors that bind Pt–DNA include human upstream binding factor (hUBF), TATA-binding protein (TBP), and Y-box binding protein (YB-1). High-mobility group box protein 1 (HMGB1), a very abundant non-histone chromosomal protein, binds cisplatin–DNA adducts tightly and with selectivity. HMGB1 is implicated to play a role in the mechanism of action of cisplatin in a variety of ways. Structure specific recognition protein 1 (SSRP1), an HMG-domain containing protein, is a subunit of FACT (facilitates chromatin transcription), which is a critical chromatin remodeling factor involved in transcription of nucleosomal DNA. SSRP1 was one of the first HMG-domain proteins known to bind platinated DNA.Upstream binding factor
The interaction between HMG-domain proteins and Pt–DNA adducts has been thoroughly studied.71,72 One member of this class of proteins, the ribosomal RNAtranscription factor hUBF, binds the cisplatin 1,2-d(GpG) cross-link with a Kd of 60 pM, the highest known affinity of any protein toward a Pt–DNA lesion.73 In an in vitro transcription assay with RNA polymerase I, treatment of DNA with cisplatin inhibited ribosomal RNA synthesis by sequestering hUBF.74,75TATA-binding protein
The TATA-binding protein is a critical transcription factor for all three eukaryotic RNA polymerases (pol I, II, and III).76 This protein binds DNA at promoter sites in the minor groove, bending the double helix toward the major groove and causing a structural change similar to that of a cisplatin intrastrand cross-link (see Fig. 3a).77TBP binding to the 1,2-{Pt(NH3)2}2+-d(GpG) adduct is similar to that of a promoter binding in terms of affinity, with Kd ∼ 0.3–10 nM. The kinetics are also similar, with relatively slow on and off rates. TBP binds the 1,2-d(GpG) cross-link of cisplatin better than the 1,3-d(GpTpG) adduct.78 | ||
| Fig. 3 Protein recognition and binding to Pt–DNA adducts. (a) Overlay of X-ray crystal structures of TBP-bound DNA (1TGH, blue) and DNA containing a cisplatin 1,2-d(GpG) intrastrand cross-link (1AIO, burgundy). (b) X-Ray crystal structure of HMGB1 domain A bound to a cisplatin 1,2-d(GpG) intrastrand cross-link (1CKT). An intercalated phenylalanine residue plays a key role in substrate recognition. PDB accession codes are given in parentheses. | ||
Y-box binding protein
Another transcription factor that binds cisplatin-modified DNA is YB-1, a protein that recognizes an inverted CCAAT sequence termed the Y-box.79 This protein is important both for signaling of DNA damage and for cell proliferation. YB-1 binds selectively to 1,2-d(GpG), 1,2-d(ApG), and 1,3-d(GpTpG) cross-links of cisplatin,80 and is overexpressed in the nuclei of cisplatin-resistant cell lines.81,82mRNA for YB-1 is increased approximately 6-fold as a response to cisplatin treatment.83High-mobility group box protein 1
HMGB1 has been implicated to have a regulatory effect on many cellular processes involving DNA, including chromatin remodeling, recombination, replication, and transcription.84,85 The relationship between HMGB1 levels and cisplatin sensitivity is reviewed elsewhere.11 Interest in the role of HMGB1, and HMG-domain proteins in general, in mediating cisplatin cytotoxicity has stimulated much research into the interactions between the HMG domain and Pt–DNA adducts. HMGB1 contains two tandem HMG domains, A and B, and a C-terminal acidic tail. The binding affinity of domain A for the 1,2-{Pt(NH3)2}2+-d(GpG) adduct depends on the flanking nucleotide sequence, with Kd values ranging between 1.6–517 nM. The range of binding affinities for domain B is slightly weaker, between 48–1300 nM.86 The full-length protein binds the cisplatin intrastrand cross-link primarily through the A domain with a dissociation constant of 120 nM.87 HMGB1 also recognizes the interstrand cross-link of cisplatin, with approximately 5-fold lower affinity.88 The structure of a complex between a 16mer duplex DNA containing a centralized 1,2-d(GpG) cisplatin intrastrand cross-link and the A domain of HMGB1 was solved by X-ray crystallography (Fig. 3b).89 The HMG domain binds the adduct in the widened minor groove to the 3′ side of the platinated strand. A phenylalanine residue intercalates into a hydrophobic notch created by the cisplatin cross-link; binding of the domain is dramatically reduced when this residue is mutated to alanine. These data provide insight into the recognition of Pt–DNA adducts by all HMG-domain-containing, and possibly other, proteins.Structure specific recognition protein 1
SSRP1 was discovered from expression screening of a human B-cell cDNA library as a protein that binds to cisplatin modified DNA.72 This protein, along with Spt16, comprise the FACT heterodimer, which alleviates the nucleosomal barrier to transcription.90 FACT binds cisplatin globally-modified DNA and the 1,2-d(GpG) cross-link with specificity over undamaged DNA or DNA treated with trans-diamminedichloroplatinum(II).91 Isolated SSRP1 did not form a high-affinity complex with cisplatin–DNA adducts, demonstrating the requirement for Spt16 in recognition of the platinum damage, but the truncated HMG domain of SSRP1 did recognize the 1,2-d(GpG) cross-link. The affinity of this critical transcriptional mediator for cisplatin-DNA damage suggests that binding of SSRP1 and FACT to platinum cross-links may be important to the mechanism of transcription inhibition by this drug.Inhibition of transcription by platinum antitumor complexes
A key indication that cisplatin–DNA adducts inhibit transcription was uncovered when it was reported that G2 arrest of L1210 leukemia cells was required for apoptosis and that loss of DNA replication viability did not correlate with cell death.92–94 Prior to these results, inhibition of DNA replication had been widely considered to be a key to the mechanism of cisplatin cytotoxicity.95–97 These new data suggested that cells arrested in G2 phase because they could not synthesize the mRNA necessary to pass into mitosis, implicating transcription inhibition as a critical determinant in the pathway of apoptosis triggered by cisplatin. Since these reports, numerous systems employing both site-specifically and globally platinated DNA templates, with both recombinant proteins and living cells, have been designed to study inhibition of transcription by cisplatin and other platinum anticancer agents. Taken together, the data clearly demonstrate that the ability of a platinum complex to block RNA synthesis correlates directly with its efficacy as an antitumor agent.98Reconstituted systems
Initial studies of transcription inhibition by platinum antitumor agents utilized DNA containing site-specific Pt–DNA adducts transcribed by purified mammalian RNA polymerase II (pol II) and E. coliRNA polymerase (RNAP).99–102 Data from these experiments demonstrated that 1,2-d(GpG) and 1,2-d(ApG) adducts of cisplatin blocked both polymerases almost completely when placed on the DNA template strand, whereas transcription was only slightly inhibited when the lesions were placed on the non-template strand. The 1,2-{Pt(NH3)2}2+-d(GpG) cross-link reduced binding affinity of E. coli RNAP and increased the apparent Km of the enzyme by a factor of 4–5.100 Furthermore, 1,3-d(GpTpG) cross-links of both cis- and trans-diamminedichloroplatinum(II) strongly blocked elongation by both RNA polymerases.103 Modest inhibition was also observed when the 1,3-cross-link was located on the non-template, or coding, strand. Bifunctional Pt cross-links were much more effective at impeding transcription progression than monofunctional cisplatin adducts. Furthermore, arrested transcription elongation complexes were identified as substrates of the RNAtranscript cleavage reaction mediated by TFIIS, indicating that the stalled elongation complex is not released from template DNA.102 Other studies using globally platinated DNA probes and T7 or SP6 RNA polymerases showed that transcription was halted primarily at 1,2-d(GpG) or d(ApG) Pt adduct sites, and to a lesser extent at the cisplatin ICL locations,104 but no inhibition was observed due to monofunctional adducts of [Pt(dien)Cl]+ or cis-[Pt(NH3)2(H2O)Cl]+.105 Interstrand cross-links of trans-DDP were similarly effective at blocking these enzymes.106Use of an immobilized DNA template allows for a high degree of control over transcriptional experiments. Such systems have been utilized in more recent investigations of RNA polymerase inhibition by Pt–DNA adducts to provide additional mechanistic insight. In the first of these reports, site-specific 1,2-Pt-d(GpG) and 1,3-Pt-d(GpTpG) adducts of both cisplatin and oxaliplatin were incorporated into DNA strands that were subjected to both promoter-dependent and -independent transcription by T7 RNAP in a reconstituted system.107 All four adducts strongly block transcription by the enzyme, with the oxaliplatin 1,3-d(GpTpG) adduct providing the greatest inhibition, followed by cisplatin 1,3-d(GpTpG), cisplatin 1,2-d(GpG), then oxaliplatin 1,2-d(GpG) cross-links in decreasing order. It was also discovered that UTP is incorrectly incorporated into the RNA strand opposite the platinated guanosine residue and that stalled polymerases can resume transcriptional activity upon removal of the platinum adduct by cyanide treatment.
Studies in cell extracts and cell culture
Other investigations of platinated DNA templates were performed either in live cells or using cell extracts. The first such report utilized a plasmid containing a β-galactosidase (β-gal) reporter gene transfected into HeLa, CHO, or human lymphoblastoid cell lines.108 Transcriptional activity was monitored colorimetrically by addition of the β-gal substrate ortho-nitrophenol-β-galactoside. Plasmids treated with cisplatin inhibited transcription 2–3-fold more readily than plasmids treated with trans-DDP. In this system RNA pol II bypassed cis- and trans-DDP adducts with efficiencies of 0–16% and 60–70%, respectively, and approximately four-fold more trans-DDP relative to cisplatin was required to block gene expression by 63%. Transcription of adenovirus major late promoter containing templates by RNA pol II in cell extracts was inhibited by treatment with cisplatin in a concentration-dependent manner.109 Transcription of an undamaged template was also blocked by the addition of exogenous platinum-damaged DNA, indicating that platinum adducts may inhibit transcription initiation by hijacking essential transcription factors that bind Pt–DNA adducts. In the same study it was demonstrated that cisplatin adducts can inhibit transcription elongation as well. Site-specific 1,2-d(GpG) or 1,3-d(GpTpG) intrastrand cross-links of cisplatin were introduced into DNA and used as transcription templates. Both adducts were efficient blocks of T3 RNA polymerase, and the 1,3-d(GpTpG) cross-link inhibited transcription elongation by RNA pol II by 80%. Interestingly, pol II efficiently bypassed the 1,2-d(GpG) lesion, although this bypass may be a sequence-specific result, in light of other data99,102 that demonstrate nearly complete inhibition of pol II by the cisplatin 1,2-d(GpG) cross-link.Transcription of immobilized DNA templates containing site-specific Pt–DNA adducts in HeLa nuclear extracts revealed further details of pol II inhibition.110 The arrested enzyme remains stably associated with the Pt damage site and is capable of resuming transcription if the platinum is removed. In HeLa cell culture, stalled pol II was ubiquitylated by ubiquitin ligases at Lys-6, Lys-48, and Lys-63. However, only a portion of the modified enzyme was released from the DNA and degraded by proteasomes; the rest remained stably bound at the Pt–DNA adduct site.
Data from other studies measuring transcription fidelity in cells correlate well with those collected from in vitro experiments. Treatment of human fibroblast cells with 50 μM cisplatin resulted in a 45% decrease in mRNA levels and increased expression of p53 and p21.111 Treatment of mouse tumor cells stably transfected with the mouse mammary tumor virus promoter (MMTV) with cisplatin resulted in highly inhibited expression. MMTV has a well-characterized chromatin structure, and these experiments determined that, concomitant with reduced RNA levels, chromatin remodeling and transcription factor binding were also inhibited.112 These effects were not observed when the cells were treated with trans-DDP.
Transcription-coupled repair of Pt–DNA adducts
Transcription-coupled repair (TCR) is a sub-pathway of nucleotide excision repair (NER) that allows DNA damage sites recognized by stalled RNA polymerases to be preferentially removed.113 TCR deficiency in cells has been positively correlated with cisplatin sensitivity, whereas cells lacking proteins for global NER exhibit typical levels of resistance to platinum treatment.114,115 If transcription inhibition is a critical determinant of cytotoxicity by platinum drugs, then the mechanism by which Pt–DNA adducts elude transcription-coupled repair must be investigated. However, the TCR pathway in mammalian cells is not well understood, and there is much still to be learned about the mechanism of TCR and its role in processing Pt-DNA damage.The connection between transcription inhibition by cisplatin damage and DNA repair was investigated using immobilized DNA templates.116,117 A site-specific 1,3-{Pt(NH3)2}2+-d(GpTpG) intrastrand cross-link was incorporated into the template DNA to provide an absolute block to transcription by pol II. The fate of the stalled polymerase and repair of the cisplatin lesion were then examined both in whole cell extracts and in a reconstituted system. In repair-proficient extracts, the Pt–DNA adduct was removed by dual excision without release of pol II.116 The elongation complex stalled at the damage site was stable to detergent washes, but could be removed in an ATP-dependent process. RNA polymerase II containing a dephosphorylated carboxyl-terminal domain was more sensitive to release. In the reconstituted system, the stalled elongation complex recruited several repair proteins, including TFIIH, XPA, RPA, XPG, and XPF, in an ATP-dependent manner.117 In the presence of Cockayne syndrome group B protein (CSB), which is connected to both transcription and repair functions, the platinum lesion was excised and the RNA polymerase partially released.
RNA polymerase II is ubiquitylated in cells in response to transcription inhibition by cisplatin or UV-damage, or α-amanitin treatment.110,118 This effect is not observed in cells deficient in TCR,119 and has been demonstrated in both live cells and nuclear extracts. These results are consistent with ubiquitylation being an important step in the recognition of stalled pol II elongation complexes. Ubiquitylated pol II was partially released from template DNA and degraded by the proteasome, but the rest remained stably bound at the arrest site. The consequence of pol II removal by this mechanism has been debated: one possibility is that polymerase removal is required to allow access of repair proteins to the damage site. Another possibility is that degradation of the stalled polymerase triggers an alternative pathway to TCR.
The mechanism of transcription inhibition of Pt–DNA adducts
The evidence to date has shown that DNA adducts of platinum antitumor compounds inhibit eukaryotic transcription and strongly suggests that this process is directly correlated to its efficacy as a chemotherapy agent. More recently, effort has been focused on establishing the mechanism of this process. What is the molecular pathway linking formation of platinum–DNA adducts to disruption of RNA synthesis? Hypotheses about how cisplatin and its relatives inhibit transcription can be divided into three categories: (1) hijacking of transcription factors, (2) a physical block of the enzyme, and (3) inhibition at the stage of chromatin remodeling (Fig. 4). These hypotheses are not mutually exclusive; one or more of them may play a role in the cisplatin mechanism of action. Because previous studies suggest that platinum anticancer agents block transcription at both the initiation and elongation stages, it is likely that inhibition occurs by more than one mechanism.Transcription factor hijacking
According to the transcription factor hijacking hypothesis, Pt–DNA adducts inhibit RNA synthesis by serving as binding sites for transcription factors such as TBP that have high affinity for platinated DNA. Interactions with cisplatin adducts prevent these transcription factors from binding their native promoter sites, thus inhibiting transcription at the initiation stage. The strongest evidence for this theory comes from the observation that transcription of an undamaged DNAplasmid in human cell extracts can be inhibited in a concentration-dependent manner by introduction of an exogeneous cisplatin-modified DNA substrate.109 Furthermore, it was demonstrated that microinjection of TBP into living cells in which transcription levels had been reduced by either cisplatin- or UV-damage resulted in reversal of the inhibition.120 A similar but less dramatic effect was observed after introduction of the basal transcription factors TFIIB and TFIIH. Analysis of the X-ray crystal structures of the TATA–TBP complex121 and double-stranded DNA containing the 1,2-d(GpG) intrastrand cross-link of cisplatin reveals strong similarities between the structures of the double helix in each model (Fig. 3a).120 The bifunctional platinum adduct creates a bent DNA structure that mimics its protein-bound form. Together these data collectively suggest that transcription factor hijacking by platinum–DNA adducts prevents the assembly of transcription elongation complexes at promoter sites and inhibits the initiation of RNA synthesis.Roadblock of RNA polymerases
Data from many in vitro transcription systems indicate that platinum–DNA adducts inhibit RNA polymerases at the site of the cisplatin cross-link,102,107,117 suggesting that the DNA adducts serve as a physical impediment to transcription elongation by the enzyme. Recently an X-ray crystal structure of RNA pol II stalled at a cisplatin 1,2-d(GpG) intrastrand cross-link revealed how the DNA damage may inhibit the enzyme.122 In this structure the Pt adduct is located downstream of the pol II active site, in the +2/+3 positions along the template strand; ribonucleotide addition occurs at the +1 position of the template. Attempts to place the cross-link inside the active site of the elongation complex resulted in backtracking of the polymerase so that the damage site was again located downstream of the reaction site. This result indicates that the adduct is not stably accommodated in the +1/+2 or −1/+1 positions. From these data and from analysis of the X-ray crystal structure, the authors proposed that the cisplatin cross-link inhibits pol II due to a translocation barrier whereby the bases in the platinated dinucleotide cannot rotate properly to allow entry into the enzyme active site, stalling the protein upstream of the platinum damage site. Biochemical experiments demonstrated that the elongation complex misincorporates AMP across from the 3′ guanosine of the Pt lesion. The kinetics and manner of this process are consistent with established nontemplated AMP incorporation known as the “A-rule,”123 which provides more evidence that the Pt cross-link never enters the pol II active site. Stalling is independent of the G-A mismatch, indicating that the translocation barrier arising from the Pt cross-link, not the mismatch, is primarily responsible of pol II inhibition. Finally, the authors demonstrated that if the intrastrand cross-link is introduced and transiently stabilized upstream of the proposed translocation barrier, then lesion bypass and revived transcription occurs.The data discussed above describe a manner by which bifunctional Pt–DNA adducts can serve as a physical roadblock to pol II. This hypothesis may not apply, however, to monofunctional complexes or any non-classical platinum compounds that do not form intrastrand cross-links with DNA. We have proposed a related but different theory as to how certain monofunctional Pt complexes may inhibit transcription.61 We demonstrated that cDPCP (Fig. 1) has antitumor activity in certain systems and that transcription in HeLa cells of a plasmid treated with cDPCP is impaired at a level similar to that of cisplatin. Because only one DNA base is covalently bound by the Pt complex, there is no rotational barrier that prevents entrance of the Pt lesion into the pol II active site. We have suggested that, when the Pt–DNA adduct is in the +1 position, steric interactions between the aromatic pyridine ligand and a nearby α-helix, termed the “bridge helix,” in the enzyme would change the conformation of the guanosine base on the DNA template, possibly preventing recognition by incoming CTP and leading to stalling of the polymerase.124 That monofunctional Pt complexes lacking an aromatic or bulky ligand cis to the DNA-binding site are typically not antitumor active is evidence to support this hypothesis,23 because these complexes would not create such steric interactions with the protein. This mechanism might also apply to platinum–DNA inter- and intrastrand cross-links.
Disruption of chromatin remodeling
Another possible manner by which cisplatin–DNA adducts may interfere with transcription could occur at the level of chromatin reorganization. Proper nucleosomal positioning and mobility are critical to the fidelity of transcription.125,126 Initial transcription factor binding occurs at DNA promoter sites that are characteristically nucleosome-free, which allows the proteins to recognize and bind the naked DNA sequence. As the RNA polymerase elongation complex subsequently transcribes along the template DNA, nucleosomes are continually shifted and unwrapped by chromatin remodeling complexes such as FACT.90,126 Data on the effects of cisplatin intrastrand cross-links on nucleosome positioning and mobility suggest that platinum damage can inhibit transcription at both the initiation and elongation stages by altering the nucleosomal organization of promoter sites and reducing nucleosome mobility, respectively. | ||
| Fig. 4 Possible mechanisms of transcription inhibition by platinum antitumor agents. Platinum-modified DNA can recruit transcription factors to the damage site, preventing these proteins from binding promoter sites and blocking formation of transcriptional complexes. The Pt–DNA adduct can also serve as a physical block to RNA polymerases when the lesion is located on the transcribed DNA strand. Finally, Pt–DNA adducts can disrupt nucleosomal structure and/or mobility and block transcription by prohibiting access to DNA by transcriptional proteins. | ||
Nucleosome positioning is determined primarily by the intrinsic DNA sequence.65,127 Research in our lab has demonstrated that DNA containing a site-specific 1,2-d(GpG) or 1,3-d(GpTpG) intrastrand cross-link of cisplatin enforces a characteristic rotational positioning of the DNA strand around the octamer, such that the Pt adduct faces inward towards the histone core.67–69 This effect, if it were to occur in vivo, could disrupt native nucleosomal organization and potentially disturb protein recognition of binding sites by placing a nucleosome at the promoter position.
In contrast to these results, research from another lab concluded that platinum damage does not significantly affect nucleosome positioning. In these experiments nucleosome core particles were treated with cisplatin or oxaliplatin, and both electrophoretic mobility shift assays and X-ray crystallographic studies of crystals of NCP treated with the drugs indicated no structural changes.70,128 The difference between the two sets of experiments is that the former results describe formation of nucleosomes from site-specifically platinated DNA, where the position and structure of the adducts are known, whereas the latter involve global treatment of nucleosomes with cisplatin or oxaliplatin, which will provide a heterogeneous panoply of adducts. The latter authors suggest that platinum binds nucleosomal DNA in positions where adducts are most readily accommodated by the nucleosome structure, thus reinforcing the native positioning preference instead of modifying it. They find, however, that nucleosomes treated with cisplatin or oxaliplatin have significantly decreased heat-induced mobility. Inhibited nucleosomal sliding could also inhibit transcription by preventing access of the RNA polymerase to the DNA template. A similar mechanism has been proposed for a series of pyrrole-imidazolepolyamides that bind nucleosomal DNA, inhibit nucleosomal mobility, and block transcription from a nucleosomal template, but not from naked DNA.129–132 These polyamides reduce nucleosome sliding through a proposed blockage of DNA rotation around the histone octamer. Data from this system suggest that inhibition of nucleosome mobility is sufficient to reduce transcriptional activity. Given that platinum intrastrand cross-links cause a similar reduction in DNA sliding on the nucleosome, cisplatin may also block transcription through this twist diffusion mechanism.
From transcription inhibition to apoptosis
Many transcriptional inhibitors have been tested as antitumor agents, including compounds that bind directly to RNA polymerases133,134 and agents that block transcription by inhibiting phosphorylation of pol II by CDK9.135,136Inhibition of transcription induces a cellular response leading to the activation of p53, a tumor suppressor protein, through the ATRkinase.111,137 Induction of p53 connects blocked RNA polymerase with cell cycle checkpoints, DNA repair, and apoptosis. After a certain time point, if the transcription block persists, the cells will undergo apoptosis in either a p53-dependent or -independent manner. The mechanism of this process is not clearly understood, but several pathways have been proposed, as reviewed in detail elsewhere.137,138 We briefly discuss them here. In general, the half-lives of mRNAs encoding for pro-apoptotic proteins such as Bax or Bid are longer than for anti-apoptotic proteins such as Bcl-2 or Mcl-1; thus inhibition of transcription over an extended period of time may adjust the ratios of pro- and anti-apoptotic proteins to conditions favoring cell death.139,140 Transcription inhibition may also lead to apoptosis through either p53-dependent or -independent pathways. The role of p53 in this process is highly controversial; the p53-dependent pathway may involve translocation of the protein to mitochondria so that it can bind anti-apoptotic Bcl-2 family proteins and induce cell death.141 However, other evidence suggests that transcription blockage can induce apoptosis in cells lacking p53.142–144 A third possibility is that stalled transcription elongation complexes can block the replication machinery if the polymerase is not removed from the template DNA prior to cell entry into S phase. Finally, export of certain proteins from the cell nucleus requires constant synthesis and export of mRNAs.145 It is therefore possible that apoptosis may result from inability to shuttle key proteins from the nucleus to the cytoplasm.The proposed pathways from inhibition of transcription to apoptosis have not been proved; more research is necessary to determine whether these processes are involved in cell death. They are mentioned here in the context of platinum antitumor drug mechanisms because a common characteristic of tumor cells is an impaired ability to undergo apoptosis.146 Resistance of certain cancers to cisplatin has been thoroughly investigated and many mechanisms are in play.6 However, one aspect often ignored by the community is that, although platinum compounds can bind DNA and inhibit replication and transcription, if the signaling pathway to apoptosis is malfunctioning in the cell, tumor destruction will not occur. We have initiated studies involving platinum complexes which, in addition to binding DNA, can stimulate the induction of apoptosis from mitochondria. We hypothesize that such complexes may be capable of overcoming this resistance mechanism.
Future directions
Much progress has been made in elucidating the mechanism of action of cisplatin, one of the most successful anticancer therapeutics to date. With this information in hand, researchers can begin to take a rational approach to designing new platinum complexes that specifically target these pathways. Traditionally the focus of platinum drug design has been to prepare compounds that form intrastrand DNA cross-links like cisplatin. In the future, research should focus not only on forming a certain type of DNA adduct, but on targeting and manipulating a cellular pathway triggered by the DNA damage. In recent years, the entire field of cancer research has moved away from general cytotoxic agents and focused more on targeted therapies, where delivered agents accumulate preferentially in the tumor and spare healthy tissue.147–150 Many strategies have been devised to synthesize tumor-specific platinum complexes.151–155 By combining transcription-inhibiting design with tumor-specific accumulation, researchers should be able to significantly improve the ability of platinum complexes to treat cancer.Acknowledgements
Research in our laboratory is supported by the National Cancer Institute under grant CA034992 to S. J. L. R. C. T. is grateful for a fellowship from the Koch Fund through the Koch Institute for Integrative Cancer Research.References
- P. J. Loehrer and L. H. Einhorn, Ann. Intern. Med., 1984, 100, 704–713 CAS.
- H. M. Keys, B. N. Bundy, F. B. Stehman, L. I. Muderspach, W. E. Chafe, C. L. Suggs III, J. L. Walker and D. Gersell, N. Engl. J. Med., 1999, 340, 1154–1161 CrossRef CAS.
- M. Morris, P. J. Eifel, J. Lu, P. W. Grigsby, C. Levenback, R. E. Stevens, M. Rotman, D. M. Gershenson and D. G. Mutch, N. Engl. J. Med., 1999, 340, 1137–1143 CrossRef CAS.
- G. J. Bosl, D. F. Bajorin, J. Sheinfeld, R. J. Motzer and R. S. K. Chaganti, in Cancer: Principles & Practice of Oncology, ed. V. T. DeVita Jr, S. Hellman and S. A. Rosenberg, Lippincott Williams & Wilkins, Philadelphia, 6th edn, 2001, pp. 1491–1518 Search PubMed.
- G. J. Bosl and R. J. Motzer, N. Engl. J. Med., 1997, 337, 242–253 CrossRef CAS.
- M. Kartalou and J. M. Essigmann, Mutat. Res., 2001, 478, 23–43 CrossRef CAS.
- R. Canetta, K. Bragman, L. Smaldone and M. Rozencweig, Cancer Treat. Rev., 1988, 15, 17–32 CrossRef.
- E. Raymond, S. Faivre, S. Chaney, J. Woynarowski and E. Cvitkovic, Mol. Cancer Ther., 2002, 1, 227–235 CAS.
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CAS.
- B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS.
- G. R. Gale, C. R. Morris, L. M. Atkins and A. B. Smith, Cancer Res., 1973, 33, 813–818 CAS.
- S. P. Binks and M. Dobrota, Biochem. Pharmacol., 1990, 40, 1329–1336 CrossRef CAS.
- D. P. Gately and S. B. Howell, Br. J. Cancer, 1993, 67, 1171–1176 CAS.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef CAS.
- A. K. Holzer, G. Samimi, K. Katano, W. Naerdemann, X. Lin, R. Safaei and S. B. Howell, Mol. Pharmacol., 2004, 66, 817–823 CrossRef CAS.
- M. E. Howe-Grant and S. J. Lippard, in Metal Ions Biol. Syst., ed. H. Sigel, Marcel Dekker, New York, 1980, vol. 11, pp. 63–125 Search PubMed.
- G. L. Cohen, J. A. Ledner, W. R. Bauer, H. M. Ushay, C. Caravana and S. J. Lippard, J. Am. Chem. Soc., 1980, 102, 2487–2488 CrossRef CAS.
- M. J. Cleare and J. D. Hoeschele, Bioinorg. Chem., 1973, 2, 187–210 CrossRef CAS.
- N. Farrell, in Metal Ions Biol. Syst., ed. A. Sigel and H. Sigel, Marcel Dekker, New York, 2004, vol. 42, pp. 251–296 Search PubMed.
- L. R. Kelland, G. Abel, M. J. McKeage, M. Jones, P. M. Goddard, M. Valenti, B. A. Murrer and K. R. Harrap, Cancer Res., 1993, 53, 2581–2586 CAS.
- L. S. Hollis, A. R. Amundsen and E. W. Stern, J. Med. Chem., 1989, 32, 128–136 CrossRef CAS.
- J. M. Pérez, M. A. Fuertes, C. Alonso and C. Navarro-Ranninger, Crit. Rev. Oncol. Hematol., 2000, 35, 109–120 CrossRef CAS.
- S. M. Aris and N. P. Farrell, Eur. J. Inorg. Chem., 2009, 2009, 1293–1302 CrossRef.
- J. P. Caradonna, S. J. Lippard, M. J. Gait and M. Singh, J. Am. Chem. Soc., 1982, 104, 5793–5795 CrossRef CAS.
- A. M. J. Fichtinger-Schepman, J. L. van der Veer, J. H. J. den Hartog, P. H. M. Lohman and J. Reedijk, Biochemistry, 1985, 24, 707–713 CrossRef CAS.
- A. Eastman, Biochemistry, 1986, 25, 3912–3915 CrossRef CAS.
- P. M. A. B. Terheggen, B. G. J. Floot, E. Scherer, A. C. Begg, A. M. J. Fichtinger-Schepman and L. den Engelse, Cancer Res., 1987, 47, 6719–6725 CAS.
- Y. Mantri, S. J. Lippard and M.-H. Baik, J. Am. Chem. Soc., 2007, 129, 5023–5030 CrossRef CAS.
- F. A. Blommaert, H. C. M. van Dijk-Knijnenburg, F. J. Dijt, L. den Engelse, R. A. Baan, F. Berends and A. M. J. Fichtinger-Schepman, Biochemistry, 1995, 34, 8474–8480 CrossRef CAS.
- C. A. Lepre, K. G. Strothkamp and S. J. Lippard, Biochemistry, 1987, 26, 5651–5657 CrossRef CAS.
- A. Eastman, M. M. Jennerwein and D. L. Nagel, Chem.-Biol. Interact., 1988, 67, 71–80 CrossRef CAS.
- J. M. Pascoe and J. J. Roberts, Biochem. Pharmacol., 1974, 23, 1345–1357 CrossRef CAS.
- W. DeNeve, F. Valeriote, E. Tapazoglou, C. Everett, A. Khatana and T. Corbett, Invest. New Drugs, 1990, 8, 17–24 CAS.
- S. E. Sherman, D. Gibson, A. H.-J. Wang and S. J. Lippard, Science, 1985, 230, 412–417 CrossRef CAS.
- G. Admiraal, J. L. van der Veer, R. A. G. de Graaff, J. H. J. den Hartog and J. Reedijk, J. Am. Chem. Soc., 1987, 109, 592–594 CrossRef CAS.
- S. E. Sherman, D. Gibson, A. H.-J. Wang and S. J. Lippard, J. Am. Chem. Soc., 1988, 110, 7368–7381 CrossRef CAS.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS.
- P. M. Takahara, C. A. Frederick and S. J. Lippard, J. Am. Chem. Soc., 1996, 118, 12309–12321 CrossRef CAS.
- D. Yang, S. S. G. E. van Boom, J. Reedijk, J. H. van Boom and A. H.-J. Wang, Biochemistry, 1995, 34, 12912–12920 CrossRef CAS.
- A. Gelasco and S. J. Lippard, Biochemistry, 1998, 37, 9230–9239 CrossRef CAS.
- L. G. Marzilli, J. S. Saad, Z. Kuklenyik, K. A. Keating and Y. Xu, J. Am. Chem. Soc., 2001, 123, 2764–2770 CrossRef CAS.
- S. T. Sullivan, J. S. Saad, F. P. Fanizzi and L. G. Marzilli, J. Am. Chem. Soc., 2002, 124, 1558–1559 CrossRef CAS.
- C. J. Van Garderen and L. P. A. Van Houte, Eur. J. Biochem., 1994, 225, 1169–1179 CrossRef CAS.
- J.-M. Teuben, C. Bauer, A. H.-J. Wang and J. Reedijk, Biochemistry, 1999, 38, 12305–12312 CrossRef CAS.
- F. Coste, J.-M. Malinge, L. Serre, W. Shepard, M. Roth, M. Leng and C. Zelwer, Nucleic Acids Res., 1999, 27, 1837–1846 CrossRef CAS.
- H. Huang, L. Zhu, B. R. Reid, G. P. Drobny and P. B. Hopkins, Science, 1995, 270, 1842–1845 CAS.
- S. T. Sullivan, A. Ciccarese, F. P. Fanizzi and L. G. Marzilli, J. Am. Chem. Soc., 2001, 123, 9345–9355 CrossRef CAS.
- M. M. Jennerwein, A. Eastman and A. Khokhar, Chem.-Biol. Interact., 1989, 70, 39–49 CrossRef CAS.
- J. M. Woynarowski, W. G. Chapman, C. Napier, M. C. S. Herzig and P. Juniewicz, Mol. Pharmacol., 1998, 54, 770–777 CAS.
- B. Spingler, D. A. Whittington and S. J. Lippard, Inorg. Chem., 2001, 40, 5596–5602 CrossRef CAS.
- Y. Wu, P. Pradhan, J. Havener, G. Boysen, J. A. Swenberg, S. L. Campbell and S. G. Chaney, J. Mol. Biol., 2004, 341, 1251–1269 CrossRef CAS.
- S. Sharma, P. Gong, B. Temple, D. Bhattacharyya, N. V. Dokholyan and S. G. Chaney, J. Mol. Biol., 2007, 373, 1123–1140 CrossRef CAS.
- Y. Wu, D. Bhattacharyya, C. L. King, I. Baskerville-Abraham, S.-H. Huh, G. Boysen, J. A. Swenberg, B. Temple, S. L. Campbell and S. G. Chaney, Biochemistry, 2007, 46, 6477–6487 CrossRef CAS.
- S. G. Chaney, S. L. Campbell, E. Bassett and Y. Wu, Crit. Rev. Oncol. Hematol., 2005, 53, 3–11 CrossRef.
- L. R. Kelland, Expert Opin. Invest. Drugs, 2000, 9, 1373–1382 Search PubMed.
- C. N. Sternberg, P. Whelan, J. Hetherington, B. Paluchowska, P. H. T. J. Slee, K. Vekemans, P. Van Erps, C. Theodore, O. Koriakine, T. Oliver, D. Lebwohl, M. Debois, A. Zurlo and L. Collette, Oncology, 2005, 68, 2–9 CrossRef CAS.
- J. F. Hartwig and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 5646–5654 CrossRef CAS.
- A. P. Silverman, W. Bu, S. M. Cohen and S. J. Lippard, J. Biol. Chem., 2002, 277, 49743–49749 CrossRef CAS.
- K. S. Lovejoy, R. C. Todd, S. Zhang, M. S. McCormick, J. A. D’Aquino, J. T. Reardon, A. Sancar, K. M. Giacomini and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8902–8907 CrossRef CAS.
- L. S. Hollis, W. I. Sundquist, J. N. Burstyn, W. J. Heiger-Bernays, S. F. Bellon, K. J. Ahmed, A. R. Amundsen, E. W. Stern and S. J. Lippard, Cancer Res., 1991, 51, 1866–1875 CAS.
- S. Zhang, K. S. Lovejoy, J. E. Shima, L. L. Lagpacan, Y. Shu, A. Lapuk, Y. Chen, T. Komori, J. W. Gray, X. Chen, S. J. Lippard and K. M. Giacomini, Cancer Res., 2006, 66, 8847–8857 CrossRef CAS.
- M. Hayer-Zillgen, M. Brüss and H. Bönisch, Br. J. Pharmacol., 2002, 136, 829–836 CrossRef CAS.
- E. Segal, Y. Fondufe-Mittendorf, L. Chen, A. Thåström, Y. Field, I. K. Moore, J.-P. Z. Wang and J. Widom, Nature, 2006, 442, 772–778 CrossRef CAS.
- R. D. Kornberg and Y. Lorch, Nat. Struct. Mol. Biol., 2007, 14, 986–988 CrossRef CAS.
- A. J. Danford, D. Wang, Q. Wang, T. D. Tullius and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12311–12316 CrossRef CAS.
- M. Ober and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 6278–6286 CrossRef CAS.
- M. Ober and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 2851–2861 CrossRef CAS.
- B. Wu and C. A. Davey, Chem. Biol., 2008, 15, 1023–1028 CrossRef CAS.
- W. M. Scovell, N. Muirhead and L. R. Kroos, Biochem. Biophys. Res. Commun., 1987, 142, 826–835 CrossRef CAS.
- S. L. Bruhn, P. M. Pil, J. M. Essigmann, D. E. Housman and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2307–2311 CAS.
- D. K. Treiber, X. Zhai, H.-M. Jantzen and J. M. Essigmann, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5672–5676 CAS.
- P. Jordan and M. Carmo-Fonseca, Nucleic Acids Res., 1998, 26, 2831–2836 CrossRef CAS.
- X. Zhai, H. Beckmann, H.-M. Jantzen and J. M. Essigmann, Biochemistry, 1998, 37, 16307–16315 CrossRef CAS.
- B. P. Cormack and K. Struhl, Cell (Cambridge, Mass.), 1992, 69, 685–696 CrossRef CAS.
- F. Coin, P. Frit, B. Viollet, B. Salles and J.-M. Egly, Mol. Cell. Biol., 1998, 18, 3907–3914 CAS.
- Y. Jung, Y. Mikata and S. J. Lippard, J. Biol. Chem., 2001, 276, 43589–43596 CrossRef CAS.
- D. K. Didier, J. Schiffenbauer, S. L. Woulfe, M. Zacheis and B. D. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7322–7326 CrossRef CAS.
- T. Ise, G. Nagatani, T. Imamura, K. Kato, H. Takano, M. Nomoto, H. Izumi, H. Ohmori, T. Okamoto, T. Ohga, T. Uchiumi, M. Kuwano and K. Kohno, Cancer Res., 1999, 59, 342–346 CAS.
- T. Ohga, K. Koike, M. Ono, Y. Makino, Y. Itagaki, M. Tanimoto, M. Kuwano and K. Kohno, Cancer Res., 1996, 56, 4224–4228 CAS.
- H. Yahata, H. Kobayashi, T. Kamura, S. Amada, T. Hirakawa, K. Kohno, M. Kuwano and H. Nakano, J. Cancer Res. Clin. Oncol., 2002, 128, 621–626 CrossRef CAS.
- H. Uramoto, H. Izumi, T. Ise, M. Tada, T. Uchiumi, M. Kuwano, K. Yasumoto, K. Funa and K. Kohno, J. Biol. Chem., 2002, 277, 31694–31702 CrossRef CAS.
- A. D. Baxevanis and D. Landsman, Nucleic Acids Res., 1995, 23, 1604–1613 CrossRef CAS.
- J. O. Thomas and A. A. Travers, Trends Biochem. Sci., 2001, 26, 167–174 CrossRef CAS.
- S. U. Dunham and S. J. Lippard, Biochemistry, 1997, 36, 11428–11436 CrossRef CAS.
- Y. Jung and S. J. Lippard, Biochemistry, 2003, 42, 2664–2671 CrossRef.
- J. Kasparkova, O. Delalande, M. Stros, M.-A. Elizondo-Riojas, M. Vojtiskova, J. Kozelka and V. Brabec, Biochemistry, 2003, 42, 1234–1244 CrossRef CAS.
- U.-M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, Nature, 1999, 399, 708–712 CrossRef CAS.
- D. Reinberg and R. J. Sims III, J. Biol. Chem., 2006, 281, 23297–23301 CrossRef CAS.
- A. T. Yarnell, S. Oh, D. Reinberg and S. J. Lippard, J. Biol. Chem., 2001, 276, 25736–25741 CrossRef CAS.
- C. M. Sorenson and A. Eastman, Cancer Res., 1988, 48, 4484–4488 CAS.
- C. M. Sorenson and A. Eastman, Cancer Res., 1988, 48, 6703–6707 CAS.
- C. M. Sorenson, M. A. Barry and A. Eastman, J. Natl. Cancer Inst., 1990, 82, 749–755 CrossRef CAS.
- H. C. Harder, R. G. Smith and A. F. Leroy, Cancer Res., 1976, 36, 3821–3829 CAS.
- N. P. Johnson, J. D. Hoeschele, N. B. Kuemmerle, W. E. Masker and R. O. Rahn, Chem.-Biol. Interact., 1978, 23, 267–271 CrossRef CAS.
- A. L. Pinto and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4616–4619 CAS.
- K. E. Sandman, S. S. Marla, G. Zlokarnik and S. J. Lippard, Chem. Biol., 1999, 6, 541–551 CrossRef CAS.
- Y. Corda, C. Job, M.-F. Anin, M. Leng and D. Job, Biochemistry, 1991, 30, 222–230 CrossRef CAS.
- Y. Corda, M.-F. Anin, M. Leng and D. Job, Biochemistry, 1992, 31, 1904–1908 CrossRef CAS.
- Y. Corda, C. Job, M.-F. Anin, M. Leng and D. Job, Biochemistry, 1993, 32, 8582–8588 CrossRef CAS.
- S. Tornaletti, S. M. Patrick, J. J. Turchi and P. C. Hanawalt, J. Biol. Chem., 2003, 278, 35791–35797 CrossRef CAS.
- E. Larsen, K. Kwon, F. Coin, J.-M. Egly and A. Klungland, DNA Repair, 2004, 3, 1457–1468 CrossRef CAS.
- M.-A. Lemaire, A. Schwartz, A. R. Rahmouni and M. Leng, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 1982–1985 CAS.
- V. Brabec, V. Boudný and Z. Balcarová, Biochemistry, 1994, 33, 1316–1322 CrossRef CAS.
- V. Brabec and M. Leng, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5345–5349 CAS.
- Y. Jung and S. J. Lippard, J. Biol. Chem., 2003, 278, 52084–52092 CrossRef CAS.
- J. A. Mello, S. J. Lippard and J. M. Essigmann, Biochemistry, 1995, 34, 14783–14791 CrossRef CAS.
- C. Cullinane, S. J. Mazur, J. M. Essigmann, D. R. Phillips and V. A. Bohr, Biochemistry, 1999, 38, 6204–6212 CrossRef CAS.
- Y. Jung and S. J. Lippard, J. Biol. Chem., 2006, 281, 1361–1370 CAS.
- M. Ljungman, F. Zhang, F. Chen, A. J. Rainbow and B. C. McKay, Oncogene, 1999, 18, 583–592 CrossRef CAS.
- J. S. Mymryk, E. Zaniewski and T. K. Archer, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2076–2080 CrossRef CAS.
- J. Q. Svejstrup, Nat. Rev. Mol. Cell Biol., 2002, 3, 21–29 CrossRef CAS.
- T. Furuta, T. Ueda, G. Aune, A. Sarasin, K. H. Kraemer and Y. Pommier, Cancer Res., 2002, 62, 4899–4902 CAS.
- J. T. Bulmer, N. J. Zacal and A. J. Rainbow, Cancer Chemother. Pharmacol., 2005, 56, 189–198 CrossRef CAS.
- A. Tremeau-Bravard, T. Riedl, J.-M. Egly and M. E. Dahmus, J. Biol. Chem., 2004, 279, 7751–7759 CAS.
- J.-P. Lainé and J.-M. Egly, EMBO J., 2006, 25, 387–397 CrossRef CAS.
- K.-B. Lee, D. Wang, S. J. Lippard and P. A. Sharp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4239–4244 CrossRef CAS.
- D. B. Bregman, R. Halaban, A. J. van Gool, K. A. Henning, E. C. Friedberg and S. L. Warren, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11586–11590 CrossRef CAS.
- P. Vichi, F. Coin, J.-P. Renaud, W. Vermeulen, J. H. J. Hoeijmakers, D. Moras and J.-M. Egly, EMBO J., 1997, 16, 7444–7456 CrossRef CAS.
- Z. S. Juo, T. K. Chiu, P. M. Leiberman, I. Baikalov, A. J. Berk and R. E. Dickerson, J. Mol. Biol., 1996, 261, 239–254 CrossRef CAS.
- G. E. Damsma, A. Alt, F. Brueckner, T. Carell and P. Cramer, Nat. Struct. Mol. Biol., 2007, 14, 1127–1133 CrossRef CAS.
- B. S. Strauss, BioEssays, 1991, 13, 79–84 CrossRef CAS.
- D. Wang, D. A. Bushnell, K. D. Westover, C. D. Kaplan and R. D. Kornberg, Cell (Cambridge, Mass.), 2006, 127, 941–954 CrossRef CAS.
- K. Luger, Chromosome Res., 2006, 14, 5–16 CrossRef CAS.
- J. L. Workman, Genes Dev., 2006, 20, 2009–2017 CrossRef CAS.
- N. Kaplan, I. K. Moore, Y. Fondufe-Mittendorf, A. J. Gossett, D. Tillo, Y. Field, E. M. LeProust, T. R. Hughes, J. D. Lieb, J. Widom and E. Segal, Nature, 2009, 458, 362–366 CrossRef CAS.
- B. Wu, P. Dröge and C. A. Davey, Nat. Chem. Biol., 2008, 4, 110–112 CrossRef CAS.
- J. M. Gottesfeld, C. Melander, R. K. Suto, H. Raviol, K. Luger and P. B. Dervan, J. Mol. Biol., 2001, 309, 615–629 CrossRef CAS.
- J. M. Gottesfeld, J. M. Belitsky, C. Melander, P. B. Dervan and K. Luger, J. Mol. Biol., 2002, 321, 249–263 CrossRef CAS.
- R. K. Suto, R. S. Edayathumangalam, C. L. White, C. Melander, J. M. Gottesfeld, P. B. Dervan and K. Luger, J. Mol. Biol., 2003, 326, 371–380 CrossRef CAS.
- R. S. Edayathumangalam, P. Weyermann, J. M. Gottesfeld, P. B. Dervan and K. Luger, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6864–6869 CrossRef CAS.
- H. M. Sobell, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5328–5331 CrossRef CAS.
- D. A. Bushnell, P. Cramer and R. D. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1218–1222 CrossRef CAS.
- M. V. Blagosklonny, Cell Cycle, 2004, 3, 1537–1542 Search PubMed.
- S. K. Radhakrishnan and A. L. Gartel, Cancer Res., 2006, 66, 3264–3270 CrossRef CAS.
- F. A. Derheimer, C.-W. Chang and M. Ljungman, Eur. J. Cancer, 2005, 41, 2569–2576 CrossRef CAS.
- A. L. Gartel, Biochim. Biophys. Acta, 2008, 1786, 83–86 CAS.
- L. T. Lam, O. K. Pickeral, A. C. Peng, A. Rosenwald, E. M. Hurt, J. M. Giltnane, L. M. Averett, H. Zhao, R. E. Davis, M. Sathyamoorthy, L. M. Wahl, E. D. Harris, J. A. Mikovits, A. P. Monks, M. G. Hollingshead, E. A. Sausville and L. M. Staudt, GenomeBiology, 2001, 2, 1–11.
- E. Yang, E. van Nimwegen, M. Zavolan, N. Rajewsky, M. Schroeder, M. Magnasco and J. E. Darnell, Jr, Genome Res., 2003, 13, 1863–1872 CAS.
- N. D. Marchenko, A. Zaika and U. M. Moll, J. Biol. Chem., 2000, 275, 16202–16212 CrossRef CAS.
- G. I. Shapiro, D. A. Koestner, C. B. Matranga and B. J. Rollins, Clin. Cancer Res., 1999, 5, 2925–2938 CAS.
- M. Alonso, C. Tamasdan, D. C. Miller and E. W. Newcomb, Mol. Cancer Ther., 2003, 2, 139–150 CAS.
- Z. N. Demidenko and M. V. Blagosklonny, Cancer Res., 2004, 64, 3653–3660 CrossRef CAS.
- H. M. O'Hagan and M. Ljungman, Exp. Cell Res., 2004, 297, 548–559 CrossRef CAS.
- D. Hanahan and R. A. Weinberg, Cell (Cambridge, Mass.), 2000, 100, 57–70 CAS.
- W. N. Hait, Cancer Res., 2009, 69, 1263–1267 CrossRef CAS.
- A. N. Gordon, M. Tonda, S. Sun and W. Rackoff, Gynecol. Oncol., 2004, 95, 1–8 CrossRef CAS.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O'Shaughnessy, J. Clin. Oncol., 2005, 23, 7794–7803 CrossRef CAS.
- I. Collins and P. Workman, Nat. Chem. Biol., 2006, 2, 689–700 CrossRef CAS.
- J. R. Rice, J. L. Gerberich, D. P. Nowotnik and S. B. Howell, Clin. Cancer Res., 2006, 12, 2248–2254 CrossRef CAS.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS.
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |