Characterization of metal–peptide complexes in feed supplements of essential trace elements
Alexandros
Yiannikouris
*a,
Cathal
Connolly
b,
Ronan
Power
a and
Ryszard
Lobinski
c
aAlltech Inc, 3031 Catnip Hill Pike, Nicholasville, KY 40356, USA
bAlltech Ireland, Sarney Summerhill Road, Dunboyne, County Meath, 78930, Ireland
cLaboratoire de Chimie Analytique Bio-inorganique et Environnement, CNRS UMR 5254, Hélioparc, 2 avenue Président Angot, 64053, Pau, France
First published on 16th April 2009
Abstract
Metal chelates with biomolecules are increasingly used in animal supplementation to increase the bioavailability of essential trace elements. However, the transfer of the chelates is not well understood and speciation studies may bring a comprehensive insight to further investigate the biological uptake mechanism(s) implicated. An analytical method was developed for the characterization of the water-soluble metal complexes in animal feed supplements obtained by reaction of a metal salt with a non-GMO soybean enzymatic digest. The method was based on fractionation of the extract by size-exclusion chromatography followed by the analysis of the metal-containing fraction by reversed-phase nanoHPLC with parallel ICP MS and electrospray MS/MS detection. The metal complexes were identified in the mass spectra owing to the Cu characteristic isotopic pattern; the complexation was corroborated by the presence of a peak corresponding to the non-metallated peptide. The study demonstrated the feasibility of SEC-ICP MS to produce characteristic metal (Cu, Zn, Mn, Fe) distribution patterns, which can be of interest to test batch-to-batch reproducibility and to determine the origin of the supplement. The use of the method could be extended to animal feeds prepared using the metal-chelated complexes. Electrospray MS/MS allowed the identification of a number of Cu complexes with peptides . Four different structure conformations were modeled by means of molecular mechanics investigations to assess the chelation stability.
Introduction
A number of essential minerals, such as copper (Cu), zinc (Zn), selenium (Se), iron (Fe), and manganese (Mn), are necessary in animals to maintain body function and to optimize growth, reproductive performance, and immune response. These minerals have been traditionally supplemented to animal diets to compensate for deficiencies, to promote optimum health, and to prevent serious disease.1,2 However, little research has been conducted to determine the most biologically available chemical forms of trace elements for either human or animal use.3 Evidence from the field of animal nutrition suggests that trace elements, when administered in the form of organic complexes are more biologically available than their inorganic counterparts.4Trace mineral supplementation has usually been achieved by the addition of simple inorganic salts such as sulfates and chlorides.5 However, it has been established that the bioavailability of trace elements can be enhanced by adding these elements in the form of a metal complex, chelated with amino acids or peptides .6–9 Indeed, the bioavailability of trace minerals strongly depends on their chemical form. The absorption mechanism of electrically uncharged organic complexes leads to a higher bioavailability of the element for the animal.10 For example, the bioavailability of inorganic Cu compounds in ruminants is very low because of their interaction with molybdenum and sulfur in the rumen.11,12 The use of Cu chelates with amino acids and/or partially hydrolyzed proteins is able to increase the rate of transfer of Cu into blood and liver.
The characterization of mineral supplements is an important task because of regulatory concerns requiring the verification of batch-to-batch reproducibility and the differentiation of supplements based on origin and manufacturer. The fine characterization of supplements is also the first step to the understanding of the absorption mechanisms that still remain elusive.13 So far most research has been devoted to Se-enriched yeast. Recent developments in multidimensional high performance liquid chromatography (HPLC) with the parallel inductively coupled plasma mass spectrometry (ICP MS) and electrospray MS/MS detection14–16 have allowed the increasingly detailed characterization of the organic forms of Se, both on the proteome17 and the metabolome level.18–20
The study of metal complexes with organic ligands is more difficult because they do not implicate covalent metal–carbon bonds as in the case of “organic” Se, but metal–nitrogen or metal–hydrogen non-covalent bonds that are coordinative and exhibit different stability constants. Examples of successful identification and characterization of metal–bioligand complexes in biological matrices are primarily limited to metal complexes with metallothioneins13,21 and with constitutive ligands in plants (e.g., phytochelatins22). Successful examples of electrospray MS data for metal–peptide complexes in biological matrices have, to our knowledge, been rarely reported. Typical obstacles include ionization suppression because of the concomitant more easily ionizable compounds present in the sample and the difficulties in the isolation of metal complexes because of their lability.
Within the past 10 years, soy foods and soybean constituents have attracted widespread research attention for their purported health benefits because of their plethora of bioactive phytochemicals, exceeding by far their potential adverse effects.23Isoflavones, saponins, phytic acids, phytosterols, trypsin inhibitors, and peptides have been characterized in soybean products for decreasing acute diarrhea, cholesterol level, menopausal symptoms, type II diabetic symptoms, osteoporosis, post-menopausal bone loss, cancer impact, and cardiovascular disease. Whole, raw soybeans have been also considered by many animal producers in the United States as an alternative source of supplemental protein.24
The fine chelation mechanisms of peptides issued from a complex matrix such as soybean have not yet been investigated and the associated amino acid sequences have not yet been characterized. The objectives of this research were (i) to develop a qualitative method to screen for the presence of metal-complexes in soybean protein hydrolysates and related feeds allowing the rapid characterization of the marketed supplements, and (ii) to develop a protocol allowing the identification of the metal–peptide complexes. The methods developed were based on HPLC with ICP MS detection, because of its sensitivity regardless of the matrix. This step allowed us to further optimize the analyte isolation and purification before its characterization through electrospray MS/MS. Better characterization of the complexes involved in the chelation of metals will ultimately bring new insight to the understanding of the intestinal absorption of organic minerals.
Experimental
Instrumentation
An Elan 6000 ICP mass spectrometer (Perkin Elmer SCIEX, Thornill, ON, Canada) or an Agilent 7500ce ICP MS equipped with a collision cell (Yokogawa Analytical Systems, Tokyo, Japan) were used alternately to detect 63Cu, 65Cu, 64Zn, 66Zn, 55Mn, 114Cd and 112Cd. The column eluate was introduced into the ICP via a cross-flow nebulizer fitted in Scott spray chamber.
Injection was done with a 2-μL injection loop for capillary-HPLC and with a 20-μL injection loop in a ‘μL pickup’ mode (5 μL of the sample injected). PEEK tubing was used for all connections with capillary tubes of 75 μm and 20 μm id (Polymicro Technologies, Phoenix, AZ, USA). The flow rate conditions for the nanoHPLC and capillaryHPLC pump are summarized in Table 1. A pre-concentration column and a C18 column equipped with a C18 guard column were used (C18 PepMap100, Dionex). A nanoflow nebulizer system described elsewhere25 was used for a sheathless interfacing of nanoHPLC. The optimization of the nanonebulization was made by adjusting the position of the needle tip in the nebulizer orifice to allow a continuous flow rate of less than 500 nL min−1. The nebulizer was fitted with a low-dead-volume drain-free vaporization chamber (3 cm3). The outlet of the capillary was fitted into the nebulizer via a zero-dead-volume connection to the nebulizer needle. The ICP MS system used was an Agilent 7500ce ICP MS equipped with a collision cell. Elution profiles were constructed using Chemstation software (Agilent).
| Time/min | Valve positioning | Acetonitrile (B) concentration % | |||
|---|---|---|---|---|---|
| 1 | 2 | Nanoflow pump (0.25 μl min−1) | Microflow pump (5.0 μl min−1) | ||
| Preconcentration | 0 | Inject | Load | 3 | 3 |
| 4 | Inject | Inject | 3 | 3 | |
| NanoHPLC separation | 10 | Load | Inject | 3 | 95 |
| 20 | Load | Inject | 50 | 95 | |
| 23 | Load | Inject | 95 | — | |
| Cleaning | 27 | Load | Inject | 95 | — |
| 30 | Load | Inject | 3 | 3 | |
| Re-equilibration | 40 | Load | Inject | 3 | 3 |
| ICP MS parameters (Agilent 7500ce) | ||
|---|---|---|
| Rf power | 1400 W | |
| Sampling depth | 7–8 mm | |
| Nebulizer gas flow | 1 L min−1 | |
| Cell gas flow H2/He | 4.5/0.5 mL min−1 (only for 56Fe) | |
| Isotopes monitored | 63Cu, 65Cu, 64Zn, 66Zn, 56Fe, 55Mn, 114Cd, 112Cd | |
| Spray chamber temperature | +2 °C | |
| Normal flow configuration | Nanoflow configuration | |
| Sampler, skimmer cones | Ni | Pt |
| Flow rate (pressure) | 0.65 mL min−1 (50 to 100 bars) | 1 and 0,25 μL min−1 (50 to 100 bars) |
| Running buffers | A: CH3COONH4, 10 mmol L−1, pH 7.5 | A: CH3COONH4, 10 mmol L−1, pH 7.5 |
| B: Acetonitrile | ||
| Mode | Isocratic | Gradient |
| Extraction lens | 4 V | 3.5 V |
| Octopole bias | −6 V | −18 V |
| Quadrupole bias | −3 V | −16 V |
| ESI QTOF MS (Applied Biosystems QStar XL) | ||
|---|---|---|
| Ionization mode | Positive | |
| Mass range | m/z 300–2000 | |
| Curtain gas | 1.38 bar N2 | |
| Entrance potential | 60 V | |
| Normal flow configuration | Nanoflow configuration | |
| Ion source | Micro-ionspray | Nano-electrospray |
| Needle voltage | 3500 V | 2050 V |
Samples and standards
Metal proteinates are the products resulting from the chelation of a soluble salt with amino acids and/or partially hydrolyzed protein. They are generally provided as an ingredient as the specific metal-proteinate, i.e., Cu-, Zn-, Mg-, Fe-, Co-, Mn- or Ca-proteinates.26 In this study, the proteinates that contained 10% metal on an elemental basis and originated from the partial hydrolysis of soybean proteins further reacted with a sulfate soluble metal salt were investigated for their chelation properties with Cu, Zn, Mn and Fe (Alltech-Ireland, Dunboyne, Ireland). Six production samples of Cu-proteinate produced under the same process were investigated for reproducibility purposes, identified with the following batch numbers: #237459-1 (A); #247487 (B); #S-235742-1 (C); #A221753 (D); #221754 (E); #205076 (F). The chelation properties of proteinates were compared with proteinate products issued from other sources of soybean material produced under different processes but containing 10% of metal on an elemental basis. The premix feed samples were prepared by adding 5, 25, 50 and 100 mg of proteinates to 5 g of pig feed or 5 g of poultry feed received from Alltech (Dunboyne, Ireland). Standard solutions of Cu (1 g L−1) were prepared by dissolving an appropriate amount of Cu sulfate in water (Sigma-Aldrich, St Quentin-Fallavier, France). Metallothionein I (MT) obtained from Sigma-Aldrich, St Quentin-Fallavier, France was dissolved in water at 1 mg mL−1 before analysis.Chemical and materials
Water was purified to 18.2 MΩ cm resistance using a Milli-Q water purification system (Millipore, Bedford, MA, USA). Per analysis grade chemicals (Sigma-Aldrich, St Quentin-Fallavier, France) were used throughout the study unless stated otherwise. Ammonium acetatebuffer solutions (pH 5.5; 7.2; 8.6) were prepared by dissolving ammonium acetate (Fluka, Buchs, Switzerland) in water to final concentrations of 2, 10, 50 mmol L−1. The pH adjustment was achieved with a solution of NH3 or HCl. The solvents were degassed by purging with helium to remove dissolved oxygen and to promote a non-oxidizing environment. Any other chemicals were purchased from Sigma-Aldrich (St Quentin-Fallavier, France) unless stated otherwise.Procedures
| ICP MS parameters (ELAN 6000) | ||
|---|---|---|
| For SEC-HPLC | For RP-HPLC | |
| Sampler, skimmer cones | Ni | Pt-Rh(10%) |
| Rf power | 1300 W | 1300 W |
| Plasma gas flow rate | 15 L min−1 | 15 L min−1 |
| Nebulizer gas flow | 0.80 L min−1 | 0.80 L min−1 |
| Oxygen flow | — | 22.4 mL min−1 |
| Isotopes monitored | 63Cu, 65Cu | 63Cu, 65Cu, 64Zn, 66Zn, 114Cd, 112Cd |
| Spray chamber temperature | +20 °C | +2 °C |
| RP-HPLC parameters | |
|---|---|
| Column | C18 Spherisorb (150 × 4.6 mm id.) |
| C18 CC guard column | |
| Injection volume | 100 μL |
| Mode | Gradient |
| Flow rate (pressure) | 0.6 mL min−1 (50–100 bar) |
| Running buffers | A: CH3COONH4, 10 mmol L−1, pH 7.5 |
| B: AcetonitrileHPLC grade | |
The protocol for the cleaning of the chromatographic column differed from the recommended procedure to remove the residual metals. The SEC columns Superdex Peptide 30 described in the instrumentation section were cleaned before the experiment using one bed volume (25 mL) of NaOH (0.5 mol L−1) at a flow rate of 0.5 mL min−1 (HR 10/30) and 3 mL min−1 (XK 26/30, used as preparative chromatography). More rigorous cleaning was achieved by using acetonitrile (ACN, 70%), NaOH (1 mol L−1), acid acetic (1 mol L−1) and HCl (0.1 mol L−1) as cleaning agent for 2 h. The cleaning agent was then washed out with water and equilibrated with the mobile phase. SEC column was cleaned by flushing with two bed volumes of 10 mmol L−1EDTA solution, followed by one bed volume of 0.5 mmol L−1 of NaOH solution to remove EDTA and avoid Cu–EDTA artifacts. When more rigorous cleaning was needed, injections of 100 μL of DTT/ß-mercaptoethanol (50/50; v/v) were performed before reconditioning the column with two bed volumes of mobile phase. The column was stored in a solution containing 20% ethanol.
The precursor proteins were identified by searching the set of proteins encoded by the soybean genome using the ExPASy Proteomics Server of the Swiss Institute of Bioinformatics. Several tools and software package were used to characterize the precursor ions, the product ions, and the peptide sequence. The TagIdent tool was used to match a short sequence tag of up to 6 amino acids; PeptideCutter software was used to estimate the enzymatic digestion pattern of an alkaline serine protease derived from Bacillus licheniformis. This enzyme most likely achieved cuts at glutamate (E) and to a lesser extent at aspartate (D)28 but was also able to make non-specific cuts.
All spectra were acquired in the mass range of 0.5 to 5.0 kDa, on a positive reflectron mode with the following settings: accelerating voltage of 20 kV; delayed ion extraction time of 100 ns; laser intensity of 2595 V; laser repetition rate 20.0 Hz; with DHB calibration matrix and 150 shots per analysis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iterations minimization method to find the lowest energy conformation for the different molecules. The docking energy for CVFF for a nonbonded interaction was calculated according to the following equation:
000 iterations minimization method to find the lowest energy conformation for the different molecules. The docking energy for CVFF for a nonbonded interaction was calculated according to the following equation: where Aij and Bij are parameters with units of kcal mol−1 angstrom12 and kcal mol−1 angstrom6 respectively, and Rij is the distance between atoms i and j in angstroms.31
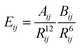
Results and discussion
Recovery of the water-soluble fraction
The recovery of the metal fraction was tested in triplicate from Cu- and Fe-proteinates using four buffer solutions: 25 mmol L−1 of acetatebuffer (pH 5.5); 25 mmol L−1 of Tris-HCl (pH 7.2); 25 mmol L−1 of Tris-HCl (pH 8.6); and methanol by total ICP MS analysis. The results are given in Table 3. An optimal Cu solubilization was found with the use of aqueous buffer solutions, especially at pH 7.2 and to a lesser extent at pH 5.5 compared with the levels obtained when methanol solutions were used. When methanol was used as extraction solvent, the quantity of solubilized Cu was 8-fold less and no detectable quantity of iron was released.| Proteinate sample | Extractant | Recovery of soluble Cu | RSD (%) |
|---|---|---|---|
| Cu | CH3COONH4, 25 mmol L−1, pH 5.5 | 59.0 ± 0.3% | 1.3 |
| Cu | Tris-HCl, 25 mmol L−1, pH 7.2 | 59.9 ± 0.1% | 1.0 |
| Cu | Tris-HCl, 25 mmol L−1, pH 8.6 | 49.3 ± 0.1% | 0.7 |
| Cu | Methanol | 8.2 ± 0.4% | 10.6 |
| Fe | CH3COONH4, 25 mmol L−1, pH 5.5 | 18.6 ± 0.1% | 1.0 |
| Fe | Tris-HCl, 25 mmol L−1, pH 7.2 | 27.2 ± 0.1% | 0.4 |
| Fe | Tris-HCl, 25 mmol L−1, pH 8.6 | 10.0 ± 0.1% | 1.0 |
| Fe | Methanol | 0.5 ± 0.1% | 52.8 |
Fractionation of the water soluble fraction by SEC–ICP MS
An insight into the chemical identity of the aqueous extract was attempted with size-exclusion LC–ICP MS. The concentration and pH of the mobile phase were optimized to maximize the recovery. Whereas the chromatograms at pH 7.2 and pH 8.6 were identical, the one obtained at pH 5.5 showed smaller peak intensity and different morphology, indicating either dissociation of the complex or its precipitation on the column. Of the buffer concentrations tested (i.e., 2, 10, 50 mmol L−1) 10 mmol L−1 was found to be optimal. At lower concentrations, the recovery was reduced and an additional peak appeared indicating an increased interaction of analyte compounds with the stationary phase. At the highest concentration tested, the chromatogram was identical, except that the background was increased. The replacement of the Tris-HCl buffer at pH 7.2 by ammonium acetatebuffer did not result in a change of either the recovery or the chromatogram morphology. The latter buffer was chosen because of its superior compatibility with electrospray ionization used in a later phase of this work.Note that Cu2+ is retained by the column. When a Cu2+ solution was injected on a clean column, the recovery was equal to 3.2 ± 1.6%. Therefore the column was periodically cleaned using a mixture of EDTA and DTT/ß-mercaptoethanol to remove excess Cu. It was found that some residual Cu deactivated the active sites of the stationary phase enabling the detection of less stable complexes. This phenomenon was similar to that described elsewhere by Ouerdane and collaborators32 for the Ni-citrate complexes.
Under optimum conditions the chromatogram showed five peaks (Fig. 1a). Peak intensity was a linear function of the total area under the curve of the sample amount taken indicating the reproducibility of extraction and interactions with the column stationary phase regardless of the Cu concentration (R2 = 0.999 and RSD < 3.25%) (inset to Fig. 1a). The elution volume indicates a smaller size of the species compared with the metallothionein complex (6.5–7 kDa) which eluted at 10–15 min.
 | ||
| Fig. 1 Elution profiles obtained under the same conditions by SEC-HPLC–ICP MS (mobile phase: CH3COONH4, 10 mmol L−1, pH 7.5, Table 2) of (a) a laboratory sample of Cu-proteinates: quantitative evaluation of Cu (n = 3) at 0.5 (light grey), 1.0 (dark grey) and 2.0 (black) μg mL−1. (a inset) Relationship between the intensity and the total Cu concentration for peak 3 at a retention time of 18.57 min (n = 3). All the values were for the apex of the corresponding peak. (b) Six production samples of Cu-proteinates differing from the laboratory sample: SEC-HPLC-ICP MS chromatogram of 63Cu coming from six different samples (noted from A to F) for evaluation of the batchwise reproducibility presented with an offset of 500 units from one to the next. | ||
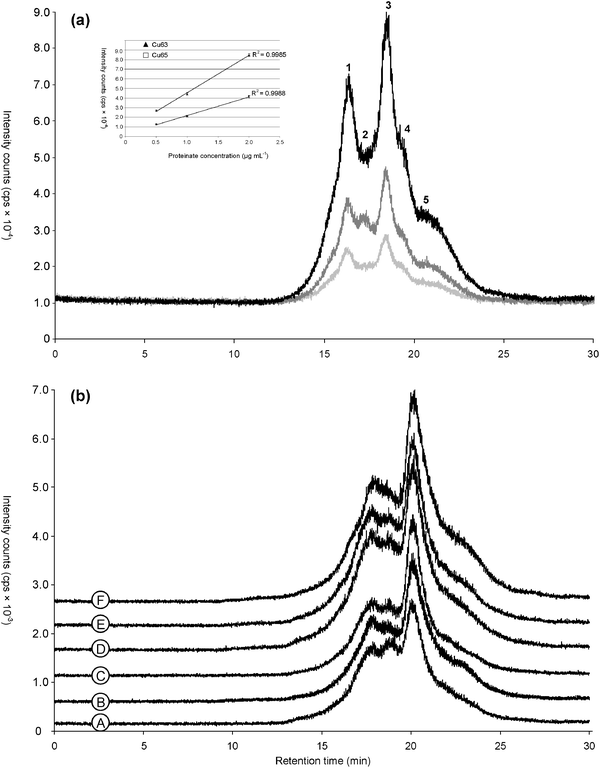
 | ||
| Fig. 2 Chromatograms obtained by SEC-HPLC–ICP MS of Cu-proteinate in two premix feed materials originated from poultry (black) and pig (grey). | ||
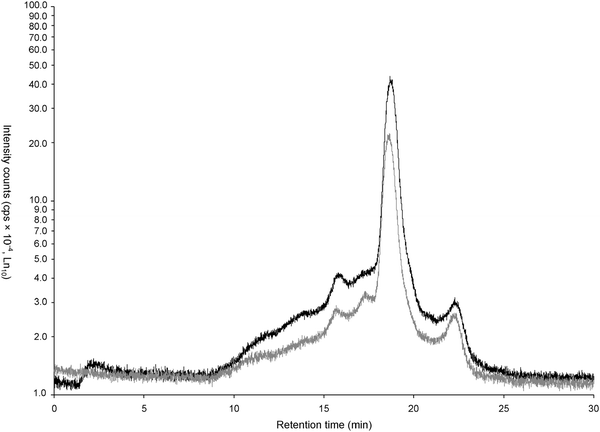
 | ||
| Fig. 3 SEC-HPLC–ICP MS chromatograms of Cu (a), Fe (b), Mn (c), Zn (d) -proteinate from soybean (black) and Cu (a), Fe (b), Mn (c), Zn (d)-proteinates extracted from an alternative soybean source (grey). | ||
Identification of the Cu-complexes formed by nanoHPLC–ESI MS/MS
The identity of the complexes formed was further studied by molecular mass spectrometry. Preliminary assays were carried out with MALDI-QTOF MS using either 2,5-dihydroxybenzoic acid (DHB) or a-cyano-4-hydroxycinnamic acid (CHCA). Under these conditions (strongly acidic) no Cu complexes were observed. A large number of peaks characteristic of a peptide mixture ranking from 500 to 1400 m/z were observed. Therefore, a softer ESI MS ionization that could be carried out under neutral pH conditions and that was likely to preserve the integrity of the Cu–peptide complexes was investigated.Analysis in the infusion mode was impossible because of a high sample complexity and the privileged ionization of sample components other than the metal complexes present. Therefore, a further fractionation of the metal-containing size-exclusion fraction by reversed-phase HPLC was considered. RP-HPLC was found to offer a Cu recovery of 89.4 ± 5.9%. The nanoHPLC–ICP MS chromatogram shows the elution of the majority of Cu between 16 and 30 min with a fairly sharp peak at between 22 and 26 min (Fig. 4a).
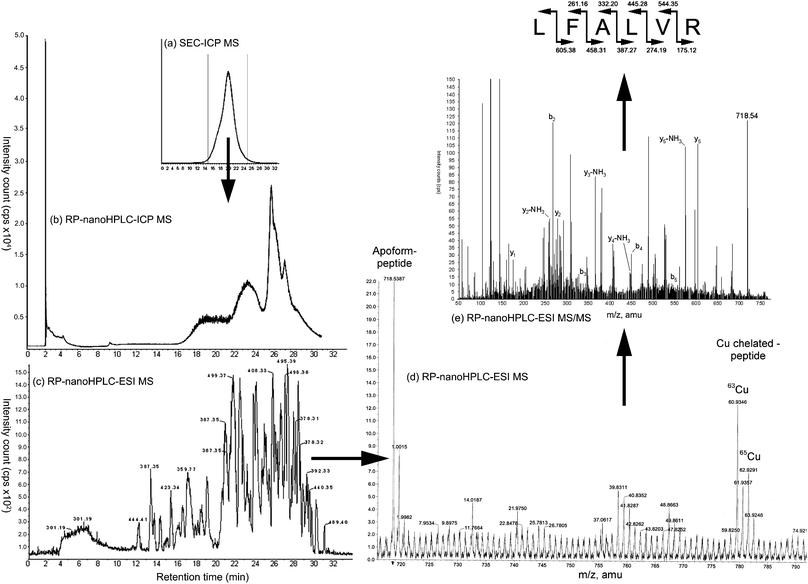 | ||
| Fig. 4 (a) Differential chromatogram obtained in RP-HPLC–ICP MS of Cu-proteinate obtained with a gradient of ammonium acetate 10 mmol L−1 pH 7.5 and acetonitrile. (b) Measurement of the Cu-proteinate fraction collected from the preparative SEC–ICP MS and chromatogram of the 63Cu, 65Cu in nanoHPLC–ICP MS. (c) Base Peak Chromatogram calculated from the Total Ion Chromatogram obtained in the nanoHPLC–ESI MS system. (d) nanoHPLC–ESI MS data for the ion extracted (m/z = 718.5387). (e) MS/MS fragmentation by collision induced dissociation of m/z = 718.53 species and related amino acid sequence identified. | ||
This data analysis strategy allowed the identification of a set of 32 peptides accompanied by metallated forms (Table 4). Five of them were doubly charged and were thus characterized by a shift of 30.5 and 31.5 m/z corresponding to the m/z ratio of 61 and 63 m/z instead. No peptide interaction with multiple Cu atoms was detected. The purity of the compound was checked by carefully examining the Extracted Ions Chromatograms (XIC) of the apo-form and the chelated form of the peptide to find the corresponding XIC peaks. The molecular mass of the peptides was in the 300–1200 Da range. The final complex was thus a non-charged molecule that was potentially able to pass through the mucus layer and the intestinal membrane without being retained in the villi of the digestive tract of animals.
| Chemical formula | Mass found | Theoretical mass (MH+) | Elution time | Sequence | Relative hydrophobicity | Charge | Isoelectric point | Hydrophilic/Hydrophobic (%) | Protein name |
|---|---|---|---|---|---|---|---|---|---|
| C11H19N3O6S1 | 321.99 | 322.36 | 15.18 | ACE | 0.0 | −1, Acidic | 3.85 | 0/33 | PUR1 |
| C16H26N4O7 | 387.34 | 387.41 | 14.1 | VGPD | 0.68 | −1, Acidic | 3.75 | 25/25 | SBP |
| C17H31N5O6 | 402.17 | 402.47 | 10.99 | VQGV | 2.26 | 0, Neutral | 6.02 | 0.50 | DMC1 |
| C16H27N5O8 | 417.23 | 418.43 | 26.36 | GGGVE | 1.73 | −1, Acidic | 3.85 | 20/20 | AOX1/AOX2 |
| C20H36N4O5S1 | 445.02 | 445.60 | 11.22 | IPCL | 13.94 | 0, Neutral | 6.02 | 0/50 | RBS1/RSB4 |
| C21H30N4O8 | 466.34 | 467.21 | 17.35 | YAVD | 4.26 | −1, Acidic | 3.75 | 25/50 | LOXX/LOX2/LOX3 |
| 467.16 | VMCD | 4.26 | −1, Acidic | 3.75 | 25/50 | TUBB1 | |||
| C17H28N6O10 | 476.20 | 477.45 | 16.15 | SAGGAD | 0.55 | −1, Acidic | 3.75 | 17/0 | CDC48 |
| C19H32N6O9 | 489.18 | 488.49 | 23.13 | GNAVE | 1.83 | −1, Acidic | 3.85 | 20/20 | TF2B |
| C20H33N7O6S1 | 499.86 | 500.22 | 20.94 | QHLC | 3.35 | +1, Basic | 7.36 | 25/25 | CAPP1/CAPP2 |
| C19H29N7O9 | 500.21 | HQTD | 0.90 | 0, Neutral | 5.28 | 50/0 | NO44 | ||
| C19H32N6O9 | 505.18 | 505.51 | 21.48 | HPHD | 1.37 | +1, Basic | 6.50 | 75/0 | CCNB2 |
| C19H30N8O11 | 547.22 | 547.50 | 22.3 | NGGGGGE | 0.31 | −1, Acidic | 3.85 | 14/0 | TF2B |
| C23H39N7O12 | 606.45 | 606.61 | 24.24 | AIGSGSD | 3.00 | −1, Acidic | 3.75 | 14/14 | PSA5 |
| C23H39N7O12 | 606.61 | NVATSD | 1.90 | −1, Acidic | 3.75 | 17/17 | NO23/NO44/NO51 | ||
| C27H38N6O10 | 607.64 | FSGEAP | 5.82 | −1, Acidic | 3.85 | 17/17 | CB22 | ||
| C25H41N7O10S1 | 631.22 | 632.71 | 22.92 | MVNPGD | 11.96 | −1, Acidic | 3.75 | 17/33 | EFTU2 |
| C28H51N7O9 | 630.77 | SVISPK | 9.56 | +1, Basic | 9.69 | 17/33 | |||
| C30H45N7O10 | 664.34 | 663.72 | 26.97 | ISNAYP | 7.61 | 0, Neutral | 6.02 | 0/33 | CYF |
| C33H45N7O8 | 668.27 | 668.34 | 21.96 | FYNPK | 10.74 | +1, Basic | 9.30 | 20/40 | GLYG4/GLYG5 |
| C35H59N9O7 | 718.54 | 718.92 | 16.2 | LFALVR | 28.87 | +1, Basic | 10.55 | 17/67 | LGBA/LGB1 |
| C29H54N8O10S2 | 737.51 | 739.94 | 19.78 | MALSCSK | 10.23 | +1, Basic | 8.55 | 14/29 | FRI2 |
| C29H51N7O11S2 | 738.90 | CVMLSGE | 15.56 | −1, Acidic | 3.85 | 14/43 | KPYC | ||
| C38H54N6O10 | 754.50 | 755.89 | 23.81 | LFFISE | 30.54 | −1, Acidic | 3.85 | 17/67 | YCF4 |
| C32H54N10O11 | 755.85 | LTGHAEK | 5.17 | +1, Basic | 7.55 | 43/14 | LGB1/2/3/A | ||
| C36H56N10O7S1 | 773.60 | 773.98 | 26.36 | MLPWAR | 25.23 | +1, Basic | 10.55 | 17/50 | NO93 |
| C34H63N9O11 | 774.94 | KASGTVVI | 16.04 | +1, Basic | 9.69 | 12/38 | LGB3 | ||
| C43H76N12O12 | 951.99 | 953.14 | 22.19 | LGPTSPIIR | 23.38 | +1, Basic | 10.55 | 11/33 | NO23/NO51 |
| C46H79N13O14S1 | 1069.72 | 1070.27 | 22.92 | NICTLKLHE | 22.61 | +1, Basic | 7.37 | 33/33 | GLYG4 |
| C46H79N13O16 | 1070.20 | VTPNNVDIAK | 14.33 | 0, Neutral | 6.23 | 20/30 | PSA5 | ||
| C46H87N17O12 | 1070.29 | RLLKKQRE | 13.56 | +3, Basic | 11.65 | 62/25 | GLCB | ||
| C48H75N15O16S2 | 1180.98 | 1182.50 | 22.01 | APVLDCHTSHC | 11.90 | +1, Basic | 6.48 | 27/18 | EF1A |
The set of 32 identified peptides shown in Table 4 involved in the chelation of Cu provides an insight into amino acids occurring in Cu-binding peptides based on the frequency of amino acids independent of the concentration of the peptides . To minimize the impact of their concentration, we selected the amino acids that were found to be most abundant according to proteinate analysis using ICP MS. Proteins eluting at 15.1 min and from 20 to 26 min were so considered.
The most frequently occurring amino acids were glycine (10.81%), followed by serine, leucine and alanine (8.11%). However, to be representative, these values should be weighed against the occurrence of amino acids coded by the soybean genome in which glutamic acid dominates (19.0%), while cysteine is the least abundant (0.9%).33–35 After correction according to the formula (AA stands for the term “amino acids”):
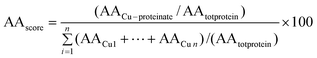
| Amino acids (AA) | Proportion of AA found in peptide implicated in chelation with Cu (%) | Average values of amino acid content found in several cultivars of soybean [Glycine max (L.) merr.] per total protein (%) | AAscore of AA corrected proportion implicated in chelation of Cu (%) |
|---|---|---|---|
| Arginine, R | 2.70 | 7.17 | 1.29 |
| Histidine, H | 4.32 | 2.88 | 5.12 |
| Isoleucine, I | 5.41 | 4.62 | 4.00 |
| Leucine, L | 8.11 | 8.21 | 3.37 |
| Lysine, K | 4.86 | 6.67 | 2.49 |
| Methionine, M | 2.70 | 1.47 | 6.26 |
| Phenylalanine, F | 2.72 | 5.20 | 1.78 |
| Threonine, T | 4.32 | 4.10 | 3.60 |
| Valine, V | 7.03 | 4.04 | 5.94 |
| Alanine, A | 8.11 | 4.55 | 6.08 |
| Aspartic acid, D | 5.41 | 12.24 | 1.51 |
| Cysteine, C | 4.86 | 0.90 | 18.50 |
| Glutamic acid, E | 5.41 | 18.97 | 0.97 |
| Glycine, G | 10.81 | 4.49 | 8.22 |
| Proline, P | 7.02 | 5.19 | 4.62 |
| Serine, S | 8.11 | 5.38 | 5.14 |
| Tyrosine, Y | 1.08 | 2.64 | 1.40 |
| Tryptophan, W | 0.54 | ND < 1.28 | 19.71 |
| Asparagine, N | 4.86 | ||
| Glutamine, Q | 1.62 |
This result confirms the dominant role of cysteine in complexing Cu; histidine and methionine evoked earlier39–41 in this context were less prominent even if the affinity for a peptide-binding residue to Cu followed the affinity sequence histidine (H) > methionine (M) > cysteine (C).42 The putative metal-binding motifs previously characterized were not systematically found. These motifs in the collection of peptide fragment recorded included the following: C(X)mC (m = 2–4); H(X)nH (n = 0–6, where for n > 3 a proline residue is generally required to provide binding geometry); H(X)pH(X)pH (n = 1–2); H(X)qM(X)rM; and M(X)sM where X corresponds to any amino acid. The refined search of these motifs using PeptideCutter and TagIdent ExPASy modules in the soybean genome retrieves only 5 from 19 potential candidates that were matched to the soybean genome and the sequence chain proposed by She and coworkers.42 The sequences HPHD and APVLDCHTSHC with the occurrence of two histidines together with one proline residue were the only ones correlating with the previous statement when poly-histidine fragments were considered. Interestingly, and because of the implication of cysteine in the peptide fragment found, the following motifs have been characterized: H(X)1C; C(X)4M; M(X)3C; C(X)1M. Furthermore, the chelating properties of di- or tri-glycine fragments for Cu were also implicated as reported in several studies,43–45 and some poly-glycine sequences were also characterized.
Molecular mechanics investigations
The elucidation of the spatial conformation of the peptides was approached using a molecular mechanics investigation and the models generated are presented in Fig. 5. The selection of the peptide was made as a function of the abundance found during the ICP MS elution and considering three possible structural interactions involving three different peptide motifs interacting with Cu. For this purpose, peptides containing histidine or methionine or polyglycine amino acid eluting at 15.1 min and 22.0 min were chosen. They were modeled using Accelrys software, which allowed statistical evaluation with a minimization iteration procedure of the most stable conformation relative to the potential energy of the peptides and the peptides interacting with a Cu atom. The designed molecules were minimized in a solvent box containing water molecules and at a pH of 7.5, which was enabled by the selection of a dielectric constant of 80. The tripeptide ACE, eluting at 15.1 min with an intense peak recorded through ICP MS was built using the template obtained from the Protein Data Bank, 2C9V.pdb belonging to a human superoxide dismutase implicated in the interaction with Cu–Zn and Zn–Zn.46 Different minimization steps were used with and without constraint. The final docking of a Cu molecule in the structure led to the calculation of a docking energy of −227 kcal mol−1 accounting for a stable conformation at pH 7.5. The tetrapeptide HPHD eluting at 21.48 min was compared with the template of the protein 2C9V.pdb. On this template, two histidine residues were conserved in positions 46 and 48 and the histidine 63 replaced by an asparagine and the last by a water molecule. Successive minimization steps were applied to the peptidic bond and on the hydrogens of the water molecule. The final minimization was achieved after docking of the Cu atom with constraint on the core amino acid sequence for 4000 iterations. The energy was evaluated at −217 kcal mol−1. After relaxation of the system over 2000 iterations, changes of the Cu position were recorded and the final new energy evaluated at −208 kcal mol−1 as of a better stability of the system. The heptapeptide NGGGGGE eluting at 22.3 min was constructed using the factor II transcription inhibitor protein of soybean where an NGGGE sequence was found in the 1QXF.pdb file47 corresponding to the 30S ribosomal protein S27E issued from a “WU-Blast2” search of alignment display. The amino acids 36 to 44 were conserved and complemented with glycineamino acid to mime the searched sequence. The structure issued from the 2C9V.pdb file was superimposed by homology using the nitrogen atoms implicated in the peptidic bond and in the Cu chelation. A Cu atom was then docked between the N atoms. All the glycine residues were constrained for 2000 iterations and then relaxed for 2000 iterations. The docking energy was found to be equal to −100 kcal mol−1. Finally, the decapeptide APVLDCHTSH eluting at 21.48 min extracted from the EF1A_SOYBN sequence was correlated to the pdb structure 1F60.pdb.48 The approaching pattern was found between the 356 and 365 amino acid positions. The serine on the N terminal position was replaced by an alanine and the penultimate alanine by a serine to match the sequence investigated. The C terminal histidine position was modified manually and the Cu molecule positioned inside the peptide. A minimization step of 6000 iterations without constraint was achieved and resulted in a docking energy of −282 kcal mol−1. The structure modeled, and its interaction with Cu, demonstrate a very stable interaction that is dependant on the length of the amino acid chain. The presence of two histidines in HPHD and APVLDCHTSH peptides enabled the lowest docking energy to be obtained. Interestingly, it must be noted that for the particular case of ACE peptide, a prime energy of −46 kcal mol−1 was calculated when the structure of the originated fragment was conserved from the 2C9V.pdb file. Allowing the structure to relax enabled the docking energy to be decreased. It is thus by the more flexible structure of the tripeptide and steric effect that we can explain the similar docking energy as the tetrapeptide containing histidine and according to the generally accepted affinity sequence: H > M > C. It can be noted that despite the length of the two latter peptides , they exhibited very low docking energy accounting for the important affinity of interaction between histidine residues and Cu as well as glycine residues. However this later had a lower affinity explained again by the previously mentioned affinity sequence favoring H-, M-, C-containing peptides .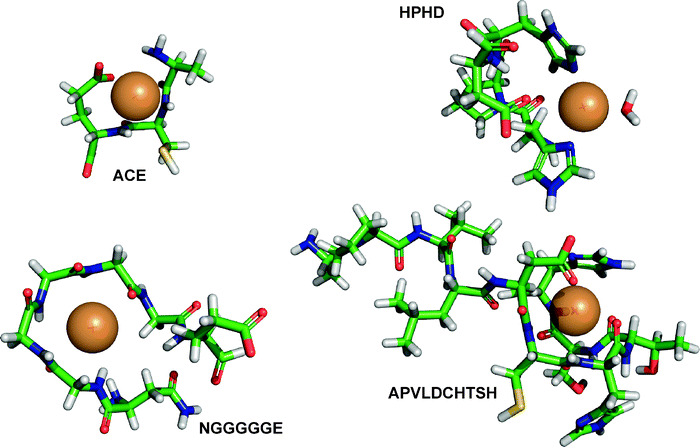 | ||
| Fig. 5 Modeling using Accelrys software and after a 10000 iterations procedure of four peptides with particular amino acid sequence responsible of the chelation of Cu in proteinates issued from soybean. | ||
Biological implications
If the use of radioisotopes in animal research has added valuable information to the understanding of Cu metabolism, the uptake and distribution mechanisms remain obscure. Furthermore, data on the influence of dietary components, the amount of Cu administered, as well as the location of the intestinal absorption and variation according to the animal species, age, nutritional, physiological, and health-status suffer from many disparities. The correct presentation of the metal, either organic or inorganic, is also of prime importance especially for understanding their transfer through the carbohydrate-rich glycoproteins composing the mucosal surface of the gastrointestinal tract. Indeed, this particular fraction is characterized by an intense negative charge due to heavily glucosylated proteins (mucins) and mucopolysaccharides that have large capacities for the binding of metal ions. The protein core comprises side chains made of oligosaccharides (N-galactosamine, sialic acid, fructose, galactose, N-acetylglucosamine). This complex network forms both soluble and insoluble phases within the gastrointestinal tract that in time bind metal species, and where the interaction strength favored M3+ > M2+ > M+ binding (M representing any metal ion), but where their mobility through mucus is inversely governed by the strength of binding and directly by their ligand exchange capacity giving as a general rule an absorption of metals with an efficiency M+ > M2+ > M3+.49 Thus, the presentation of uncharged species following the chelation of the metal to a peptide as in the proteinate study detailed herein might present an advantage for the conservation and transfer of the metal through the mucosa, this latter having a pH microclimate (5.6 to 7.16) significantly different from the lumen (acidic but buffered by the bowel) and favoring metal–ligand complexes instead of ion species that ultimately are precipitated in polymeric-ions unavailable for further absorption in this form.50Furthermore, the actually-depicted pathways necessary for the transport of trace minerals involving specific (Ctr1, Ctr2, DMT1);51,52 and non-specific membrane proteinreceptors on the apical side of epithelial cells as well as protein metallochaperones (anti-oxidant proteins such as ATX1, ATOX1, HAH1 that deliver Cu to P-type ATPases to the Golgi; the CCS Cu chaperone protein for the Cu/Zn superoxide dismutase; cytotochrome oxidase such as COX17 localized in mitochondria membrane; P-type ATPases such as ATP7A,B, which are the targets of ATX transporters)53 or binding proteins (metallothioneins) mediating the tight regulation of Cu homeostasis, antioxidant defenses, and transport throughout the cells and finally membrane proteinreceptors of the basolateral part of epithelial cells (ATP7A/B; Ctr1),52,54 still represent an incomplete picture of the absorption mechanisms of Cu. The presence of a MXC(X)2C motif in the ATX1 protein53 and a M(X)1−3M motif in Ctr156 explain the affinity for the Cu atoms.
On the other hand, the absorption of single amino acids is dependent on the electrochemical gradient of sodium across the epithelium of the epithelial cells. The basolateral membrane of the enterocytes contains additional transporters which export amino acids from the cell into blood. Knowledge of the absorption of peptides is mainly limited to the study of the H+-coupled oligopeptide transporter (PepT1), present in brush-border membranes of enterocytes in the small intestine and whose gene expression is changed in response to dietary protein level.55 This transporter is not sufficient to explain the absorption of larger peptides .
In this respect, the partial knowledge in terms of speciation of Cu from plants and feed ingredients57 is the limiting factor to the understanding of the potential routes of presentation of the minerals and further absorption at the level of the digestive membrane, that for now, are only partially described. The nature of the protein breakdown products determines the route of their epithelial absorption and thus the speciation of the peptide fraction of the proteinates needs intensive characterization. Furthermore, the possible competition between metal transporters presenting motifs such as those found in the ATX1 protein compared to the peptide motifs of the proteinates is also of major interest. In the particular case of animals, a correct mineral formulation needs to be supplied to satisfy optimal zootechnical performance. Thus the understanding of the composition of alternate sources of mineral is important, because organic-minerals enhance bioavailability and reduce mineral excretion.4,6–9,58 The next step to this research could be a direct biological approach by tracking proteinates in biological systems (epithelial cells with conserved mucin layers).
Conclusion
Size-exclusion chromatography with ICP MS detection is a valid technique to monitor the molecular mass distribution of metal-binding molecules in feed supplements for the purpose of the verification of the batch-to-batch reproducibility and differentiation between products from different sources and different manufacturers. An important benefit of this method is a detection limit below 1 μg g−1 that allows its application to premixes and feed samples. However, the main point of the article was not the absolute quantification of the different metal-proteinate samples but rather the comparison of chromatograms. In this respect, the validation resided in the demonstration of the reproducibility of analysis (which is becoming very important in this context) and the observation of sufficient differences to discriminate the various sample investigated. Adding a second chromatographic dimension and the parallel ICP MS and ESI MS/MS detection allows a fine characterization of the metal–peptide complexes formed. This latter aspect is an essential step to the better understanding of the metal-proteinate species to further investigate the routes of absorption of organic versus inorganic mineral administration.Acknowledgements
We would like to acknowledge Alltech Inc. for the financial support and Dr G. Bertin from the Alltech EU Regulatory Department, 92300 Levallois-Perret, France for setting up the project. We wish also to thank Dr G. André-Leroux from the Structural Biochemistry Unit at the Pasteur Institute, 75224 Paris, France for the technical support provided for the molecular mechanics investigations.References
- F. P. Parks and K. J. Harmston, Feed Manage., 1994, 45, 35–38 Search PubMed.
- J. W. Spears, Anim. Feed Sci. Technol., 1996, 58, 151–163 CrossRef.
- L. Harvey, Nutr. Food Sci., 2001, 31, 179–182 CrossRef.
- R. Power, in Nutritional Biotechnology in the Feed and Food Industries, Proceedings of Alltech’s 19th International Symposium, ed. T. P. Lyons and K. Jacques, Nottingham University Press, Nottingham, UK, 2004, pp. 355–364 Search PubMed.
- H. Hasman, S. Franke and C. Rensing, in Antimicrobial Resistance in Bacteria of Animal Origin, ed. F. M. Aarestrup, American Society for Microbiology Press, Washington, DC, USA, 2006, ch. 7 Search PubMed.
- Z. Du, R. W. Hemken, J. A. Jackson and D. S. Trammell, J. Anim. Sci., 1996, 74, 1657–1663 Search PubMed.
- T. Ao, J. L. Pierce, A. J. Pescatore, A. H. Cantor, K. A. Dawson, M. J. Ford and B. L. Shafer, Br. Poult. Sci., 2007, 48, 690–695 CrossRef CAS.
- W. F. Stansburry, L. F. Tribble and D. E. Orr, J. Anim. Sci., 1990, 68, 1318–1322 Search PubMed.
- G. L. Cromwell, M. D. Lindemann, H. J. Monegue, D. D. Hall and D. E. Orr, Jr, J. Anim. Sci., 1998, 76, 118–123 Search PubMed.
- R. L. Kincaid, R. M. Blauwiekel and J. D. Conrath, J. Dairy Sci., 1986, 69, 160–163 CAS.
- J. W. Spears, E. B. Kegley and L. A. Mullis, Anim. Feed Sci. Technol., 2004, 116, 1–13 CrossRef CAS.
- J. D. Ward, J. W. Spears and E. B. Kegley, J. Dairy Sci., 1996, 79, 127–132 CAS.
- H. Chassaigne and R. Lobinski, Anal. Chem., 1998, 70, 2536–2543 CrossRef CAS.
- J. Szpunar and R. Lobinski, Anal. Biochem. Chem., 2002, 373, 404–411 CAS.
- P. Giusti, D. Schaumlöffel, H. Preud’homme, J. Szpunar and R. Lobinski, J. Anal. At. Spectrom., 2006, 21, 26–32 RSC.
- J. Ruiz-Encinar, D. Schaumlöffel, Y. Ogra and R. Lobinski, Anal. Chem., 2004, 76, 6635–6642 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- V. Shulaev, Brief. Bioinform., 2006, 7, 128–139 CrossRef CAS.
- A. Saghatelian and B. F. Cravatt, Life Sci., 2005, 77, 1759–1766 CrossRef CAS.
- J. M. Halket, D. Waterman, A. M. Przyborowska, R. K. P. Patel, P. D. Fraser and P. M. Bramley, J. Exp. Bot., 2005, 56, 219–243 CAS.
- K. Polec, M. Perez-Calvo, O. Garcia-Arribas, J. Szpunar, B. Ribas-Ozonas and R. Lobinski, J. Inorg. Biochem., 2002, 88, 197–206 CrossRef CAS.
- V. Vacchina, K. Polec and J. Szpunar, J. Anal. At. Spectrom., 1999, 14, 1557–1566 RSC.
- J. Isanga and Z. Guo-Nong, Food Rev. Int., 2008, 24, 252–276 CrossRef CAS.
- J. D. Albro, D. W. Weber and T. DelCurto, J. Anim. Sci., 1993, 71, 26–32 Search PubMed.
- P. Giusti, R. Lobinski, J. Szpunar and D. Schaumlöffel, Anal. Chem., 2006, 78, 965–971 CrossRef CAS.
- R. Schroeder, R. J. Noel and S. Krebs, in Association of American Feed Control Officials Incorporated, ed. S. Krebs, AAFCO Official Publication, Oxford, IN, USA, 2008, pp. 311 Search PubMed.
- H. B. F. Dixon, A. Cornish-Bowden, C. Liébecq, K. L. Loening, G. P. Moss, J. Reedijk, S. F. Velick and J. F. G. Vliegenthart, in Biochemical Nomenclature and Related Documents, ed. C. Liébecq, Portland Press, London UK, 2nd edn, 1992, pp. 39–69 Search PubMed.
- S. K. Wierenga, M. J. Zocher, M. M. Mirus, T. P. Conrads, M. B. Goshe and T. D. Veenstra, Rapid Commun. Mass Spectrom., 2002, 16, 1404–1408 CrossRef CAS.
- C. S. Ewig, T. S. Thacher and A. T. Hagler, J. Phys. Chem. B, 1999, 103, 6998–7014 CrossRef CAS.
- W. Wang, O. Donini, C. M. Reyes and P. A. Kollman, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 211–243 CrossRef CAS.
- N. Pattabiraman, M. Levitt, T. E. Ferrin and R. Langridge, J. Comput. Chem., 1985, 6, 432–436 CrossRef CAS.
- L. Ouerdane, S. Mari, P. Czernic, M. Lebrun and R. Lobinski, J. Anal. At. Spectrom., 2006, 21, 676–683 RSC.
- B. O. Eggum and R. M. Beames, in Seed Proteins: Biochemistry, Genetics, Nutritive Value, ed. W. Gottshalk and H. P. Müller, Martinus Nijhoff/Dr W. Junk Publishers, The Hague, The Netherlands, 1983, p. 499 Search PubMed.
- I. Koshiyama, in Seed Proteins: Biochemistry, Genetics and Nutritive Value, ed. W. Gottschalk and H. P. Müller, Martinus Nijhoff/Dr W. Junk Publishers, The Hague The Netherlands, 1983, pp. 427–450 Search PubMed.
- R. F. Wilson, Seed Metabolism, in Soybeans: Improvement, Production and Uses, Agronomy Monograph 16, ed. J. R. Wilcox, ASA, Madison, WI, 2nd edn, 1987, pp. 643, 662–686 Search PubMed.
- R. M. Rainbird, J. H. Thorne and R. W. F. Hardy, Plant Physiol., 1984, 74, 329–334 CrossRef CAS.
- C. Hernandez-Sebastia, F. Marsolais, C. Saravitz, D. Israel, R. E. Dewey and S. C. Huber, J. Exp. Bot., 2005, 56, 1951–1963 CrossRef CAS.
- C. G. Zarkadas, C. Gagnon, S. Gleddie, S. Khanizadeh, E. R. Cober and R. J. D. Guillemette, Food Res. Int., 2007, 40, 129–146 CrossRef CAS.
- M. Kelly, P. Lappalainen, G. Talbo, T. Haltia, J. vand der Oost and M. Saraste, J. Biol. Chem., 1993, 268, 16781–16787 CAS.
- D. Ren, N. A. Penner, B. E. Slentz, H. D. Inerowicz, M. Rybalko and F. E. Regnier, J. Chromatogr., A, 2004, 1031, 87–92 CrossRef CAS.
- E. Balatri, L. Banci, I. Bertini, F. Cantini and S. Ciofi-Baffoni, Structure, 2003, 11, 1431–1443 CrossRef CAS.
- Y. M. She, S. Narindrasorasak, S. Yang, N. Spitale, E. A. Roberts and B. Sarkar, Mol. Cell. Proteomics, 2003, 2, 1306 CrossRef CAS.
- A. Hanaki, T. Kawashima, T. Konishi, T. Takano, D. Mabuchi, A. Odani and O. Yamauchi, J. Inorg. Biochem., 1999, 77, 147–155 CrossRef CAS.
- D. Sanna, C. G. Agoston, G. Micera and I. Sovago, Polyhedron, 2001, 20, 3079–3090 CrossRef CAS.
- J. H. Kang and R. L. Toomes, J. Chem. Phys., 2003, 118, 6059–6071 CrossRef CAS.
- R. W. Strange, S. V. Antonyuk, M. A. Hough, P. A. Doucette, J. S. Valentine and S. S. Hasnain, J. Mol. Biol., 2006, 356, 1152–1162 CrossRef CAS.
- C. Herve Du Penhoat, H. S. Atreya, Y. Shen, G. Liu, T. B. Acton, R. Xiao, Z. Li, D. Murray, G. T. Montelione and T. Szyperski, Protein Sci., 2004, 13, 1407–1416 CrossRef.
- G. R. Andersen, L. Pedersen, L. Valente, I. Chatterjee, T. G. Kinzy, M. Kjeldgaard and J. Nyborg, Mol. Cell, 2000, 6, 1261–1266 CrossRef CAS.
- M. W. Whitehead, R. P. Thompson and J. J. Powell, Gut, 1996, 39, 625–628 CAS.
- J. J. Powell, M. W. Whitehead, S. Lee and R. P. H. Thompson, Food Chem., 1994, 51, 381–388 CrossRef CAS.
- Y. Nose, B. E. Kim and D. J. Thiele, Cell Metab., 2006, 4, 235–244 CrossRef CAS.
- A. M. Zimnicka, E. B. Maryon and J. H. Kaplan, J. Biol. Chem., 2007, 282, 26471–26480 CrossRef CAS.
- T. V. O’Halloran and V. C. Culotta, J. Biol. Chem., 2000, 275, 25057–25060 CrossRef CAS.
- L. Nyasae, R. Bustos, L. Braiterman, B. Eipper and A. Hubbard, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 292, 1181–1194.
- S. A. Adibi, Gastroenterology, 1997, 113, 332–340 CrossRef CAS.
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–184 CrossRef CAS.
- R. A. Wapnir, Am. J. Clin. Nutr., 1998, 67(suppl.), 1054–1060.
- R. Guo, P. R. Henry, R. A. Holwerda, J. Cao, R. C. Littell, R. D. Miles and C. B. Ammerman, J. Anim. Sci., 2001, 79, 1132–1141 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |