Intramolecular hydrogen bonding as a determinant of the inhibitory potency of N-unsubstituted imidazole derivatives towards mammalian hemoproteins†
Lionel
Perrin
a,
François
André
a,
Caroline
Aninat
b,
Rémy
Ricoux
c,
Jean-Pierre
Mahy
*c,
Ning
Shangguan
d,
Madeleine M.
Joullié
d and
Marcel
Delaforge‡
*a
aURA 2096 du CNRS, Institut de Biologie et Technologies de Saclay, Commissariat à l’Energie Atomique, Bâtiment 528, CEA-Saclay, 91191, Gif-sur-Yvette, France. E-mail: marcel.delaforge@cea.fr; Fax: +33 1 69 08 87 17; Tel: +33 1 69 08 68 39
bINSERM U620; Université de Rennes 1, IFR 140, 2 Avenue du Pr. Léon Bernard, 35043, Rennes, France
cLaboratoire de Chimie Bioorganique et Bioinorganique, CNRS, Institut de Chimie Moléculaire et des Matériaux d’Orsay, UMR 8182, Bâtiment 420, Université Paris-Sud XI, 91405, Orsay, France. E-mail: jpmahy@icmo.u-psud.fr; Fax: +33 1 69 15 72 81; Tel: +33 1 69 15 74 21
dDepartment of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA
First published on 27th November 2008
Abstract
Enzymes involved in the mammalian microsomal metabolism of drugs are, in numerous cases, inhibited by compounds bearing an imidazolyl scaffold. However, the inhibition potency is highly dependent upon the accessibility of the imidazolyl nitrogen lone pair. In order to highlight some structural parameters of inhibitors that control this phenomenon, a series of compounds containing a nitrogen unsubstituted imidazolyl moiety with varying degrees of nitrogen lone pair accessibility was tested on human and rat hepatic cytochromes P450 and microperoxidase 8, an enzymatically active peptide derived from cytochrome c. In each case, we have shown that the accessibility of the imidazole lone pair determined the extent of inhibition. Nitrogen accessibility was tuned not only by varying the steric hindrance in the vicinity of the imidazolyl ring but also by modifying its surrounding hydrogen bonding network. Compounds in which there exists intramolecular hydrogen bonding between the imidazole moiety and an H-bond acceptor, such as an appropriately positioned amidecarbonyl group, demonstrated enhanced inhibitory effects. Conversely, imidazole moieties which are in proximity to H-bond donors, such as an amide NH group, displayed reduced potency. This trend was observed in cyclo-peptide derivatives in which the intramolecular H-bond network was adjusted through the modification of the stereochemistry of a dehydrohistidine residue. It was observed that (Z)-isomers weakly bind heme, whereas (E)-isomers demonstrated higher degrees of metal binding. Therefore, enzymatic inhibition of heme-containing proteins by compounds bearing a dehydrohistidine motif seems to be closely related to its stereochemistry and hydrogen binding propensity. At neutral pH, these differences in binding affinities can be confidently attributed to the ambident H-bond properties of imidazole nitrogen atoms. This structure-activity relationship may be of use for the design of novel imidazolyl compounds as new P450 inhibitors or drug candidates.
Introduction
Metalloproteins represent an important class of enzymes which are involved in a variety of biochemical processes such as peroxidase, catalase, monooxygenase and reductase activities, and oxygen transport and storage. These functions are related to the presence of a metal center tightly bound to amino acids such as methionine, cysteine or histidine.1 In the case of hemoproteins, four N-pyrrolyl ligands coordinate the iron in the plane of the porphyrin ring, a fifth axial position is coordinated by a histidine, methionine or cysteine residue, and the sixth by an electron rich molecule such as dioxygen, cyanide, amine, nitroso derivatives or water. In certain cases, an external molecule containing an electron rich functional group may serve as a metal ligand, leading to inhibitory effects. The extent to which such molecules possess an affinity for complexation to the heme iron determines the degree of inhibition.2,3Considering the binding features of iron in hemoproteins, as well as the importance of their biological functions, these proteins are relevant targets for studying inhibitory effects. In particular, cytochromes P450 are highly attractive since a large panel of isoforms is able to recognize a wide variety of substrates. Nitrogen rich compounds, such as imidazole derivatives, represent a large class of cytochrome P450 reversible inhibitors.2–5 Their inhibitory potency comes from their ability to form stable Fe(II)-N-imidazole complexes.6 Numerous imidazole derivatives were investigated for their ability to inhibit hemoproteins such as cytochromes P450,2,4–9 cyclo-oxygenases10 and NO-synthases.11 They are also known and used for their potent inhibition of lanosteroldemethylase activity in fungi.7,8 In addition, this inhibitory action has been fruitfully exploited to reduce the rate of metabolism of certain drugs. For example, it has been demonstrated that co-administration of ketoconazole with cyclosporine A9 or ixabepilone12 limits their metabolism and enhances their therapeutic effects. Finally, another example of heme-imidazole complexes is found in the case of monooxygenase inhibition by histamine, a natural derivative of histidine.13,14
It has been demonstrated that the stability of the reversible imidazole–iron interaction depends upon the accessibility of imidazolyl nitrogen atoms as well as the hydrophobicity of its surrounding substituents.15–18 Among the imidazole-based inhibitors, most of the molecules reported have one of their two nitrogen atoms substituted.15 In this case, the inhibition potency is enhanced by the coordination of the electron lone pair of the remaining nitrogen of the imidazole ring. This effect has been demonstrated, experimentally and theoretically, in the case of the binding of N-substituted imidazoles to the heme of P450cam.19
We have previously demonstrated that small peptides , and particularly cyclodipeptides, are recognized, and in some cases metabolized, by hepatic cytochromes P450.20,21 In the present study, we describe our efforts to demonstrate the influence of the chemical environment of imidazole derivatives on their P450 inhibition. In particular, since it is clear that the strength of the coordination of the imidazole nitrogen on the iron atom is related to the availability of its doublet, we examine the influence of its involvement in a H-bond with a neighboring hydrogen atom, which is supposed to decrease its availability and its ability to bind to the iron atom. To this end, a determination of the preferred mode of interaction between a series of dehydrohistidine-based molecules (Fig. 1) and various P450 isoforms has been performed. In this series of compounds, firstly a group of molecules which cannot establish any intramolecular hydrogen bonds was studied. This subset includes (E)-urocanic acid, its methyl ester (1) and α-bromo derivative (4), His-Phe and N-benzylimidazole. A second series of compounds which potentially develop intramolecular hydrogen bonds consists of roquefortine, isoroquefortine, phenylahistin, BS176, c-(Phe-His), c-(Phe-ΔHis), (Z)-urocanic acid, its cis methyl ester (2) and α-bromo derivatives (5), as well as the saturated derivative 3. In the latter group, two types of behavior may occur: one of the two nitrogens of the imidazole ring may act either as a hydrogen bond donor or acceptor. An example of the first case is found in roquefortine, whereas an example of the second case is illustrated by isoroquefortine.
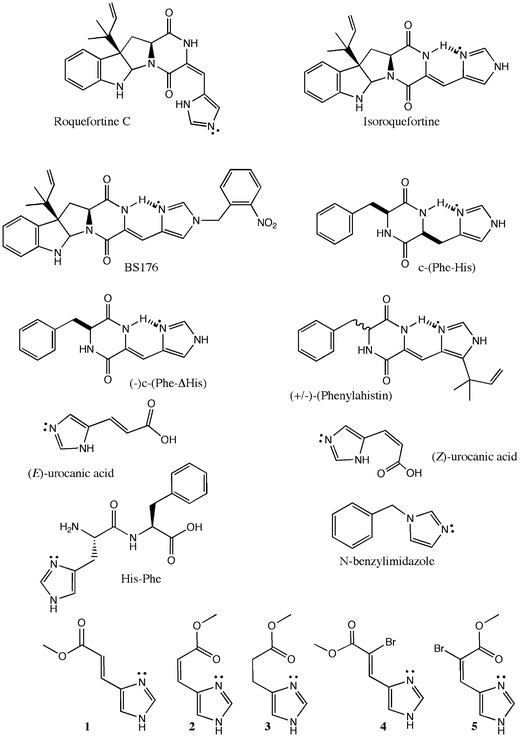 | ||
| Fig. 1 Chemical structure of the dehydrohistidine derivatives tested. Intramolecular hydrogen bonds are highlighted in bold. | ||
These differences in H-bonding properties between roquefortine and isoroquefortine are supported by NMR analysis (M Joullié, personal observations). In roquefortine, hydrogen bonding between the NH-group of imidazole and the oxygen of the neighboring carbonyl group of the diketopiperazine has been suggested,21 whereas intramolecular hydrogen bonding between the sp2 nitrogen ![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N– of the imidazole group and the NH-amide group of the diketopiperazine (Fig. 1) has been proposed for isoroquefortine.22 In terms of binding affinities, we have previously demonstrated the inhibitory action of roquefortine towards cytochromes P45021 and attributed this inhibitory action to the binding of the basic sp2 nitrogen lone pair of roquefortine to the heme iron atom (see mechanism in Fig. 2).
N– of the imidazole group and the NH-amide group of the diketopiperazine (Fig. 1) has been proposed for isoroquefortine.22 In terms of binding affinities, we have previously demonstrated the inhibitory action of roquefortine towards cytochromes P45021 and attributed this inhibitory action to the binding of the basic sp2 nitrogen lone pair of roquefortine to the heme iron atom (see mechanism in Fig. 2).
 | ||
| Fig. 2 Effect of the location of intramolecular H-bond in dehydrohistine-based cyclodipeptides. | ||
In order to gain insight into this structure–activity relationship in the case of (iso)roquefortine and to extend it to a series of analogs, a metabolism and a binding study to P450s has been undertaken. isoroquefortine should possess a lower inhibitory potential than roquefortine due to the reduced accessibility of its sp2 nitrogen. Similarly, the (Z)-isomer of urocanic acid and its derivatives 2, 3 and 5 would be expected to exhibit hydrogen bonding similar to that of roquefortine (Fig. 1) and should display markedly increased inhibitory activity relative to its (E)-isomer and its derivatives 1 and 4. In order to validate this assumption, heme spectral interaction and inhibitory studies were performed in the presence of different human isoforms of cytochrome P450 and on rat or human liver microsomes. Additionally, in order to discriminate between P450 molecular recognition and heme binding strength, the same compounds were tested on a simple hemoprotein model, microperoxidase 8 (MP8).23,24
Microperoxidase 8 is obtained by hydrolytic digestion of cytochrome c. It contains the hemeprosthetic group as well as the amino acid residues 14–21 of horse cytochrome c, including His 18, whose imidazole group acts as the fifth axial ligand of the iron. The absence of a heme iron ligand at the sixth coordination position allows this site to be occupied by a number of different ligands. In aqueous solution water occupies the sixth coordination site.23 Due to these structural characteristics, microperoxidase 8 is considered to be an effective model for the study of the redox mechanism of hemoproteins such as cytochromes, hemoglobin, myoglobin and peroxidases.23
Experimental
Chemicals
roquefortine, trans-(E)-urocanic acid, N-benzylimidazole, His-Phe, testosterone, 6β-hydroxy-testosterone, NADPH, NADP, glucose-6-phosphate (G6P), glucose-6-phosphate dehydrogenase (G6PDH), dexamethasone (DEX), clofibrate (CLO), sodium phenobarbital (PB), 3-methylcholanthrene (3-MC), and microperoxidase 8 (MP8) were obtained from Sigma Chemical. c-(Phe-His) was obtained from Bachem., BS176, compounds 1–5 and isoroquefortine were synthesized as previously described.22c-(Phe-ΔHis) and (−/+)-phenylahistin were synthesized and kindly provided by Y. Hayashi25 and cis-(Z)-urocanic acid by H. Morrison.26 All other chemicals were of the highest quality commercially available.Microsomal preparations
Human liver samples were kindly supplied by the surgery service of the Franche Comté Hospital (University of Besançon) and microsomes were prepared as previously described.27 Bactosomes®-expressed P450 3A4, 2E1, 1A2, 2C19, 2C9 and 2D6 were obtained from Tebu Co. Pooled human liver microsomes and Supersomes®-expressed P450 4A11 were obtained from Gentest Co (Woburn MA 01801, USA). Yeast-expressed human P450 1A1 was prepared as previously described and produced by SPI-Bio (Montigny le Bretonneux, France).28 Rat microsomes were prepared from a pool of livers from 4 to 6 animals untreated or treated by the various cytochrome P450 inducers as previously described.27Quantification of enzymes in microsomal fractions
Protein content in microsomal suspensions was determined by the method of Lowry et al. using bovine serum albumin as a standard.29 The P450 concentration was measured as described by Omura and Sato.30Substrate binding to P450s microsomal isoforms and to microperoxidase 8
Substrate binding to cytochrome P450 was studied by difference visible spectroscopy using 2 μM P450 from rat liver microsomes or 0.3 μM P450 from human liver microsomes in 0.1 M phosphatebuffer (pH 7.4). Bactosomes® and Supersomes®-expressed P450 were suspended in 0.1 M phosphatebuffer (pH 7.4) to obtain a P450 concentration of 0.13 μM. The solution was equally divided between two cuvettes (1 mL cuvette for microsomes and 100 μL cuvette for Bactosomes® and Supersomes®). Low amounts of human liver were used due to their high protein concentrations and turbidity. After recording the baseline, aliquots (0.5–1 μL) of substrate solutions were added to the sample cuvette, and the same volume of solvent (DMSO) was added to the reference cuvette. Difference spectra were recorded between 350 and 520 nm at room temperature, with a Perkin Elmer λ18 UV spectrophotometer equipped with a turbidity accessory. Ks and ΔODmax were calculated by linear regression from double reciprocal plots of 1/ΔOD versus 1/[substrate].31 Microperoxidase 8 preparations at 2 μM were performed in 20 mM phosphatebuffer solutions at pH 6, 7.4 or 8.1 containing 20% methanol in order to obtain a homogenous solution.23 Spectral interactions were measured in a differential mode as for hepatic microsomal preparations. Control measurements were done in simple beam spectrometry in order to evaluate the absolute high spin to low spin transition upon addition of imidazole derivative.23,24Cytochrome P450 activities
Incubations of human liver microsomal suspensions (1 mg protein/mL) were done in the presence of 100 μM of testosterone and 0.5 mM of NADPH in a 0.1 M phosphatebuffer (pH 7.4). Experiments were performed at 37 °C in the presence of 50 μM of each tested compound. Incubations were stopped after 15 min by the addition of an equivalent volume of acetonitrile. The final mixture was cooled and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes. HPLC analysis was performed as previously indicated; testosterone and 6β-hydroxytestosterone were detected at 254 nm.21
000 rpm for 10 minutes. HPLC analysis was performed as previously indicated; testosterone and 6β-hydroxytestosterone were detected at 254 nm.21
Inhibition of microperoxidase 8 activity
The peroxidase activity of MP8 was assayed by measurement of the kinetics of oxidation of o-dianisidine by H2O2.32 The assay system contained o-dianisidine (100 μM) and MP8 (0.22 μM) in phosphatebuffer (0.1 M, pH 7.4) at 25 °C in the presence or absence of the imidazole derivatives. The reaction was initiated by addition of hydrogen peroxide (90 μM). The concentration of oxidized o-dianisidine was determined by the change in absorbance at 500 nm using a molar extinction coefficient of 6360 M−1 cm−1.Molecular modeling
Quantum mechanics calculations have been carried out at the DFT-B3LYP level of theory.33–35 C, H, O and N atoms have been represented by an all-electron triple-ζ quality basis set: 6-311G(d,p).36 A relativistic electron core potential and its associated basis set have been used for Br.37 A d polarization function has been added for Br (α = 0.389). All calculations have been achieved with the Gaussian 98 suite of programs.38 Geometries have been fully optimized without any symmetry restrictions. For each molecule, all major conformers have been optimized and compared. Only the most stable conformers of the configuration of interest are presented. The nature of the extrema has been checked by an analytical calculation of the frequencies.Results
Recognition of imidazole derivatives by P450
Spectral evaluation of the binding of substrates, using human liver microsomes, revealed a type II interaction for all the substrates tested except for BS176, and trans-(E)-urocanic acid (Table 1). This type II interaction, which is characterized by a spectral absorption around 430 nm, is characteristic of the formation of a heme–imidazole complex. The dissociation constants can be calculated for compounds with values between 0.4 and 20 μM (Table 1). These affinities are in the same range as those of N-substituted imidazoles such as miconazole for which Ks values were determined to be lower than 1 μM.39| Compound | Spectral interaction | |||
|---|---|---|---|---|
| ΔODmax | K s/μM | Spectrum type | % inhibition of testosterone hydroxylation | |
| Spectral interactions were measured at room temperature using 1 mg prot/mL (0.3 nmol P450) of a pool of 10 human liver microsomes in phosphatebuffer. Spectrum type II was characterized by a substrate dependent formation of a peak around 430 nm and a trough at 390 nm in the native oxidized cytochrome P450 state. ΔODmax values were expressed as nmol P450 and Ks values were determined from double reciprocal plots from spectra obtained using 0.5 to 100 μM substrate. Incubations of 100 μM testosterone were performed in the presence of 50 μM imidazole derivatives. | ||||
| roquefortine | 0.024 | 0.4 | II | 88 |
| isoroquefortine | 0.006 | 9.0 | II | 66 |
| BS176 | n.m. | n.m. | n.m. | <5 |
| c-(Phe-His) | 0.053 | n.m. | II | 5 |
| c-(Phe-ΔHis) | 0.030 | n.m. | II | 50 |
| (±)-Phenylahistin | 0.011 | 9.0 | II | 54 |
| trans-(E)-Urocanic acid | n.m. | n.m | n.m | <5 |
| cis-(Z)-Urocanic acid | 0.047 | 20.0 | II | <5 |
| Compound 1 | 0.015 | 1.4 | II | 19 |
| Compound 2 | 0.018 | 4.6 | II | 46 |
| Compound 3 | 0.017 | 1.4 | II | 27 |
| Compound 4 | 0.016 | 1.0 | II | 19 |
| Compound 5 | 0.019 | 0.95 | II | 71 |
| His-Phe | 0.037 | n.m. | II | 25 |
| N-Benzylimidazole | 0.084 | 2.7 | II | 48 |
The strongest amplitude spectrum is observed with N-benzylimidazole (Fig. 3) for which the two imidazole nitrogen atoms are chemically non-equivalent and the accessible nitrogen is in its sp2 hybridization state. For all the other compounds measured, the protonation state of the two nitrogen atoms generally exchanges very rapidly, but interaction spectra led to the opposite conclusion. In the case of stereoisomer couples (roquefortine/isoroquefortine (Fig. 1) or trans/cis-urocanic acids), there are important discrepancies between the two stereoisomers. roquefortine contains an (E)-dehydrohistidine residue and exhibits a stronger affinity as well as spectral amplitude relative to isoroquefortine. This may be the consequence of an intramolecular hydrogen bond between the imidazole NH-group and a carbonyl of the diketopiperazine moiety. In the case of isoroquefortine, such an intramolecular hydrogen bond is not possible, and as a consequence, a lower interaction spectrum was observed with human liver microsomes. The spin shift constant measured (Ks) for the (E)-isomer roquefortine was at least 20 times lower than that of its (Z)-isomer, namely isoroquefortine. This has been confirmed using liver microsomes from rat treated with various inducers or using expressed human liver microsomes (Table 2). In addition, only roquefortine was recognized by all microsomal preparations or isoforms tested. In humans, isoroquefortine was recognized only by P450 3A4 and produced a type I spectral signature, with a minimum around 420 nm, characteristic of the low-spin hexacoordinate P450 Fe(III)H2O species in the reference cuvette and a minimum around 390 nm, that is characteristic of the formation of the high-spin Fe(III)P450 species in the sample cuvette. roquefortine had the smallest Ks values for all the microsomal preparations tested, including human liver or yeast-expressed cytochrome P450s (Table 2).
 | ||
| Fig. 3 Spectral interaction of 100 μM N-benzyl imidazole (dashed line), roquefortine (thin line) or isoroquefortine (bold line) on 2 μM microperoxidase 8. Difference spectra were obtained by addition of 100 μM imidazole derivatives to a 2 μM solution of microperoxidase 8 in a 20 mM phosphate buffered solution containing 20% methanol. | ||
| isoroquefortine | roquefortine | |||||
|---|---|---|---|---|---|---|
| ΔODmax | K s/μM | Spectrum type | ΔODmax | K s/μM | Spectrum type | |
| a Difference spectra were obtained by addition of increasing amounts of substrate to microsomal suspension containing 2 μM P450 from rats, either untreated (UT-rat) or treated with 3-methylcholanthrene (3MC-rat), dexamethasone (DEX-rat), phenobarbital (PB-rat), or clofibrate (CLO-rat), 0.13 μM P450 from yeast-expressed human P450 or 0.3 μM P450 from human liver microsomes. Spectrum type I was characterized by a substrate dependent peak formation at 390 nm and a through at 420 nm and type II by a peak around 430 nm and a trough at 390 nm in the native oxidized cytochrome P450 states. ΔODmax values and Ks values are means of two to four measurements. b In a few cases, two values were obtained from a biphasic curve; n.m. not measurable; n.d. not determined. | ||||||
| Human liver microsomes | ||||||
| 0.006 | 9.0 | II | 0.024 | 0.43 | II | |
| Rat liver microsomes | ||||||
| UT-rat | 0.048 | 101.0 | II | 0.058 | 0.63 | II |
| DEX-rat | 0.050 | 67.0 | I | ΔODmax1 = 0.033b | K s1 = 0.17 | II |
| ΔODmax2 = 0.208 | K s2 = 2.35 | |||||
| 3MC-rat | 0.050 | 6.0 | II | 0.077 | 0.69 | II |
| CLO-rat | 0.0084 | 26.3 | II | 0.067 | 0.46 | II |
| PB-rat | n.m. | n.m. | I or II | 0.082 | 2.55 | II |
| P450 isoforms | ||||||
| 1A2 | n.m. | n.m. | n.m. | 0.0105 | 0.91 | II |
| 2E1 | n.m. | n.m. | n.m. | n.m. | n.m. | n.m. |
| 3A4 | 0.010 | 1.6 | I | 0.010 | 1.60 | II |
| 2C9 | n.m. | n.m. | n.m. | 0.010 | 0.83 | II |
| 2C19 | n.m. | n.m. | n.m. | 0.011 | 0.71 | II |
| 4A11 | n.m. | n.m. | n.m. | n.m. | n.m. | n.m. |
| 2D6 | n.m. | n.m. | n.m. | 0.007 | 4.80 | II |
Comparison of the interaction mode of (Z)- and (E)-urocanic acids showed that the (Z)-isomer was the only one recognized by rat and human liver microsomes (Table 1). In this case, the spectral interaction mode was interpreted as a type II.
Cytochrome P450 inhibitory effect
Upon incubation using human liver microsomes in the presence of 50 μM imidazole derivatives and 100 μM testosterone, all the compounds, except BS176, cis- and trans-urocanic acid, and c-(Phe-His), exhibited inhibitory properties (Table 1). Observation of the amplitude of interaction shows that inhibition ranges are not only related to the presence of the imidazole moiety but also to the stereochemistry of the double bond. roquefortine demonstrated the most potent inhibitory properties whereas the derivatives of cis urocanic acid (compounds 2, 3 and 5) were more efficient inhibitors than their trans analogs 1 and 4.Interaction with microperoxidase 8
Microperoxidase 8 is a simple model of hemoprotein which is derived from cytochrome c. The binding of imidazole to the heme functionality of microperoxidase 8 has already been reported and discussed.23,24N-Benzylimidazole was used as a reference compound for the formation of a low spin N-imidazole–iron complex (absorption at 409 nm) from the high spin aqua-complex (broad absorption peak around 394 nm) (Fig. 3). In differential mode visible spectroscopy, all the tested compounds except BS176 and phenylahistin gave a 394 to 409 nm shift (Table 3). This spectral shift was concentration-dependent. His-Phe, c-(Phe-His) and compound 3, which have mobility between the amide function and the imidazole moiety and can form an intramolecular C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HN(Im) bond (Fig. 2), gave similar high spin to low spin transition ratios with respect to N-benzylimidazole, whereas c-(Phe-ΔHis), which forms an intramolecular NH⋯N(Im) bond, exhibited lower interaction spectrum. (Z)- and (E)-urocanic acid and compounds 4 and 5 showed similar spectral transitions of approximately 50% of that of N-benzylimidazole. The strongest differences between E and Z configurations were observed in the roquefortine series. roquefortine led to around 60% transition spectra whereas isoroquefortine showed around 5% (pH 7.4). Similarly the cis-urocanic methyl ester 2 led to a 4 times stronger interaction spectrum than its trans isomer 1. BS176, which is a N-substituted isoroquefortine, was unable to significantly modify the microperoxidase 8 spin state.
O⋯HN(Im) bond (Fig. 2), gave similar high spin to low spin transition ratios with respect to N-benzylimidazole, whereas c-(Phe-ΔHis), which forms an intramolecular NH⋯N(Im) bond, exhibited lower interaction spectrum. (Z)- and (E)-urocanic acid and compounds 4 and 5 showed similar spectral transitions of approximately 50% of that of N-benzylimidazole. The strongest differences between E and Z configurations were observed in the roquefortine series. roquefortine led to around 60% transition spectra whereas isoroquefortine showed around 5% (pH 7.4). Similarly the cis-urocanic methyl ester 2 led to a 4 times stronger interaction spectrum than its trans isomer 1. BS176, which is a N-substituted isoroquefortine, was unable to significantly modify the microperoxidase 8 spin state.
| Compound | Spectral interactiona | Inhibition of peroxidase activityd | |
|---|---|---|---|
| ΔOD409–394 nm | Percentb | ||
| a Difference spectra were obtained by addition of 100 μM imidazole derivatives to a 2 μM solution of microperoxidase 8 in a 20 mM phosphate buffered solution containing 20% methanol. b Percentage of the spectral interaction obtained with N-benzylimidazole which possesses only one accessible tertiary nitrogen. c n.d. no detectable peak shift. d Percentage of the initial velocity of peroxidase activity obtained using 100 μM o-dianisidine as substrate, 90 μM H2O2, 0.22 μM microperoxidase 8, 100 μM imidazole derivatives in 0.1 M phosphatebuffer, pH 7.4 at 25 °C. Peroxidase activity was 23 nmol o-dianisidine consumed min−1 nmol microperoxidase−1. Values are the mean of two to four independent determinations. | |||
| N-Benzylimidazole | 0.28 | 100 | 58 |
| roquefortine | 0.17 | 61 | 76 |
| isoroquefortine | 0.014 | 5 | 0 |
| BS176 | <0.005 | n.d.c | 60 |
| c-(Phe-His) | 0.29 | 103 | 80 |
| c-(Phe-ΔHis) | 0.057 | 20 | 0 |
| (±)-Phenylahistin | n.d.c | n.d.c | 0 |
| trans-(E)-Urocanic acid | 0.16 | 57 | 18 |
| cis-(Z)-Urocanic acid | 0.19 | 68 | 69 |
| Compound 1 | 0.03 | 11 | 25 |
| Compound 2 | 0.12 | 43 | 39 |
| Compound 3 | 0.29 | 105 | 55 |
| Compound 4 | 0.07 | 37 | 30 |
| Compound 5 | 0.12 | 44 | 13 |
| His-Phe | 0.34 | 121 | 41 |
The concentration of imidazole–iron complexes may be related to the decrease of the peroxidase activity, since N-benzylimidazole, His-Phe, c-(Phe-His), compound 3, roquefortine and (Z)-urocanic acid inhibited more than 60% of the initial rate of o-dianisidine peroxidation. isoroquefortine, c-(Phe-ΔHis) and (E)-urocanic acid and its derivatives 1, 2, 4 and 5 showed either no or low inhibition of such activity (Table 3). BS176, which is unable to form a significant concentration of microperoxidase complex, is still able to inhibit microperoxidase activity. We have observed that this compound can be oxidized under such conditions, it could then act as a competitive inhibitor for the oxidation of ortho-dianisidine.
Cytochrome P450 metabolism of imidazole derivatives
Liver microsomes from control or dexamethasone pretreated rats in combination with a NADPH-generating system were used for the metabolism study. Under these conditions, only substituted cyclodipeptides were metabolized. isoroquefortine, and phenylahistin, were significantly metabolized (turnover values being around 2 nmol metabolized min−1 nmol P450−1), whereas c-(Phe-His), c-(Phe-ΔHis) and His-Phe were not. Metabolism of urocanic acid derivatives was not studied.Using human liver microsomes, more than 60% of isoroquefortine was metabolized in three major monohydroxylated metabolites, the main one being hydroxylated on the isoprenyl function (see figures A and B in the ESI† ) whereas roquefortine yielded almost no metabolite (see figure A in ESI† ). This isoroquefortinemetabolism is only observed in the presence of NADPH and was inhibited by ketoconazole, a classical P450 3A inhibitor. Other P450 inhibitors such as furafylline (1A2), quinidine (2D6) and sulfaphenazole (2C9) had no significant effects on the formation of metabolites.
Molecular modeling
In order to obtain more information concerning the relative stability of the different tautomers of roquefortine, urocanic acids and their derivatives, the energy-minimized structures of these molecules have been determined by quantum mechanics at the DFT-level. Fig. 4 indicates the most stable isomers of each protonation state and their relative energy in kcal mol−1. In the gas phase, the more favorable conformation of isoroquefortine is slightly more stable than that of roquefortine by 2.8 kcal mol−1. This can be explained by a lesser cyclic constraint in isoroquefortine (H-bond based on a 6 membered ring) than in roquefortine (H-bond based on a 7-membered ring). However, considering the precision of the calculation level, these two isomers have to be considered as isoenergetic. For both isomers, the more stable tautomer is the one that can develop an intramolecular hydrogen bond. The loss of this hydrogen bond leads to structures with increased energies of 7.3 kcal mol−1 (isoroquefortine) and 12.6 kcal mol−1 (roquefortine). In these two molecules, the imidazole ring has a different role with respect to the establishment of a hydrogen bond. In isoquefortine, imidazole acts as an H-bond acceptor, whereas it acts as a donor in roquefortine. Hence, roquefortine can act as a Lewis base and efficiently bind a metal center whereas isoroquefortine cannot.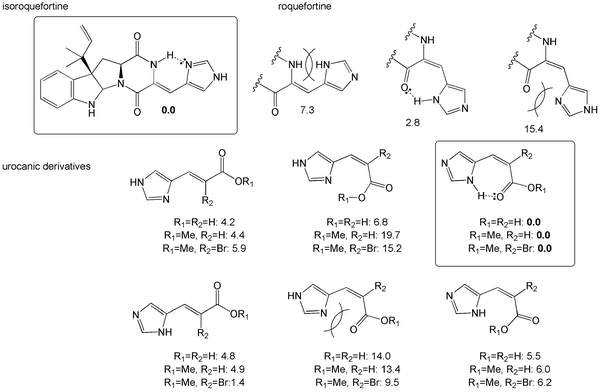 | ||
| Fig. 4 Relative energies of tautomers of roquefortine/isoroquefortine and urocanic acids. The relative energies are given with respect to the more stable isomer and are expressed in kcal mol−1. | ||
The same analysis can be performed in the case of urocanic acids and their methyl ester and α-brominated derivatives. Whereas the two tautomers of (E)-urocanic acid are isoenergetic, one isomer of (Z)-urocanic acid is much more stable than the others. This isomer is characterized by an H-bond between the imidazole (acting as an H-bond donor) and the carboxylic function (acting as the complementary acceptor). Hence the imidazole ring of this isomer can behave as a Lewis base and efficiently bind the iron atom of heme. Noteworthily, the reverse situation, in which the imidazole ring acts as an acceptor and the carboxylic as a donor is slightly less favorable. The same computational study applied to the urocanic acid methyl esters and the α-brominated derivatives leads to very similar results. In each case, the Z conformation in which the imidazole group acts as an H-bond donor and the carbonyl group as an H-bond acceptor is the most stable configuration.
Finally, the conformational sampling of the dihydro isomer 3 revealed the absence of an intramolecular H-bond. This result shows that the energy that could be gained through H-bonding cannot compensate the cost associated with the formation of a 7-membered ring.
Discussion
Imidazole derivatives are largely used as hemoproteininhibitors, and the most effective compounds generally contain N-substituted imidazoles.15 A few unsubstituted imidazole derivatives exhibit weak inhibitory effects mainly related to the rapid proton exchange between the two imidazole nitrogens. To our knowledge, this report represents the first study of the hemoprotein inhibitory properties of dehydrohistidine-based derivatives in which nitrogen atoms are not substituted. Our studies demonstrate that these compounds interact with the heme iron of cytochrome P450 and microperoxidase 8. Formation of the iron–imidazole complex is sufficiently stable to perturb the catalytic cycle of such hemoproteins. Inhibitory potency is dependent upon the formation of intramolecular hydrogen bonds between either imidazole nitrogen and the surrounding CO or NH groups of amide functions. Comparison of the inhibitory effects between c-(Phe-His) and c-(Phe-ΔHis) (Table 3) clearly demonstrates that this intramolecular interaction is associated with the planarity imposed on the molecule by the Cα–Cβ double bond of the dehydro-residue. The (E)-configuration allows strong hydrogen bonding between the diketopiperazine carbonyl and the imidazole NH group. Therefore, the imidazolesp2 nitrogen is accessible to bind the heme iron. In contrast, in the (Z)- configuration, the basic nitrogen atom of imidazole is inaccessible due to hydrogen bonding with a vicinal NH group, which lowers the binding potency to the heme iron atom. This interpretation of the effect of the molecular environment on the binding of ligand to heme is strongly supported by quantum mechanical optimized structures of the different isomers of the molecules herein studied.Such discrepancies in the affinities of (Z)- and (E)-dehydrohistidines arise from differences in imidazole nitrogen accessibility. As indicated by NMR and molecular modeling, (Z)-isomers possess an imidazole nitrogen doublet, which is involved in a hydrogen bond with a NH group of the diketopiperazine (Fig. 2 and 4). This conformation greatly minimizes the availability of the imidazole nitrogen doublet for hemeiron binding. The accessible nitrogen doublet of the imidazole shows only a low affinity for iron association.15–18 In contrast, in (E)-isomers such as roquefortine, the hydrogen atom of the protonated imidazole nitrogen is bonded to the oxygen of the diketopiperazine carbonyl group (Fig. 2 and 4), making the lone pair of the remaining nitrogen atom available for binding to heme iron.
In general, (Z)-dehydrohistidine isomers are preferentially formed by dehydration of histidine derivatives.40 In the same way, natural dehydro-amino acids are predominantly found in the (Z)-configuration.41–44 Interestingly, some methods for the preparation of dehydro-diketopiperazines have been recently patented.45 Alternatively, examples of natural (E)-dehydrohistidine are few, and roquefortine is an exception that has been of interest due to its antibacterial activities.10 The first total synthesis of roquefortine was recently reported using a novel elimination strategy.46
In the urocanic acid series, cis isomers exhibit higher inhibition strength towards P450s and MP8. This result is in agreement with the structure of the most stable tautomers of these cis-compounds in which the imidazole ring acts as an H-bond donor with respect to the intramolecular carbonyl group. Experimentally, the inhibition potency of these compounds is reduced by changing the stereochemistry of the double bond from cis to trans (Z to E). In trans isomers, no intramolecular H-bond can be developed, hence both imidazole tautomers are in equilibrium, which results in a decrease of binding strength. Finally, compound 3 highlights the sterical constraints induced by α,β-saturation on the nitrogen lone pair accessibility. Indeed, the planarity of the dehydro-compounds causes the Cα–H or Cβ–H to point towards a direction similar to that of the imidazole lone pair and, hence, decreases the binding strength of the molecule compared to compound 3 in which the planarity is lost.
Our results suggest intriguing possibilities for the development of hemoproteininhibitors, which may be deactivated by simple structural modification. Such structural modifications can easily be obtained under light exposure or saturation reduction. In the case of urocanic acid isomers, further research is in progress in an attempt to correlate hemoprotein inhibition properties to in vivo modifications of physiological parameters and precisely address the impact of the substitution on the α and/or β position in the dehydro series.
Acknowledgements
Part of this work was supported by the AC 009E Environnement et Santé program of the French Ministère de l’Ecologie, de l’Energie , du Développement durable et de l’Aménagement du Territoire. We also wish to thank Ms S. Blondel for her excellent technical assistance. The synthetic work was supported by NSF (CHEM 0515443).References
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, Science Book University, Mill Valley, 1994 Search PubMed.
- P. R. Ortiz de Montellano and M. A. Correia, in Cytochrome P450: Structure, Mechanism and Biochemistry, ed. P. R. Ortiz de Montellano, Plenum Press, New York, 1995, pp. 305–364 Search PubMed.
- S. D. Zaric, D. M. Popovic and E. W. Knapp, Biochemistry, 2001, 40, 7914–7928 CrossRef CAS.
- B. Testa and P. Jenner, Drug Metab. Rev., 1981, 12, 1–117 CAS.
- M. Murray and A. J. Ryan, Xenobiotica, 1983, 13, 707–714 CAS.
- W. Zhang, Y. Ramamoorthy, T. Kilicarslan, H. Nolte, R. F. Tyndale and E. M. Sellers, Drug Metab. Dispos., 2002, 30, 314–318 CrossRef CAS.
- H. Van den Bossche, P. Marichal, J. Gorrens and M. C. Coene, Br. J. Clin. Pract. Suppl., 1990, 71, 41–46 Search PubMed.
- E. Albengres, H. Le Louet and J. P. Tillement, Drug Saf., 1998, 18, 83–97 CrossRef CAS.
- M. A. Abraham, P. P. Thomas, G. T. John, V. Job, V. Shankar and C. K. Jacob, Transplant. Proc., 2003, 35, 215–216 CrossRef CAS.
- P. Cozzi, G. Branzoli, C. Carganico, C. Ferti, A. Pillan, D. Severino and R. Tonani, Eur. J. Med. Chem., 1991, 26, 423–433 CrossRef CAS.
- D. J. Wolff, G. A. Datto, R. A. Samatovicz and R. A. Tempsick, J. Biol. Chem., 1993, 268, 9425–9429 CAS.
- S. Goel, M. Cohen, S. N. Cömezoglu, L. Perrin, F. André, D. Jayabalan, L. Iacono, A. Comprelli, V. T. Ly, D. Zhang, C. Xu, W. G. Humphreys, H. McDaid, G. Goldberg, S. B. Horwitz and S. Mani, Clin. Cancer Res., 2008, 14, 2701–2709 CrossRef CAS.
- L. J. Brandes, G. M. Queen and F. S. LaBella, Cancer Chemother. Pharmacol., 2000, 45, 298–304 CrossRef CAS.
- L. L. Von Moltke, D. J. Greenblatt, S. X. Duan, J. S. Harmatz, C. E. Wright and R. I. Shader, J. Clin. Psychopharmacol., 1996, 16, 104–112 CrossRef CAS.
- T. D. Rogerson, C. F. Wilkinson and K. Hetarski, Biochem. Pharmacol., 1977, 26, 1039–1042 CrossRef CAS.
- K. K. Hajek, N. I. Cook and R. F. Novak, J. Pharmacol. Exp. Ther., 1982, 223, 97–104 CAS.
- M. Murray and L. Zaluzny, Biochem. Pharmacol., 1988, 37, 415–420 CrossRef CAS.
- C. F. Wilkinson, K. Hetnarski, G. P. Cantwell and F. J. Di Carlo, Biochem. Pharmacol., 1974, 23, 2377–2386 CrossRef CAS.
- A. Verras, I. D. Kuntz and P. R. Ortiz de Montellano, J. Med. Chem., 2004, 47, 3572–3579 CrossRef CAS.
- M. Delaforge, G. Bouille, M. Jaouen, C. K. Jankowski, C. Lamouroux and C. Bensoussan, Peptides, 2001, 22, 557–565 CrossRef CAS.
- C. Aninat, Y. Hayashi, F. André and M. Delaforge, Chem. Res. Toxicol., 2001, 14, 1259–1265 CrossRef CAS.
- B. M. Schiavi, D. J. Richard and M. M. Joullié, J. Org. Chem., 2002, 67, 620–624 CrossRef CAS.
- H. M. Marques, M. S. Shongwe, O. Q. Munro and T. J. Egan, S. Afr. J. Chem., 1997, 50, 166–180 CAS.
- D. A. Baldwin, H. M. Marques and J. M. Pratt, J. Inorg. Biochem., 1986, 27, 245–254 CrossRef CAS.
- Y. Hayashi, S. Orikasa, K. Tanaka, K. Kanoh and Y. Kiso, J. Org. Chem., 2000, 65, 8402–8405 CrossRef CAS.
- T. Mohammad and H. Morrison, OPPI Briefs, 2000, 32, 581–584 Search PubMed.
- P. Kremers, P. Beaune, T. Cresteil, J. de Graeve, S. Columelli, J. P. Leroux and J. E. Gielen, Eur. J. Biochem., 1981, 118, 599–606 CrossRef CAS.
- M. A. Peyronneau, J. P. Renaud, G. Truan, P. Urban, D. Pompon and D. Mansuy, Eur. J. Biochem., 1992, 207, 109–116 CrossRef CAS.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- T. Omura and R. Sato, J. Biol. Chem., 1964, 239, 2370–2378 CAS.
- J. B. Schenkman, S. G. Sligar and D. L. Cinti, Pharmacol. Ther., 1981, 12, 43–71 CrossRef CAS.
- Y. Kawamura-Konishi, A. Asano, M. Yamazaki, H. Tashiro and H. Suzuki, J. Mol. Catal. B: Enzym., 1998, 14, 181–190 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98 (Revision A.11), Gaussian Inc., Pittsburgh PA, 2001 Search PubMed.
- K. Morita, T. Ono and H. Shimakawa, J. Pharmacobiodyn., 1988, 11, 106–114 CAS.
- H. Kanzaki, D. Imura, T. Nitoda and K. Kawazu, J. Biosci. Bioeng., 2000, 90, 86–89 CAS.
- R. Cardillo, C. Fuganti, G. Gatti, D. Ghiringhelli and P. Grasseli, Tetrahedron Lett., 1974, 15, 3163–3166 CrossRef.
- C. G. Shin, M. Hayakawa, K. Mikami and J. Yoshimura, Tetrahedron Lett., 1977, 18, 863–866 CrossRef.
- B. Kopp-Holtwiesche and H. J. Rehm, J. Environ. Pathol. Toxicol. Oncol., 1990, 10, 41–44 Search PubMed.
- O. P. Larsen, J. C. Frisvad and S. R. Jensen, Phytochemistry, 1992, 31, 1613–1615 CrossRef.
- E. Couadouros, A. Magos and A. Strongilos, Methods of preparation of dehydro-diketopiperazines in liquid phase or of solid supported phase. WO 2005/003102 A1, 2005.
- N. Shangguan, W. J. Hehre, W. S. Ohlinger, M. P. Beavers and M. M. Joullié, J. Am. Chem. Soc., 2008, 130, 6281–6287 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC separation and MS-MS spectra of roquefortine and its metabolites. See DOI: 10.1039/b817743k |
| ‡ Marcel Delaforge, CEA-Saclay, iBiTec-S SB2SM, CNRS URA2096, bât. 528, 91191 Gif-sur-Yvette, France. |
| This journal is © The Royal Society of Chemistry 2009 |