The role of copper in cysteine oxidation: study of intra- and inter-molecular reactions in mass spectrometry†
Michel
Prudent
and
Hubert H.
Girault
*
Laboratoire d’Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland. E-mail: Hubert.girault@epfl.ch; Fax: +41 21 693 36 67; Tel: +41 21 693 31 45
First published on 24th December 2008
Abstract
Cysteine-containing peptide oxidation was studied both by using an inert platinum electrode and a sacrificial electrode (copper or zinc) generating metallic ions in electrospray ionization mass spectrometry (ESI-MS). Using peptides containing one, two and three cysteines, we have compared the different chemical and electrochemical oxidation pathways of cysteine (RS−IIH) to cystine (RS−IS−IR) and to sulfenic, sulfinic and sulfonic acid (RS0OH, RSIIO2H and RSIVO3H, respectively). In the absence of copper ions, intra-molecular reactions were the most abundant, whereas inter-molecular reactions were found to be enhanced by the presence of copper ions. These cations favor the formation of 2 : 1 (peptide : copper) complexes compared to 1 : 1 complexes, thus enhancing the formation of inter-molecular bridges. This study highlights the importance of the position of cysteine inside a peptide during disulfide bridge formation.
Introduction
Misfolding of proteins and their aggregation are responsible for neurological disorders, such as Alzheimer’s (AD), Parkinson’s (PD) or prion (PrP) diseases. The formation of plaques and other aggregates are the hallmark of these neurodegenerative disorders. Reactive oxygen species (ROS) are involved in cell damage, protein misfolding, etc. and are implicated in a large number of degenerative diseases and aging. An imbalance between the production of ROS and the antioxidant defenses results in the oxidative stress responsible for this damage. In the presence of hydrogen peroxide, ROS are generated by either Haber–Weiss or Fenton reactions. The latter involves transition metal ions, especially copper and iron in their low oxidation states.Cysteine residues play a crucial role in these structures. The oxidation of the thiol (RS−IIH) residues of the cysteines is particularly sensitive and forms reversible covalent bonds, which are major mechanisms that control the function of proteins.1 These oxidations lead to the formation of intra-molecular and inter-molecular disulfide bridges (RS−IS−IR: cystine) or sulfenic, sulfinic and sulfonic acid, RS0OH, RSIIO2H and RSIVO3H, respectively. Their interactions with transition metal ions are of biological importance2 and can produce toxicity or lethality. Cysteines are, for instance, present in copper binding sites for copper homeostasis, which is known to be involved in two human disorders of copper transport, i.e. Menkes and Wilson’s diseases.3–5Thiol residues are oxidized in the presence of molecular oxygen6 and these reactions are catalyzed by transition metal ions, such as iron or copper.7,8 Kachur et al. have described the mechanism of copper-catalyzed auto-oxidation of cysteine,9 where the cuprous biscysteine complex (RS−CuI−SR) was identified as the catalytic species.10 Copper ions exist mainly as cupric and cuprous ions and interact with cysteine according to the hard–soft acid–base classification forming stable complexes. Cu2+ is an intermediate acid that prefers binding to intermediate ligands such as N-imidazole (histidine residue) and Cu+ is a soft acid that prefers anchoring to soft ligands such as S-thiolate (cysteine residue) and also N-imidazole.11 In mass spectrometry (MS), where ions are transferred from a liquid to a gas phase, the binding sites of Cu2+ in proteins are identical12,13 to those observed in solution, but arginine becomes the preferred anchorage for Cu+.13–15 Cerda and Wesdemiotis noticed that the Cu+ affinity for amino acids is increased when soft donor groups such as RSH are present compared to that of protons.14
Mass spectrometry, especially coupled with an electrospray ionization (ESI) source, is a method of choice for studying biomolecules and their interactions with metal ions, as well as for studying peptide or protein oxidation. Commonly, these modifications induce mass shifts and their locations are analyzed by tandem mass spectrometry.16–18 In proteins, few amino acids can be oxidized, i.e.tyrosine, tryptophan, histidine, methionine and cysteine. All the different routes of cysteine oxidation have been studied by MS, i.e. the formation of disulfide bridges19–21 or the formation of sulfenic and derivative acids,21–24 and these oxidations can be not only chemical but also electrochemical.25,26 Permentier et al. have studied the on-line electrochemical oxidation and cleavage of proteins in MS,17,27 because the electrospray itself can be used directly for oxidation.21,28,29 Thus, reactions or tagging of biomolecules can be achieved by taking advantage of these properties30–32 and sacrificial electrodes can supply the production of metal ions.33–35 Indeed, in positive mode, the electrode used to supply the spray current is an anode that can electrogenerate metallic ions, such as Zn2+ or Cu2+/+. In our laboratory, sacrificial electrodes and microchips have been coupled together with ESI-MS in order to study on-line complexation of peptides with metallic ions.36 In particular, sacrificial copper electrodes lead to the electrogeneration of either copper(I) or copper(II) ions depending on the ligands present in aqueous solution.13,37
Taking advantage of the electrochemical aspects of ESI, the complexation of copper ions, in different oxidation states, with non-cysteinyl peptides have been previously studied by using a sacrificial copper electrode.13 Herein we study, with the same device, cysteine oxidation of peptides containing, one, two or three cysteines to compare the different oxidation pathways. The experiments were carried out first with a platinum electrode to observe the oxidation of the peptidesper se, and then with a copper electrode to study the role of copper ions. The three-cysteine-containing peptide, showing the different trends of oxidation, was also tested with a zinc electrode, which is a cation that cannot transfer electrons. In addition, the effect of the electrode material was pointed out. This work highlights both the influence of copper ions on cysteine oxidation and the influence of the number and position of the cysteines along the peptidic chain.
Experimental section
Chemicals
The one- and two-cysteine-containing peptides AIKCTKF (M1, monoisotopic mass m = 809.5 u) and ALRCTCS (M2, m = 752.3 u) were synthesized at the Institut de Biochimie (Lausanne University, Switzerland) and the three-cysteine-containing peptide KCTCCA (M3, m = 627.2 u) was bought from Bachem (Bubendorf, Switzerland). Cupric sulfate pentahydrate (CuSO4·5H2O) and zinc chloride (ZnCl2) were from Fluka (Büchs, Switzerland). Methanol was from Riedel-de-Haën (Seelze, Germany). Deionized water (18.2 MΩ cm) was prepared using a Milli-Q system from Millipore (Bedford, MA). The peptides were used as received and diluted at a final concentration of 50 μM in 50/50 (v/v) MeOH–H2O. When the salts were used, they were mixed with the peptide in order to get a final concentration of 50 μM of peptide and 50 μM of copper or zinc. All the solutions were prepared daily.Microspray interface and MS setup
Metal ion on-line complexation was carried out using a microspray interface36,37 with a microchannel of 45 μm × 120 μm × 1 cm, made in polyimide and developed by DiagnoSwiss SA (Monthey, Switzerland).38,39 A reservoir of polycarbonate (ø = 6.5 mm, h = 5 mm) was glued at the inlet of the microchannel. The sample was loaded (V = 100 μL) in the reservoir in which a metallic electrode was immersed. This electrode was made either of platinum wire (blank, copper(II) or zinc(II) salt experiments), or of copper or zinc plate. All electrodes were sanded and rinsed with methanol before each experiment.An LCQ DUO ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) was used in positive ionization mode. The heated capillary was kept at 200 °C. The commercial ESI interface was removed and the microchip was mounted on a plate fixed on the probe slide adapter of the mass spectrometer. The MS power supply was fixed at 3.5 kV. The ion optics parameters were kept constant for each peptide studied and optimized on the protonated peptide.
The experiments were followed with time until reaching a steady-state conversion rate of the oxidation of cysteines. The conversion rates χ were calculated as follows:
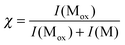
The nomenclature considered below for the covalent dimers formed by a disulfide bridge is 2M in which the loss of 2H is included. Mox is used for intra-molecular oxidation.
Results and discussion
One-cysteine-containing peptide
AIKCTKF (M1) was analyzed successively with a Pt and a Cu electrode. Scheme 1 summarizes the different molecules formed according to the sulfur oxidation states. With a Pt electrode, the cysteine moiety of AIKCTKF was slightly oxidized (approximately 10%) to cystine forming a covalent dimer (2M1) at m/z = 1617.5 Th (see Table 1, MS not shown). Mass shifts of +16, +32 and +48 Da evidenced the formation of cysteine sulfenic, sulfinic and sulfonic acid, respectively. These oxidations can be induced either by the oxygen present in solution during the ESI process or by electrochemical oxidation of the cysteine moiety at the Pt electrode in contact with the solution (see last section).17,26 | ||
| Scheme 1 Reactions of AIKCTKF observed in microchip ESI-MS in the presence or absence of copper, classified according to the sulfur oxidation state of the reacted cysteine. The relative abundances are pointed out by using different font sizes. Copper complexes were observed either with a Cu electrode or with the addition of a copper(II) salt, which is indicated on the arrows. Numbers stand for the m/z in Th. By default z = 1 otherwise the net charge is written in brackets. | ||
| m/z/Th | Pt | Cua | ||
|---|---|---|---|---|
| a The relative abundances, added in brackets, were taken when the oxidations reached a steady state. − −: not observed; −: weak; +: less abundant; ++: abundant. | ||||
| AIKCTKF | M1 | 810.3 | ++ | + |
| Oxidation to cystine (2M1) | 540.3/809.4/1617.6 | −/−/− | +/++/+ | |
| Oxidation to RSOH/RSO2H/RSO3H | 826.3/842.3/858.3 | −/−/− | − −/− −/+ | |
| Copper adducts on M1 | 934.1 | − − | 2 Cu+ (−) | |
| 871.1/872.2 | − −/− − | 1 Cu2+ (−)/1 Cu+ (++) | ||
| Copper adducts on dimer | 839.7/840.3 | − −/− − | 1 Cu2+ (+)/1 Cu+ (++) | |
| 1678.3/1679.3 | −/− | 1 Cu2+ (−)/1 Cu+ (−) | ||
| ALRCTCS | M2 | 753.3 | + | + |
| Oxidation to cystine (M2ox) | 751.1 | ++ | ++ | |
| Oxidation to RS2O/RS2O2 | 767.3/783.2 | +/+ | +/++ | |
| Dimer (2M2) | 1503.3 | − − | — | |
| Copper adducts on M2 | 939.0 | − − | − − | |
| 877.1 | − − | − − | ||
| 815.2 | − − | 1 Cu+ (−) | ||
| Copper adducts on dimer | − − | − − | − − | |
| KCTCCA | M3 | 628.3 | ++ | + |
| Oxidation to cystine (M3ox) | 626.3 | + | ++ | |
| Oxidation to RSOH/RSO2H/RSO3H | 644.1/660.1/676.1 | − −/−/+ | −/Very weak/− | |
| 642.1/658.1/674.1 | Very weak | −/−/− | ||
| Dimer (2M3ox/M3+M3ox) | 1249.2/1251.2 | Very weak | −/− | |
| Copper adducts on M3 | 875.9 | − − | 4 Cu+ (−) | |
| 813.9 | 3 Cu+ (+) | |||
| 750.0 | 2 Cu2+ (−) (or 2 Cu+ (−) on M3ox) | |||
| 688.1 | − − | |||
| Copper adducts on M3+M3ox | 1560.7 | − − | − − | |
| 1498.7 | ||||
| 1436.9 | ||||
| 1375.0 | ||||
| 1313.1 | ||||
The same phenomena were observed when using a Cu electrode (see Fig. 1), but the dimer formation was found to be more important (see Table 1). This abundant dimer is present as multiply charged species: [2M1 + 3H]3+ at m/z = 540.3 Th, [2M1 + 2H]2+ at m/z = 809.4 Th and [2M1 + H]+ at m/z = 1617.6 Th. The conversion rate of the cystine formation increased with time until reaching a plateau at χ = 65% after 40 min of electrospray. According to the MS/MS data shown in Fig. 2, two AIKCTKF were linked via the formation of a disulfide bridge. Indeed, different fragments corresponding to the presence of a bond between the two cysteines, with a typical loss of 34 Da (H2S) and addition of 32 Da (S) on M1 are observed at relative abundances of about 20%. This fragmentation is typical of a disulfide bridge. The other fragments also corroborate the disulfide bridge formation, as shown in the inset.
 | ||
| Fig. 1 Mass spectrum of AIKCTKF 50 μM in 50/50 (v/v) MeOH–H2O electrosprayed with a copper electrode. * m/z = 858.3 Th, cysteine oxidation to RSO3H. | ||
 | ||
| Fig. 2 Tandem mass spectrum of 2M1 (m/z = 1617.6 Th) at 30% of relative collision energy. | ||
The electrooxidation of the Cu electrode produces either Cu2+ or Cu+ cations in solution.13,37 These cations can here react with M1 and 2M1. The addition of Cu+ on M1 (m/z = 872.2 Th) and on 2M1 (m/z = 840.3 Th for z = 2 and m/z = 1679.3 Th for z = 1‡) were the most abundant metal complexes observed. After cysteine oxidation, Cu+ can still bind to lysine. Two Cu+ ions on M1 were observed at m/z = 934.1 Th and a small quantity of Cu2+ is linked to M1 and 2M1 (see Table 1). The MS/MS experiments performed on [M1 + CuI]+ and [M1 + 2CuI]+ show that copper ions were anchored to the cysteine and to the lys3. The experiments performed with a Pt electrode in the presence of 50 μM of CuSO4 revealed almost the same copper additions but with lower abundances (see Electronic Supplementary Information, ESI† ). It has to be pointed out that copper ions do not induce cysteinyl peptide fragmentation as in the case of non-cysteinyl peptides, where the formation of copper complexes produces metastable ions that break up in the gas phase. These fragmentations were either related to peptide oxidation by Cu2+ or by metastable ions.13 This observation is identical for the multi-cysteine-containing peptides reported below.
When Cu2+ ions were added in solution and the experiments were performed with a Pt electrode (see ESI† ), copper(I) ions were also observed. This reduction is produced either in the gas phase or in solution by reductants, e.g.solvent or analyte.41–44 So, the presence of Cu+ when using a copper electrode can be explained by two processes.37 First, electrogenerated copper(II) ions can be reduced in solution or in the gas phase. Second, copper metal atoms are oxidized to copper(I) ions that react with specific ligands, such as cysteine, according to a Mattsson and Bockris mechanism,45 as discussed previously.37 We can conclude that, with a copper electrode, both copper(I) and copper(II) ions are electrogenerated.37 Copper(II) catalyzes the oxidation of cysteines to cystine as corroborated by the high abundance of 2M1 (in different charged states) and copper(I) binds to free cysteines or other residues specific to this ion, such as lysine.
Two-cysteine-containing peptide
Intra-molecular cysteine oxidation is typically observed by a negative mass shift of 2 Da of the native peak. The oxidation of the cysteine residues of ALRCTCS (M2) to cystine was fast enough to be observed in the absence of copper ions, even at the beginning of the experiments. The mass spectra obtained when using a Pt electrode showed the monomer (M2) at m/z = 753.3 Th oxidized during the experiment to M2ox at m/z = 751.1 Th (see Scheme 2 and Table 1). Here, the oxidation did not produce a dimer but an intra-molecular cystine between the two cysteines of M2 (no dimer was observed and the isotopic distribution at m/z = 751.1 Th clearly showed the presence of a 1+ species and did not exhibit any doubly charged species such as [2M2 + 2H]2+). This oxidation reached a conversion rate of ∼70% after 15 min of electrospray. In addition, peaks at m/z = 767.3 Th and m/z = 783.2 Th corresponding to a mass shift of +16 and +32 Da of M2ox indicate that the sulfur atoms of cystine are further oxidized to sulfoxide and/or sulfone (not distinguishable, top and middle right on Scheme 2), as observed by Permentier et al. where the oxidation of disulfide bridges was observed in an electrochemical cell coupled to MS.17 As reported in the previous section, these oxidations stem either from the presence of oxygen or from oxidation at the Pt electrode.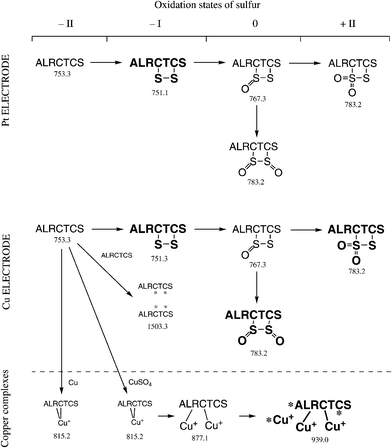 | ||
| Scheme 2 Reactions of ALRCTCS in microchip ESI-MS in presence or absence of copper. * possible binding sites. Descriptions are the same as Scheme 1. | ||
When using a copper electrode to induce the electrospray (see Fig. 3), the same oxidations of M2 were observed but the reactions were faster. The formation of sulfoxide and/or sulfone was slightly enhanced compared to oxidation in the absence of copper ions. A peak at m/z = 815.1 Th shows the binding of one Cu+, which anchors to M2 between the arginine and the cys4 according to the tandem mass spectrum (data not shown). As for the dimer 2M2, it can be detected at m/z = 1503.3 Th with a low abundance. The MS/MS experiment of M2ox (Fig. 4) confirms that the oxidation took place between the two cysteines belonging to the same peptide. This fragmentation also shows a few fragments related to the presence of a dimer. Indeed, a fragment at m/z = 1161.3 Th corresponding to M2ox bound to an oxidized y4-ion (see right inset in Fig. 4), indicates the formation of two disulfide bridges. A zoom on M2ox at m/z = 751.3 Th (Fig. 3) does not show any isotope characteristic of a doubly charged ion. However, the broad-based peak profile could hide the presence of [2M2 + 2H]2+ possibly selected during the MS/MS measurements. This dimer was probably formed in solution and isolated during the selection of M2ox (at m/z = 751.3 Th). No copper complexes were observed on M2ox.
 | ||
| Fig. 3 Mass spectrum of ALRCTCS 50 μM in 50/50 (v/v) MeOH–H2O electrosprayed with a copper electrode. | ||
 | ||
| Fig. 4 Tandem mass spectrum of M2ox (m/z = 751.3 Th) at 40% of relative collision energy. The dimer (inset in top right) can be selected as a doubly charged species during the ion selection (see text for details). | ||
Three-cysteine-containing peptide
Scheme 3 summarizes all the compounds and pathways observed for the reaction of KCTCCA (M3) in the presence or absence of copper ions. According to the number of cysteines, the two behaviors described before, i.e. intra-molecular and inter-molecular reactions were present. M3 at m/z = 628.3 Th was oxidized to cystine (M3ox at m/z = 626.3 Th) when the solution was sprayed with a Pt electrode and the presence of dimers was very dim. This oxidation was stable and the conversion rate of M3 to M3ox was around 30%. The cystine is formed between the vicinal cys4 and the cys5 according to the MS/MS performed on M3ox at m/z = 626.3 Th (data not shown). In addition, the oxidation of the thiol of the cysteine to sulfinic and sulfonic acid can take place on one of the three cysteines (top right on Scheme 3).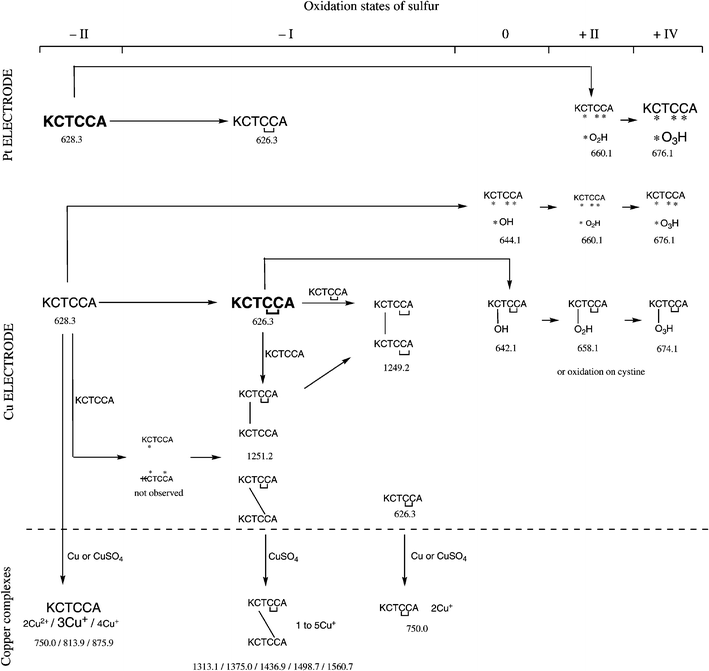 | ||
| Scheme 3 Reactions of KCTCCA in microchip ESI-MS in presence or absence of copper. * possible binding sites. Descriptions are the same as Scheme 1. | ||
When a Cu electrode was used (Fig. 5), the same oxidation was observed to reach a conversion rate of 82% after 22 min of experiment. The position of the oxidation remained identical as before, i.e. between cys4 and cys5, and the copper ions influenced only the conversion rate of oxidation and not the reaction site. The presence of copper ions also induced the formation of the dimers 2M3ox and M3 + M3ox at m/z = 1249.2 and 1251.2 Th, respectively. The dimer 2M3 was not observed. The MS/MS of 2M3ox (Fig. 6a) breaks it up in two parts, amongst other fragments, with typical mass shifts of −34 Da and +32 Da of M3. This fragmentation was also observed with the dimer of the one-cysteine-containing peptide (Fig. 2). The structure deduced from the tandem mass spectrum corresponds to the one shown as an inset and in Scheme 3. As before, this oxidation was catalyzed by copper ions electrogenerated during the electrospray process. These copper ions (up to four Cu+) linked to M3 and the most abundant was [M3 + 3Cu+ − 2H]+ at m/z = 813.9 Th. The addition of 2Cu+ is also possible on M3ox at m/z = 750.0 Th but cannot be distinguished from the addition of 2Cu2+ on M3. No addition of copper ions was obtained on the dimers, except when using a copper(II) salt where copper ions bound to the dimer M3 + M3ox were observed (see Scheme 3 and ESI† ). As for the fragmentation of M3 + M3ox at m/z = 1251.2 Th (Fig. 6b), the disulfide bridge linking the two peptides is positioned between cys2 of M3ox and cys5 of M3, as shown as an inset and at the bottom of Scheme 3. Of course, a bridge between the two cys2 should be also present in order to produce the structure of 2M3ox shown in Fig. 6a. The loss of 34 Da and the addition of 32 Da were observed in both fragments M3ox and M3, as illustrated in the magnified part of the spectrum. Of course, the fragments marked with an asterisk can stand for other ions of symmetrical structure but imply the same locus of oxidation.
 | ||
| Fig. 5 Mass spectrum of KCTCCA 50 μM in 50/50 (v/v) MeOH–H2O electrosprayed with a copper electrode. * cysteine oxidation to RSOH, RSO2H and RSO3H; ■ M3ox + 56 (see text for details). | ||
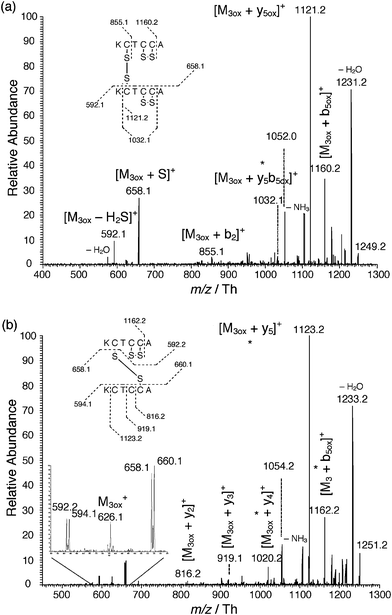 | ||
| Fig. 6 Tandem mass spectra of (a) 2M3ox (m/z = 1249.2 Th) and (b) M3 + M3ox (m/z = 1251.2 Th) at 27% of relative collision energy. * stand for compounds where alternative structures can be considered. | ||
KCTCCA reactions with Zn2+ and other reactions
Finally, the reactions of KCTCCA were followed in the presence of Zn2+ electrogenerated or added as a zinc salt to study exclusively the complexation reactions in the absence of any auto-oxidation. Fig. 7a shows the mass spectrum obtained when KCTCCA was sprayed with a Zn electrode. The peaks at m/z = 689.9 Th and m/z = 751.9 Th correspond to the addition of one and two 64Zn2+ ions on M3, respectively. The complex with M3ox ([M3ox + ZnII − H]+) was also observed with a low relative abundance. The peptide was oxidized to form M3ox but the dimers, observed in the presence of copper, were not observed in the presence of Zn2+. Moreover, oxidation to sulfenic, sulfinic or sulfonic acid did not take place. However, the behavior was sightly different when the experiments were performed with a Pt electrode in the presence of ZnCl2 (Fig. 7b). The addition of one and two Zn2+ were weaker than in the case of the Zn electrode. As before, the formation of dimers was not achieved. However, mass shifts of +16, +32 and +48 Da of M3ox are observed, showing the oxidation of the free cysteine, which was not the case with a Zn electrode. The relative abundances of these modifications increased with time and cysteinesulfonic acid was the most abundant species (the mass spectra shown were taken at equivalent time in order to compare the influence of the two kinds of electrodes). The redox potential of Zn2+/Zn0 is very low (E0Zn2+/Zn = −0.76 V vs. SHE) and Zn0 is easily oxidized. In the ESI source and hence in the well where the electrode was immersed, Zn will be the first species oxidized. Thus, the oxidation of the free cysteine (cys2) can be performed with a Pt electrode but not with a Zn electrode, the redox potentials of these oxidations being higher than that of Zn. As for the abundance of the zinc adducts, the presence of the counter ions can decrease the yield of complexation, as it was pointed out before.37 | ||
| Fig. 7 Mass spectra of KCTCCA 50 μM in 50/50 (v/v) MeOH–H2O electrosprayed with (a) a Zn electrode, t = 9 min, and (b) a Pt electrode in presence of ZnCl2 50 μM, t = 7 min. * adduct of Na+; ■ M3 + 56 at m/z = 684.2 Th and also M3ox + 56 in (a). The dimers are not formed. The insets show the absence of RSOxH when a Zn electrode is used (see text for details). | ||
As a last remark, a positive mass shift of 56 Da of M3 and of M3ox (see square marker in Fig. 5 for the former) and in less abundance of M2 was observed. This addition was identified on the cysteine moiety and more precisely on cys6 of M2, cys2 of M3ox and cys4 of M3 according to the MS/MS data (not shown). This modification was important with KCTCCA because one of the three cysteines was still free to be modified. But, in the case of ALRCTCS, the intra-molecular reaction was important and this modification disappeared during the course of the experiment. At the beginning of the oxidation process, a peak at m/z = 809.4 Th was present, corresponding to a mass shift of +56 Da. Then, when the conversion rate of oxidation was high, both M2 (m/z = 753.3 Th) and M2 + 56 disappeared, which corroborates the link with the reduced form of the cysteine. The same phenomenon happened with the Cu electrode, where M3 + 56 was observed at the beginning of the analysis and, when M3 was oxidized, M3ox + 56 was enhanced. The site of modification was clearly identified on the free thiol moietyof cysteine, but the type of modification has not been identified yet.
Inter-molecular and intra-molecular oxidation of cysteines catalyzed by copper ions
Common oxidations on proteins during the analyses by ESI-MS are localized on tyrosine, tryptophan, methionine and cysteine. In the present study, only cysteines are present in the sequence of the chosen peptides and are the loci of the oxidations. Oxygen leads to the oxidation of cysteines to cystine and other derivative products with higher sulfur oxidation states. The microchips used operate in the air, and during the transition between the liquid and the gas phase the contact with oxygen is enhanced. The Taylor cone and the droplets expulsed from it,29,46 locus of this transition, increase the area in contact with the air.In addition, the applied potential inherent to the electrospray process in positive ionization mode allows the oxidation of these thiol moieties.21,26 Especially in the case of the two-cysteine-containing peptide, the abundance of M2ox increased during the experiment. The sample was placed in a well in contact with the electrode. This well, acting as an electrochemical cell, accumulates electrochemically oxidized peptides, which explains the evolution with time. These accumulations were achieved in the well because of the low flow rate governed by the height of the solution in the well.32 The oxidation potential depends on several parameters such as the pH and the solvent. Indeed, in positive ionization mode, an oxidation has to occur in order to maintain the ESI process, normally being the solvent. Here, the redox potential of cysteine is low enough to be oxidized and observed in ESI-MS. The evidence of the electrochemical oxidation of cysteine was also pointed out by using a zinc electrode, where Zn0 was preferentially oxidized and disabled some cysteine oxidation (Fig. 7). In addition, this electrochemical oxidation has been observed in thioether crowns.47
Transition metal ions catalyze the cysteine oxidation by generating reactive intermediates such as radicals.9,10 Copper plays the role of electron carrier from thiol to oxygen where the complexation of copper(II) is the first step in the oxidation process. In both experiments with the copper electrode and with the copper(II) salt, the oxidations were important, even though they were rather higher in the presence of copper(II) salt. The complexes observed in MS were made of Cu+. Cuprous ions mainly bind to the reduced peptides, cysteines being the major anchorage and not available after oxidation. The complexation with 2M1 was possible because of the two lysines, another binding site for Cu+, and, for M3 + M3ox, two cysteines were still free in addition to the lysines and the N-termini. The presence of Cu+ ions was explained before and it is the consequence of both the peptide oxidation and the copper etching process.37 With the Cu electrode, it was also observed that a part of the copper electrogenerated was Cu2+ ions.37 In this case, these ions were responsible for the oxidation of cysteine.
The reactivity of cysteine and the compounds formed do not only depend on the presence of a catalyst but also on their position, their number in the structure, as pointed out by Multhaup and coworkers,48 and the coordination with the catalyst. Moreover, the pKa of the cysteine and its reactivity can be influenced by the surrounding environment. Intra-molecular reactions are enhanced, as it was shown by the two- and three-cysteine-containing peptides. Indeed, in these experiments, the formation of covalent dimers via disulfide linkages is weaker than the oxidation of the peptide itself. Moreover, the oxidations induced by electrogenerated copper ions reached a plateau after 45 min for AIKCTKF, 11 min for ALRCTCS and 25 min for KCTCCA. The slower reaction is the inter-molecular cystine formation. The entropy favors intra-molecular reactions. Inter-molecular reactions were enhanced in the presence of copper ions, as it was shown by the study of the one- and three-cysteine-containing peptides. Kachur et al. have described cysteine auto-oxidation where the complexes between cysteines and copper ions are involved in the pathway as an intermediate species.9 Copper(II) is known to bridge two cysteines to form a RS−CuII−SR complex, which can then produce the disulfide bridge.10 These copper ions bound to cysteine can bridge two peptides together and help the formation of a dimer and not only oxidation to intra-molecular cystine. The formation of a Cu2+ complex is not only related to the hard–soft acid–base classification of the ligand but also to the structure that plays an important role in the complex stability. According to the structure of the peptides studied here, inter-molecular copper complexes are the easiest to form and their enthalpy will be higher. In the case of intra-molecular reactions, the thiol moietiesare too close to correctly coordinate Cu2+ ions. Thus, Cu2+ ions enhance the inter-molecular reactions coming from an inner sphere electron transfer. In the case of Zn2+, only intra-molecular oxidations were observed. Indeed, Zn2+ is not an electron carrier and could not help the oxidation and the formation of the disulfide bridges. Although Zn2+ is an intermediate acid that could bind to cysteine, it cannot oxidize them.
Conclusions
Oxidation of cysteine-containing peptides to different oxidation states of the sulfur atoms to form disulfide bridges, sulfenic, sulfinic and sulfonic acid or the oxidation of the sulfur atoms of cystine to sulfoxide and/or sulfone were studied by MS in presence of copper ions. These reactions were achieved both chemically (by oxygen or catalyzed by copper ions) and electrochemically. According to the electrochemical aspect of the ESI source, the thiol moietycan be oxidized to the different products cited above. Finally, the importance of the position and the number of cysteine residues in a peptide has been highlighted. Intra-molecular reactions governed by entropy are favored in comparison with inter-molecular reactions. However, in the presence of copper ions, more inter-molecular disulfide linkages were observed, which results in the enhancement of 2 : 1 peptides : copper complexes. In the presence of Zn2+, oxidation to covalent dimers was not observed in spite of the coordination properties.Acknowledgements
The Fonds National Suisse pour la Recherche Scientifique is thanked for financial support through the projects “Analytical tools for fast phosphoproteome analysis” (Grant no 200020-113413/1). The authors also thank Prof. Clémence Corminboeuf for discussion, and Fernando Cortés Salazar and Manuel A. Méndez for their suggestions.References
- S. A. Lipton, Y. B. Choi, H. Takahashi, D. Zhang, W. Li, A. Godzik and L. A. Bankston, Trends Neurosci., 2002, 25, 474–480 CrossRef CAS.
- N. M. Giles, A. B. Watts, G. I. Giles, F. H. Fry, J. A. Littlechild and C. Jacob, Chem. Biol., 2003, 10, 677–693 CrossRef CAS.
- D. J. Waggoner, T. B. Bartnikas and J. D. Gitlin, Neurobiol. Dis., 1999, 6, 221–230 CrossRef CAS.
- A. Myari, N. Hadjiliadis, N. Fatemi and B. Sarkar, J. Inorg. Biochem., 2004, 98, 1483–1494 CrossRef CAS.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS.
- A. P. Mathews and S. Walker, J. Biol. Chem., 1909, 6, 21–28 CAS.
- A. P. Mathews and S. Walker, J. Biol. Chem., 1909, 6, 299–312 CAS.
- C. A. Elvehjem, Biochem. J., 1930, 24, 415–426 CAS.
- A. V. Kachur, C. J. Koch and J. E. Biaglow, Free Radic. Res., 1999, 31, 23–34 CrossRef CAS.
- L. Pecci, G. Montefoschi, G. Musci and D. Cavallini, Amino Acids, 1997, 13, 355–367 CAS.
- R. M. Roat-Malone, Bioinorganic Chemistry, John Wiley & Sons, Hoboken, 2002, 1st edn Search PubMed.
- H. Lavanant, E. Hecquet and Y. Hoppilliard, Int. J. Mass Spectrom., 1999, 187, 11–23 Search PubMed.
- M. Prudent and H. H. Girault, J. Am. Soc. Mass Spectrom., 2008, 19, 560–568 CrossRef CAS.
- B. A. Cerda and C. Wesdemiotis, J. Am. Chem. Soc., 1995, 117, 9734–9739 CrossRef CAS.
- B. K. Bluhm, S. J. Shields, C. A. Bayse, M. B. Hall and D. H. Russell, Int. J. Mass Spectrom., 2001, 204, 31–46 Search PubMed.
- K. L. Schey and E. L. Finley, Acc. Chem. Res., 2000, 33, 299–306 CrossRef CAS.
- H. P. Permentier and A. P. Bruins, J. Am. Soc. Mass Spectrom., 2004, 15, 1707–1716 CrossRef CAS.
- J. L. E. Reubsaet, J. H. Beijnen, A. Bult, R. J. van Maanen, J. A. D. Marchal and W. J. M. Underberg, J. Pharm. Biomed. Anal., 1998, 17, 955–978 CrossRef CAS.
- R. Mihalca, Y. E. M. van der Burgt, A. J. R. Heck and R. M. A. Heeren, J. Mass Spectrom., 2007, 42, 450–458 CrossRef CAS.
- S. S. Thakur and P. Balaram, Rapid Commun. Mass Spectrom., 2007, 21, 3420–3426 CrossRef CAS.
- H. P. Permentier, A. P. Bruins and R. Bischoff, Mini-Rev. Med. Chem., 2008, 8, 46–56 CrossRef CAS.
- V. Shetty, D. S. Spellman and T. A. Neubert, J. Am. Soc. Mass Spectrom., 2007, 18, 1544–1551 CrossRef CAS.
- L. J. Men and Y. S. Wang, Rapid Commun. Mass Spectrom., 2006, 20, 777–784 CrossRef CAS.
- Y. S. Wang, S. Vivekananda, L. J. Men and Q. B. Zhang, J Am. Soc. Mass Spectrom., 2004, 15, 697–702 CrossRef CAS.
- B. Malfoy and J. A. Reynaud, J. Electroanal. Chem., 1980, 114, 213–223 CrossRef CAS.
- J. A. Reynaud, B. Malfoy and P. Canesson, J. Electroanal. Chem., 1980, 114, 195–211 CrossRef CAS.
- H. P. Permentier, U. Jurva, B. Barroso and A. P. Bruins, Rapid Commun. Mass Spectrom., 2003, 17, 1585–1592 CrossRef.
- G. J. Van Berkel and V. Kertesz, Anal. Chem., 2007, 79, 5510–5520 CrossRef CAS.
- R. B. Cole, Electrospray Ionization Mass Spectrometry: Fundamentals Instrumentation & Applications, John Wiley & Sons, New York, NY, 1997, 1st edn Search PubMed.
- T. Y. Zhang, S. P. Palii, J. R. Eyler and A. Brajter-Toth, Anal. Chem., 2002, 74, 1097–1103 CAS.
- L. Dayon, C. Roussel, M. Prudent, N. Lion and H. H. Girault, Electrophoresis, 2005, 26, 238–247 CrossRef CAS.
- M. Prudent, J. S. Rossier, N. Lion and H. H. Girault, Anal. Chem., 2008, 80, 2531–2538 CrossRef CAS.
- A. T. Blades, M. G. Ikonomou and P. Kebarle, Anal. Chem., 1991, 63, 2109–2114 CrossRef CAS.
- G. J. Van Berkel, K. G. Asano and P. D. Schnier, J. Am. Soc. Mass Spectrom., 2001, 12, 853–862 CrossRef CAS.
- G. J. Van Berkel and V. Kertesz, J. Mass Spectrom., 2001, 36, 1125–1132 CrossRef CAS.
- T. C. Rohner and H. H. Girault, Rapid Commun. Mass Spectrom., 2005, 19, 1183–1190 CrossRef CAS.
- M. Prudent, C. Roussel and H. H. Girault, Electrochem. Commun., 2007, 9, 2067–2074 CrossRef CAS.
- J. S. Rossier, C. Vollet, A. Carnal, G. Lagger, V. Gobry, H. H. Girault, P. Michel and F. Reymond, Lab Chip, 2002, 2, 145–150 RSC.
- V. Gobry, J. Van Oostrum, M. Martinelli, T. C. Rohner, F. Reymond, J. S. Rossier and H. H. Girault, Proteomics, 2002, 2, 405–412 CrossRef CAS.
- MS-product, http://prospector.ucsf.edu/cgi-bin/msform.cgi?form = msproduct, Accessed 1st semester 2008.
- C. L. Gatlin, F. Turecek and T. Vaisar, J. Am. Chem. Soc., 1995, 117, 3637–3638 CrossRef CAS.
- L. Gianelli, V. Amendola, L. Fabbrizzi, P. Pallavicini and G. G. Mellerio, Rapid Commun. Mass Spectrom., 2001, 15, 2347–2353 CrossRef CAS.
- H. Lavanant, H. Virelizier and Y. Hoppilliard, J. Am. Soc. Mass Spectrom., 1998, 9, 1217–1221 CrossRef.
- D. Schröder, M. C. Holthausen and H. Schwarz, J. Phys. Chem. B, 2004, 108, 14407–14416 CrossRef.
- E. Mattsson and J. O. Bockris, Trans. Faraday Soc., 1959, 55, 1586–1601 RSC.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1964, 280, 383–397 Search PubMed.
- M. Prudent, M. A. Méndez and H. H. Girault, Anal. Sci., 2008, 24, 1399–1404 CrossRef CAS.
- G. Multhaup, A. Schlicksupp, L. Hesse, D. Beher, T. Ruppert, C. L. Masters and K. Beyreuther, Science, 1996, 271, 1406–1409 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Results of the experiments performed with CuSO4. See DOI: 10.1039/b817061d |
| ‡ The m/z value shown in Fig. 1 corresponds to the highest peak of the isotopic distribution, which explains such a difference of 1 Th. |
| This journal is © The Royal Society of Chemistry 2009 |