An electrospray/inductively coupled plasma dual-source time-of-flight mass spectrometer for rapid metallomic and speciation analysis: instrument design
Duane A.
Rogers
,
Steven J.
Ray
and
Gary M.
Hieftje
*
Chemistry Department, Indiana University, 800 E Kirkwood Ave, Bloomington, IN 47405, USA. E-mail: Hieftje@indiana.edu; Fax: +1 812 855 0985; Tel: +1 812 855 2189
First published on 2nd December 2008
Abstract
A time-of-flight mass spectrometer (TOFMS), which employs inductively coupled plasma (ICP) and electrospray ionization (ESI) sources simultaneously, has been designed, constructed, and evaluated for comprehensive elemental speciation analysis. The simultaneous operation of both sources with a single mass spectrometer is an improvement over existing techniques. The mass analyzer shares a third-stage vacuum system, extraction region, acceleration region, field-free region, and two-stage reflectron between both sources. Most of the other components, such as first and second-stage vacuum systems, pre-extraction ion optics, microchannel plate detectors, and data acquisition are independent, to provide the greatest degree of flexibility in source operation and signal optimization. A detailed description is given of the design and optimization of the orthogonal acceleration and spontaneous drift geometry, energy discrimination, and the reflectron and preliminary performance data are presented.
Introduction
In recent years atomic spectrometry has had a growing influence on the biological sciences.1–3 Metallomics has thus emerged as a subset of the field of proteomics.4–6 Metals and metalloids have been found to play a significant role in cancer,7–10 Alzheimer’s disease,11–14 Parkinson’s disease,15–17 lung disease,18–20 and a range of other disorders.21–23 Therefore, there is an unprecedented need for new analytical techniques capable of quickly producing multi-dimensional elemental analysis of biological systems.In order to determine the full role of a metal or metalloid in a biological system it is first necessary to perform effective elemental speciation analysis: the quantitative determination of the various oxidative, molecular, and complex forms of an element.24 Most laboratories perform elemental speciation by means of hyphenated techniques that include an atomic detector (e.g.HPLC-ICP-MS). However, the presence of novel or unexpected species can lead to the misidentification of chromatographic peaks.25,26 The identity of the unknown species must then be determined by repeating the separation with a molecular detector (e.g.ESI-MS) and correlating the unidentified atomic chromatographic peaks with those from the molecular detector.27–31
While ICP-MS can provide elemental, isotopic, and quantitative information, it necessarily loses molecular details in the atomization and ionization processes. In the case of ESI-MS, the quantitative capabilities are limited and analyte-specific32,33 and isotopic information is often difficult to glean because it is convoluted with multiple charge states, adducts, clusters, and metastable species. However, ESI-MS is capable of producing intact molecular ions and tunable fragmentation through collision-induced dissociation.34 Therefore, the ICP and ESI provide complementary data and can be used together to provide what has come to be known as comprehensive elemental speciation.35,36
Unfortunately, the scheme of using multiple separations for comprehensive elemental speciation necessitates multiple, repetitive separation steps, which increases sample requirements, preparation time, and the potential for human error. In order to overcome this source of error and reduce sample requirements and preparation time, some researchers have split the flow from a single separation between an ESI-MS and an ICP-MS.37,38
In the work to be described here, a dual-source time-of-flight mass spectrometer has been designed and constructed. It enables both an electrospray and an inductively coupled plasma source to be operated simultaneously, allowing a single chromatographic separation to supply both sources at once by means of a stream-splitting valve. In addition, the design of the dual-source time-of-flight mass spectrometer (ds-TOFMS) reduces redundant components, thus simplifying instrument operation and signal synchronization. Fig. 1 illustrates the dual-source time-of-flight mass spectrometer.
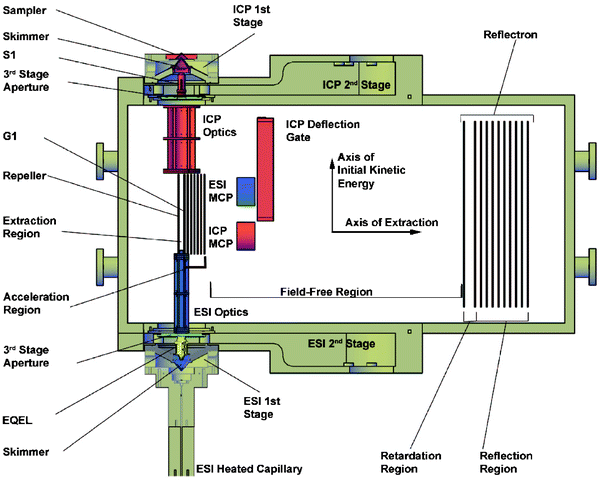 | ||
| Fig. 1 Schematic diagram of the dual-source time-of-flight mass spectrometer. A cutaway cross section is shown for the three differentially pumped vacuum stages to expose the relevant components. All components are discussed in the text. MCP is the microchannel plate detector for each channel. | ||
Instrument design
ICP source and interface
The ionization source for the elemental channel is an atmospheric pressure, argon inductively coupled plasma. The ICP uses a commercial rf generator, impedance matching network and torch housing from LECO Corp. (St. Joseph, MI, USA). The first vacuum stage is the interface from the LECO Renaissance®TOFMS, evacuated by a 65 m3 h−1 mechanical roughing pump (Leybold AG, Pfäffikon, Switzerland, model D65B) to a pressure of less than 2 Torr (typically ca. 1 Torr; 1 Torr = 133.3 Pa). Pressure in the first and second vacuum stages is monitored by separate Pirani gauges (HPS Division, MKS Instruments, Inc., Boulder, CO, USA, model 325 Moducell). The plasma is supported by an 18-mm tangential-flow quartz torch (Precision Glass Blowing, Centennial, CO).Aqueous samples are nebulized by a CETAC U-6000AT+ ultrasonic nebulizer (Omaha, NE, USA). The resulting aerosol then passes through a CETAC membrane desolvator, as part of the U-6000AT+ assembly, followed by an additional CETAC membrane desolvator (model MDX–100, Omaha, NE, USA). It was found that the additional membrane desolvation unit improved sensitivity. The specific operating conditions of the ICP source and interface are compiled in Table 1.
| Sample introduction | Ultrasonic nebulizer with desolvation |
|---|---|
| Sample uptake rate | 1 ml min−1 |
| Plasma power | 1.35 kW |
| Plasma frequency | 40.68 MHz |
| Central gas flow | 0.8–1.2 l min−1 |
| Intermediate gas flow | 0.8–1.15 l min−1 |
| Outer gas flow | 15 l min−1 |
| Membrane desolvator #1 gas flow | 3.75 l min−1 |
| Membrane desolvator #2 gas flow | 4.1 l min−1 |
| Sampling position | 7 mm above load coil |
| Sampler orifice diameter | 1 mm |
| Skimmer orifice diameter | 0.5 mm |
| Sampler material | Nickel |
| Skimmer material | Nickel |
| ICP First-stage pressure | 1–2 Torr |
| ICP Second-stage pressure | 2.2 mTorr |
| Third-stage pressure | <3.5 μTorr |
The plasma is extracted into the mass spectrometer by a water-cooled nickel sampling cone with a 1 mm orifice followed by a water-cooled nickel skimmer cone with an orifice of 0.5 mm. The skimmer-sampler distance is 10 mm. The second vacuum stage was constructed in house and equipped with a 330 l s−1 turbomolecular pump (model TPU 330, Pfeiffer Balzers GmbH, Germany), controlled by a TCP 380 power supply (Pfeiffer Balzers GmbH, Germany). In the second vacuum stage, ions are focused by a single ion optic (S1) through a 1 mm diameter conductance-limiting orifice plate into the third vacuum stage.39,40 The third-stage orifice plate is fabricated from 304 stainless steel and is electrically isolated from the instrument housing by means of a PEEK insulator. A potential is applied to the third-stage-orifice plate by a Kepco power supply (model 188–0030, 1.5 kV max, 10 mA, Kepco, Flushing, NY, USA).
ESI source and interface
The electrospray source was constructed in house from 50 μm I.D. fused silica tubing, a syringe pump and a 28 Ga stainless steel hypodermic needle. The tip of the hypodermic needle was cut flat using a wire electrical discharge machine to prevent metal burrs at its tip. The fused silica tubing was then passed through the hypodermic needle so the tips are flush. A 3 kV potential is applied to the needle and it is positioned within 1 cm of the heated-capillary interface. The needle current is monitored with a homemade current-to-voltage converter and a Fluke 76 digital multimeter (Everett, WA, USA). The needle potential is optimized for stable needle current in the 90–150 nA regime. The beam from a He-Ne laser is directed 1–3 mm forward of the ESI tip to permit the spray to be visualized and to confirm that an even, stable spray is formed.The electrospray aerosol is sampled by a heated, stainless-steel capillary interface that was designed and constructed in-house. The heated capillary has an ID of 380 μm, an OD of 1.55 mm, and is 20 cm long (Small Parts, Inc., Miramar, FL, USA). The capillary is held in place by an aluminum block that is heated by a set of two cartridge heaters (model CH26661, 120 V, 250 W, Fast Heat, Elmhurst, IL, USA) controlled in turn by a DIN controller unit (model CN76000, Omega Engineering, Inc., Stamford, CT, USA). The temperature is monitored by an iron–constantan thermocouple (Omega Engineering, Inc., Stamford, CT, USA).
Following the heated capillary, the ESI ion beam enters the first vacuum stage, maintained at 0.5 Torr by a 60 m3 h−1 mechanical roughing pump (model D90A, Trivac, Leybold AG, Pfäffikon, Switzerland). The ion beam enters the second vacuum stage through a 0.5 mm LECO skimmer, held in place by an insulating Delrin® spacer. The skimmer is held at potential by a Fluke 415B high voltage power supply (Fluke model 415B, 3100 V max, 10 mA, Everett, WA, USA) to increase the ion throughput . The second vacuum stage was constructed in house and equipped with a 330 l s−1 turbomolecular pump (model TPU 330, Pfeiffer Balzer GmbH, Germany), controlled by a TCP 380 power supply (Pfeiffer Balzer GmbH, Germany). Pressure in the first and second vacuum stages is monitored by separate Pirani gauges (model 325 Moducell, HPS Division, MKS Instruments, Inc., Boulder, CO, USA).
In the second vacuum stage, ions are focused onto the third stage orifice plate by an EQEL optic, described by Barnes and coworkers.41 The potential necessary for the internal electrode of the EQEL optic is controlled by a 244 Keithley power supply (2200 V max, 10 mA, Keithley Instruments Inc., Cleveland, OH, USA) and the potential for the barrel electrode of the EQEL is controlled by a model 240A Keithley Instruments power supply (1111 V max, 10 mA, Keithley Instruments Inc., Cleveland, OH, USA). The third stage orifice plate was fabricated from 304 stainless steel and is electrically isolated from the instrument housing by a PEEK insulator. A potential is applied to the third stage-orifice plate by a Bertan power supply (model 214, 1000 V max, 15 mA, Bertan Associates, Inc., New York, NY, USA). The specific operating conditions of the ESI source and interface are given in Table 2.
| Interface temperature | 80 °C |
| Sample solution flow rate | 0.75–2 μl min−1 |
| Heated-capillary length | 14.6 cm |
| Heated-capillary inner diameter | 380 μm |
| Heated-capillary material | 304 stainless steel |
| Heated capillary potential | 250 V |
| Skimmer material | 304 stainless steel |
| ESI First-stage pressure | 1 Torr |
| ESI Second-stage pressure | <0.4 mTorr |
| Third-stage pressure | <3.5 μTorr |
Third stage vacuum chamber
After passing through their respective third-stage orifice plates, the ion beams from both sources enter a shared third-stage vacuum chamber. The third-stage vacuum chamber was constructed in house with 19 mm 304 stainless steel walls, and internal dimensions of 41.9 cm (W) × 87.6 cm (L) × 30.5 cm (H). Operating pressure in the third vacuum stage is maintained below 3.5 μTorr (typically ca. 1 μTorr) by a 520 l s−1 turbomolecular pump (Model TMU 521P, Pfeiffer Vacuum, Asslar, Germany), operated by a DCU controller unit, in conjunction with a 250 l s−1 turbomolecular pump (model TMH 260 SPEZ, Pfeiffer Balzers GmbH, Germany), operated by a TCP 380 power supply (Pfeiffer Balzers GmbH, Germany). Pressure in the third stage is continuously monitored by a cold-cathode pressure gauge (Series 423 I–MAG, HPS Division, MKS Instruments, Inc., Boulder, CO, USA). All of the pressure gauges are read out on an MKS gauge controller (model 937, HPS Division, MKS Instruments, Inc., Boulder, CO, USA).Ion beam arrangement
To reduce crosstalk between the two ion beams, the two sources are oriented in an anti-parallel geometry with a 10 cm vertical offset. As will be discussed later, the spontaneous-drift geometry utilizes the ions’ initial kinetic energy to direct ions onto each respective detector. Because the ion beams are arranged in an anti-parallel geometry, the initial kinetic energy of each ion beam carries the extracted ions away from the detector of the other channel. Since the extraction region is only 1 cm wide (along the axis of extraction) with a 10 cm vertical offset, space–charge effects between the two channels are effectively eliminated in this region. Simion simulations were performed to confirm this assertion.Initial testing revealed that operation of the ICP channel raised the background on the ESI detector. This ion-beam crosstalk was eliminated by baffling the two channels along the length of the field-free region with a 304 stainless steel plate held at the acceleration potential. It is thought that ICP ions impacting walls in the field-free region produced secondary ions and neutrals that impacted the ESI detector.
Ion optics
It has long been realized that resolution in orthogonal-acceleration time-of-flight mass spectrometers is strongly affected by the spatial distribution of ions in the extraction region42 (i.e. source region); by reducing the distribution of ions along the axis of acceleration in the extraction region, resolution can be significantly improved. It has been demonstrated by others that electrostatic (dc) quadrupoles can effectively focus an incoming ion beam into a slit image in the extraction region.40 Accordingly, both channels here utilize separate, independent quadrupoles of slightly different design to focus ions entering the third vacuum stage into the extraction region.ICP ion optics in the third vacuum stage
Following the third-stage orifice plate, ions from the ICP pass through a single ring electrode, which serves to focus them onto the front of the quadrupole optic (cf.Fig. 2a) and to reduce the angular dispersion of the ion beam. The ring optic is fabricated from 304 stainless steel, has an outer diameter of 73 mm, an inner diameter of 30 mm, and is 3.18 mm thick.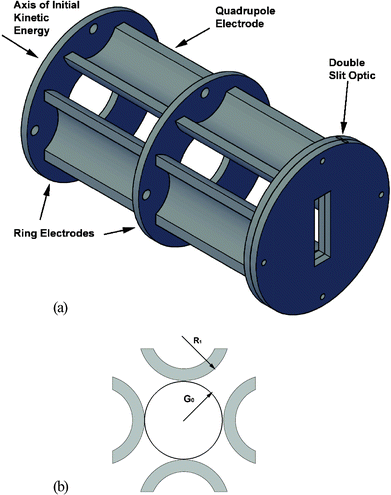 | ||
| Fig. 2 (a) Illustration of the ICP ion optics train, consisting of two ring electrodes, two sets of dc-quadrupoles, and two slit optics. (b) Illustration of the relationship of the quadrupole radius of curvature to the lateral spacing, based on Eqn (1). | ||
Space–charge effects caused by the high ion flux in the ICP beam are reduced by focusing the ion beam into a taller slit image in the vertical direction. Accordingly, the distance between opposing dc quadrupole rods (2G0) was set at 30 mm, which dictated the radius of curvature for the rods (R1) to be 17.2 mm, according to Eqn (1).43 In Eqn (1), the ideal hyperbolic quadrupole fields are approximated by using cylindrical electrodes, where G0 is half the distance between opposing rods and R1 is the radius of curvature of each cylindrical rod (cf. Fig. 2b). For fabrication convenience, each rod was cut in half lengthwise and arranged with the rounded edge toward the ion beam. This arrangement produces approximately hyperbolic fields, as described in Eqn (1), while accommodating the necessary mounting hardware. The quadrupoles responsible for focusing the ion beam in the vertical axis are termed Y quads while those responsible for horizontal focusing are termed X quads. The dc-quadrupoles are biased by a separate power supply (model 205B–01R, 1 kV max, 10 mA, Bertan Associates, Inc., New York, NY, USA), which produces four independent voltages via a variable voltage divider that was constructed in house.
| R1 = 1.147G0 | (1) |
| S1 optic | −2.0 kV |
| Third stage aperture plate | −1.2 kV |
| Ring optic 1 | −431 V |
| Quadrupole X1 | −269 V |
| Quadrupole Y1 | −201 V |
| Ring optic 2 | −20 V |
| Quadrupole X2 | −293 V |
| Quadrupole Y2 | −181 V |
| Slit optic 1 | −356 V |
| Slit optic 2 | −20 V |
Each component of the ICP ion optic train is positioned 0.18 mm from the subsequent optic by a 3.18 mm sapphire bead (Small Parts, Inc., Miramar, FL, USA) recessed in a 2.95 mm diameter hole on each opposing optic. The entire optic train is then held together on a #10 threaded Delrin® rod and a nylon nut on each end of the optic train. In this arrangement, the Delrin® rod serves to hold the optics laterally while the sapphire beads prevent the optics from moving vertically or horizontally.
ESI ion optics in the third vacuum stage
Similar to the ICP ion optics, the ESI channel uses a combination of ring electrodes and dc-quadrupoles to focus the ESI ion beam into a narrow slit image in the extraction region (cf. Fig. 3a). However, because the ion fluxes are significantly lower in the ESI channel, smaller dimensions can be used. Following the third stage aperture, the ion beam from the ESI source is first focused by a 3-ring Einzel lens. Each ring electrode of the Einzel lens (Einzel lens element 1, 2, and 3) has an OD of 28.6 mm, an ID of 12.7 mm, and is 3.18 mm thick. Next, the ion beam is focused by a double set of dc-quadrupole rods separated by a single ring electrode. Each of the quadrupole rods has a radius of curvature of 7.15 mm and is 6.38 cm long. For fabrication convenience, each rod was cut in half lengthwise and arranged with the rounded edge toward the ion beam. This arrangement maintains hyperbolic fields as described by Eqn (1) while accommodating the necessary mounting hardware. In the final arrangement, each rod is positioned 12.6 mm from the opposing rod, in accordance with Eqn (1).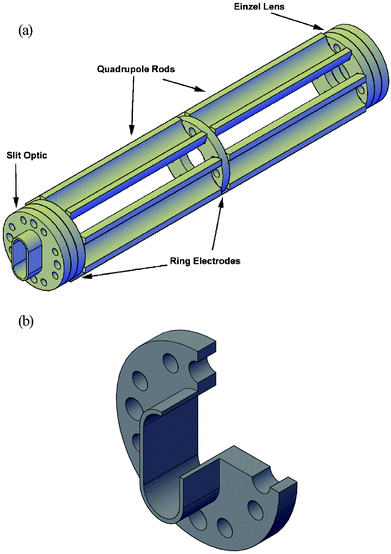 | ||
| Fig. 3 (a) Illustration of the ESI ion optics train, consisting of an Einzel lens, two sets of dc-quadrupoles, three independent ring electrodes, and an extended slit optic. (b) Cutaway illustration of the ESI slit optic used to improve ion beam collimation. | ||
Between the first and second set of ESI quadrupole rods is a single ring electrode (ring optic 1) with identical dimensions to those of the ESI-channel Einzel lens. After the second set of quadrupoles are two more ring electrodes followed by a slit electrode.
Similar to the ring electrodes, the slit electrode (cf.Fig. 3b) has an OD of 28.6 mm, but the slit extends into the extraction region. The slit opening measures 15.2 mm tall and 6.35 mm wide, with the long dimension perpendicular to the axis of extraction. The slit optic is 9.53 mm long. The additional length extends the optic 0.5 mm into the extraction region and serves to better collimate the ion beam. In later generations, the resolution of the ESI channel will be improved by narrowing the slit and extending it farther into the extraction region.
Each component of the ion optic train is positioned 0.17 mm from the subsequent optic by using 3.18 mm sapphire beads (Small Parts, Inc., Miramar, FL, USA) recessed in 2.71 mm diameter holes on each opposing optic, just as in the ICP optics train. These beads are not shown in either Fig. 2 or 3. The entire optic train is then held together by means of a #4 nylon threaded rod and a nylon nut on each end of the optic train. A full list of optimized third-stage ESI ion optic potentials is given in Table 4.
| Skimmer | 10–20 V |
| EQEL outer optic | −250 V |
| EQEL inner optic | 0 V |
| Third stage aperture plate | −45 V |
| Einzel lens 1 | −207 V |
| Einzel lens 2 | −948 V |
| Einzel lens 3 | −502 V |
| Quadrupole X1 | −1219 V |
| Quadrupole Y1 | −1198 V |
| Ring optic 1 | −418 V |
| Quadrupole X2 | −398 V |
| Quadrupole Y2 | −406 V |
| Ring optic 2 | −927 V |
| Ring optic 3 | −649 V |
| Slit optic | −577 V |
Spontaneous drift considerations
After passing through independent differentially pumped interfaces and independent pre-extraction ion optics, ions from both sources enter a shared extraction region. In the current arrangement, a single extraction pulse is used to extract ions from both ion beams into the acceleration region. Therefore, the duty cycle of both channels is limited by the mass range of the ESI channel. In later generations, a gating scheme, applied separately to each input channel, will be employed to improve the duty cycle of the ICP channel by 1.5 to 3 fold.In the extraction region, ions are accelerated in a direction orthogonal to their initial kinetic energy. Therefore, the energy imparted to the ions by the extraction, acceleration, reflectron, energy discrimination, or post-acceleration fields is independent of their initial directed energy. The final angle (θ) at which the ions exit the acceleration region is described by Eqn (2), where vext is the velocity ions obtain in the extraction and acceleration regions (along the axis of acceleration) and vi is the initial velocity the ions have before extraction (along the axis of their initial kinetic energy, which is perpendicular to the axis of acceleration). Similarly, Eext and Ei represent the kinetic energies along the axis of acceleration and the axis of initial kinetic energy, respectively.
| tan(θ) = vext/vi = Eext1/2/Ei1/2 | (2) |
Kinetic energy bandpass
The kinetic energy bandpass of the current instrument design was calculated from Simion 7.0 simulations and is shown in Fig. 4a. Simion is a program that has been shown to be useful for modeling the trajectory of ions through electric and magnetic fields and can be tailored to specific ion-optic arrangements.45–47 Before construction began on the ds-TOFMS, the entire instrument was modeled with Simion to optimize the theoretical performance of the individual ion optics for the given source conditions. Under experimental conditions optimized for the highest resolution, only ions with energies of 3.5–8.5 eV are sampled with the greatest efficiency. Ions with energies outside this range impact the sides of the acceleration region, resulting in only partial coverage of the detector surface. In ICP-MS, it is well known that ion energies of heavier elements (i.e. 238U) can exceed 14 eV.48–51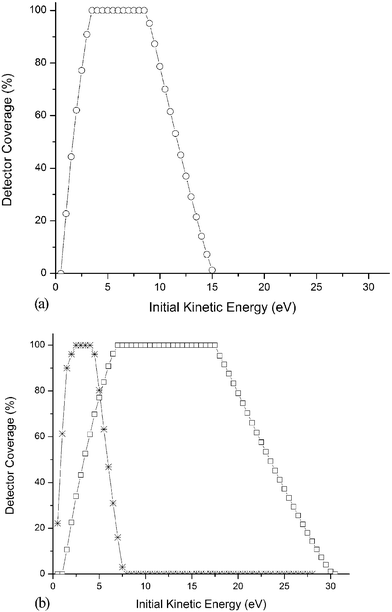 | ||
| Fig. 4 (a) Current energy bandpass of the ds-TOFMS, obtained when EExt = 310 V and EAccel = 1330 V. Since ions with energies outside of the optimal range impact the surface of the extraction region and fail to reach the detector, sampling efficiency can be considered in terms in terms of detector surface coverage. (b) The energy bandpass can be tailored by altering the extraction and acceleration fields while maintaining the necessary ratio for optimal space focusing. Modified energy bandpass obtained when Uext = 155 V cm−1 and Uaccel = 164 V cm−1 (–*–) or Uext = 620 V cm−1 and Uaccel = 650 V cm−1 (–□–). | ||
To adjust the energy bandpass of the instrument, two options exist: (1) change the extraction angle (θ) or (2) modify the extraction region. In order to alter the extraction angle, either Eext or Ei in Eqn (2) must be adjusted. The Eext term can be changed by increasing the extraction field (UE) and the acceleration field (UA). Similarly, the Ei term can be changed by adjusting the input ion optics and the bias potential on the repeller and first extraction grid. However, it is significantly more difficult to properly focus the ions in the extraction region while making these modifications.
To optimize resolution at the first space-focus plane, a specific ratio of extraction to acceleration fields (UE : UA) must be maintained.52–55 Therefore, when UE is increased, UA must be raised proportionately, and the energy imparted to the extracted ions along the extraction axis goes up. Subsequently, the extraction angle is increased without a significant effect on resolution. However, as noted by others,56 the problems associated with very high acceleration potentials are not trivial.
As shown in Fig. 4b, ions with lower kinetic energies can be sampled by applying a lower UE. Unfortunately, the overall bandpass is then also reduced. Alternatively, with greater UE, a broader range of kinetic energies can be sampled with a wider bandpass. However, a greater range of low-energy ions fail to reach the detector.
The most effective way to increase the bandpass, without sacrificing low-energy ions, is to extend the extraction region along the axis of initial kinetic energy. The ds-TOFMS has the capability of a 14 cm long extraction region. However, in the current embodiment, the extraction region was held to 7.6 cm for preliminary characterization. If the extraction region is extended to 14 cm, ions with initial kinetic energies as high as 24 eV can be effectively sampled.
Extraction/acceleration optimization
The ds-TOFMS uses a pulsed extraction region followed by an electrostatic acceleration region. In the extraction region, a high-voltage pulse generator (Directed Energy, Inc., model PVX-4140, Fort Collins, CO, USA) is used to drive a single repeller plate for the molecular and elemental channels simultaneously. The repeller is a 15.2 cm × 24.1 cm × 1.5 mm 304 stainless steel sheet with a mirror finish that is pulsed to +310 V to push the ions into the acceleration region. The repeller trigger and subsequent deflection timing and detection gate are driven by a single master trigger with up to eight independent timing delays (Berkeley Nucleonics Corp., San Rafael, CA, Model 565 Pulse/Delay Generator).The extraction region is 1 cm wide along the axis of extraction, with the repeller on one side and the first acceleration grid (G1) on the other. The acceleration region is comprised of a grid at each end and 5 intermediate ring electrodes spaced 5.1 mm apart. The total length of the acceleration region is 40 cm. A biasing power supply (typically held at ground potential) is connected to the first grid (G1) and the acceleration potential (−1330 V) is applied to the last grid (3000 V max, 10 mA, Thorn EMI Gencom, Inc., model 3000R, Fairfield, NJ, USA). A linear field is formed by using a seven-stage voltage divider consisting of one 1 MΩ 0.5 W precision resistor between each pair of electrodes.
Because of the broader mass range sampled on the ESI channel, it is necessary to lengthen the extraction pulse width to fully push the ions out of the extraction region. If the pulse is too short, heavy ions are not fully extracted before the pulse terminates. As a result, they receive less than the expected level of energy. Yet, if the extraction pulse is too long compared to the repetition rate of mass-spectral generation, the slowest moving ions entering the extraction region do not have enough time to fill the region before the next extraction event. Importantly, ions that exit the final slit optic while the repeller voltage is high are unable to enter the acceleration region and thus do not appear in the mass spectra and do not contribute to the background. Overall, the pulse width is governed by the largest m/z of the ESI channel while the duty cycle is set by both the largest m/z and the lowest initial kinetic energy that must be sampled.
Conveniently, the TOFMS duty cycle benefits from the multiple charges that typically accompany a protein ion produced by ESI. In data to be shown later, the lowest charge state observed for intact myoglobin (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 950 Da) was +11. Therefore, the largest ESI m/z observed is only 6.5 times greater than 238U on the ICP channel, rather than 71 times greater if myoglobin were singly charged. The disparity in mass range of the two channels is further compensated by the quadratic relationship of m/z with time. Therefore, monitoring the +11 charge state of myoglobin extends the mass analysis window by a factor of only 2.5 versus238U.
950 Da) was +11. Therefore, the largest ESI m/z observed is only 6.5 times greater than 238U on the ICP channel, rather than 71 times greater if myoglobin were singly charged. The disparity in mass range of the two channels is further compensated by the quadratic relationship of m/z with time. Therefore, monitoring the +11 charge state of myoglobin extends the mass analysis window by a factor of only 2.5 versus238U.
Later generations of the instrument will incorporate ion gating on the ESI channel to interdigitate the maximum number of ICP spectra within the time needed to acquire a single ESI spectrum. Theoretically, interdigitation should increase the ICP channel duty cycle, and subsequently its sensitivity, by 1.5 to 3 fold, depending on the mass range of the ESI.
Single reflectron
A single two-stage reflectron is used in the ds-TOFMS for both source channels. The first section, the retardation region, eliminates approximately 60% of the ions’ kinetic energy (along the axis of extraction). This retardation region is 2 cm long with grids on each end. The first grid is held at the acceleration potential (−1330 V) while the potential on the second grid is optimized at −430 V (model 415B, 3100 V max, 10 mA, Fluke, Everett, WA, USA).The second section, the reflection region, uses nine ring electrodes (1 cm apart) along its length followed at the far end by a solid 304 stainless steel reflection plate with a mirror finish. A linear field is established in this region by separating the first ring electrode from the retardation grid, and each subsequent ring electrode from the next with a 1 MΩ, 0.5 W precision resistor and applying the reflection potential to the final reflection plate with a high voltage power supply (model 245, 2200 V, 10 mA, Keithley Instruments Inc., Cleveland, OH, USA).
Deflection of unwanted ions
In TOFMS all ions present in the incoming beam are sampled simultaneously. As a result, ions from the matrix and plasma are often several orders of magnitude more abundant than analyte species. Argon ions from the ICP, for example, can saturate the MCP detector and preamplifier, obscure adjacent ions, and ultimately shorten the life of the MCP.To overcome this problem in the ds-TOMF, an ion gate is positioned at the first space-focus plane. The extracted ions pass through this deflection optic shortly after the acceleration region, and before most of the field-free region or the reflectron. Because the ions are mass resolved at the first space-focus plane, it is possible to selectively deflect a particular m/z from the passing beam by employing a short pulse on the deflection gate. However, at the first space-focus plane the ions have not had the benefit of energy compensation, and the individual m/z’s are narrowly separated. Therefore, deflection of a single low m/z is not trivial.
The design used for the deflection optic uses interdigitated wire electrodes similar to those first described by Cravath57 and then again by Bradbury and Neilson58 seven years later. Similar deflection gates have become common in recent years as the field of time-of-flight mass spectrometry has expanded.59–61 The specific gate constructed for the ds-TOFMS instrument uses a total of 60 copper wires (150 μm diameter, 18.4 cm long) running parallel along a single plane. The even-numbered wires are pulsed positive relative to the acceleration potential while the other half are pulsed negative relative to the flight tube (typically 75–150 V). Each wire is run horizontally across the flight path and spaced 1 mm from its neighbors. The ions are accordingly steered vertically off-axis from the detector.
There are several benefits of the interdigitated electrodes over a plate-based deflection system. First, the rise and fall times of the high-voltage pulse generator are shorter, since the voltage imposed on each electrode is only about half of that necessary for a plate deflector. Also, because the electric fields from opposing wires cancel each other within 1–2 mm, fringing fields are relatively unimportant. Finally, because the wire network is very thin in the direction of the ion flight, a narrower range of masses can be selectively deflected.
Because of the high abundance of 40Ar+ relative to any other species in the ICP, its peak is broader and a much longer deflection window must be used to adequately deflect it. As a result, neighboring peaks are affected. A 200 ns deflection window for Ar+ has been shown to affect m/z as high as 50. Further refinement of the high voltage pulse generator and gate might reduce the affected mass range.
In the current configuration a 200 ns pulse is used to deflect Ar+ while a 45 ns pulse is sufficient to deflect H2O+. Four independent deflection pulses can be produced by the BNC model 555 master trigger; the final high voltage pulse is produced by a LECO high voltage differential pulse generator (LECO Corp., St. Joseph, MI, USA). The positive voltage for the pulse generator is provided by a Series 230 Bertan HV power supply floated above the acceleration potential (model 203–03F, 3 kV max, 5 mA, Bertan Associates, Inc., New York, NY, USA), while the negative voltage is provided by a separate Series 230 Bertan HV power supply, which is floated below the acceleration potential (model 203–01F, 1 kV max, 15 mA, Bertan Associates, Inc., New York, NY, USA). Fig. 5 reveals that 100 V applied to the deflection optics is enough to eliminate 98% of the 40Ar+ signal.
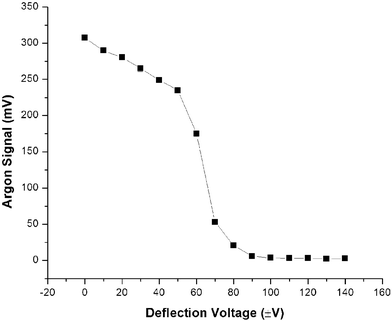 | ||
| Fig. 5 Efficiency of deflection gate for 40Ar+. | ||
In the case of the ESI channel, the total ion flux is insufficient to saturate the detector, so no deflection is necessary.
Energy discrimination
As described by Mahoney and co-workers,62 it is often necessary to utilize energy discrimination (ED) to reduce background noise in ICP-TOFMS. Due to the abundance of background ions in the ICP beam (i.e. Ar+), significant space–charge effects arise. As a result, some of the ions exit the last ion optic at an angle and “leak” into the acceleration region; those ions are subsequently accelerated toward the detector. However, since leakage ions are not synchronized with the extraction pulse, they are not temporally resolved, which results in an elevated, continuous background across the full mass spectrum. Of course, the leakage ions do not gain additional energy from the extraction pulse. Therefore, by applying a positive potential to a grid located approximately 1 cm in front of the detector and 5 mm after the last flight tube grid, ions with a kinetic energy less than that of the positive grid are prevented from reaching the detector.As shown in Fig. 6a, when the ED potential is raised, the background noise drops. However if the potential is increased further, an unacceptable fraction of the analyte flux is also prevented from reaching the detector. Furthermore, the presence of a positive potential between the field-free region and the detector can degrade resolution. Therefore, the minimum ED potential necessary is used. In the example illustrated in Fig. 6, the background is minimized at an ED potential of 50 V, resulting in the highest signal-to-noise ratio. Depending on the operating conditions and the potential of the slit optic, the optimal ED potential typically lies between 50–100 V.
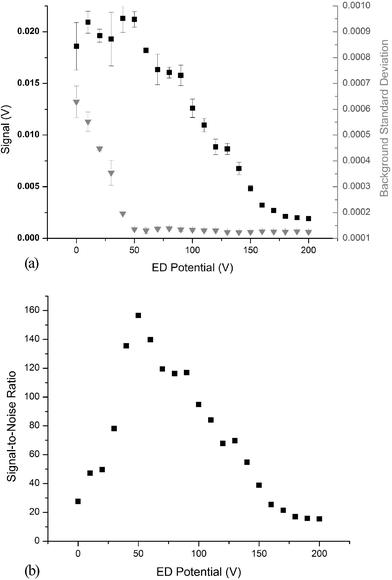 | ||
| Fig. 6 (a) Effect of energy discrimination potential on 238U+ signal (–■–, left vertical axis) and the standard deviation of the background (–▼–, right vertical axis). Error bars represent one standard deviation for N = 3 measurements. (b) Effect of energy discrimination potential on signal-to-noise ratio for 238U+. | ||
In the case of the ESI channel, the total ion flux is substantially lower than that of the ICP channel. As a result, leakage ions are not a significant contribution to the ESI background, so no energy discrimination is needed.
Detectors and detection electronics
Post-acceleration employed with a microchannel-plate detector (MCP) improves detection efficiency, particularly for large molecules.63 Each channel in the ds-TOFMS employs a 25 mm active area advanced performance MCP detector operated in a chevron configuration (extended dynamic range microchannel-plate detector, model 3025, Burle Industries, Inc., Lancaster, PA, USA). The ICP channel MCP (model 205–05B, 5 kV max, 10 mA, Bertan Associates, Inc., New York, NY, USA) uses a 2000 V post acceleration potential while the ESI detector (model 205-10R, 10 kV max, 10 mA, Bertan Associates, Inc., New York, NY, USA) uses 7250 V. A fan-cooled voltage divider for each detector was constructed from 2 W ceramic resistors to provide the necessary 1000 V drop across each plate and to provide improved stability over long periods.The ESI channel uses a commercial Galileo Electro-Optics Corp. Anode (Sturbridge, MA, USA), while the ICP uses an anode made in house to a close approximation of the Galileo design. Single ion events at either detector produced peak widths of 8 ± 2 ns.
For signal acquisition, the ESI channel employs an Ortec model 9308 picosecond time analyzer (Oak Ridge, TN, USA) in conjunction with a Tennelec TC454 four-channel constant fraction discriminator (Oak Ridge, TN, USA). Mass spectra were recorded with Ortec PTA32 Histogram Software (Version 2.01, Oak Ridge, TN, USA). For analogue signals the ICP channel is amplified by an Ortec VT120 non-inverting fast timing preamplifier (Oak Ridge, TN, USA) and read out with a Tektronix TDS7254 digit oscilloscope (Richardson, TX, USA), with a 2.5 GHz bandpass. When the oscilloscope is operated at 20 Gsamples s−1 with a record length of 800000 samples, a 40 μs mass window can be recorded to provide full elemental mass coverage. After acquisition, ESI and ICP spectra were manipulated and mass calibrated with Origin 7.5 SR6 software (OriginLab Corp., Northampton, MA, USA)
Preliminary characterization and optimization
Initial characterization and optimization of the instrument were performed by using DC glow discharges based upon the cell design described by McClenathan and Hieftje.64 Two glow discharge cells were operated in place of the ICP and ESI sources to demonstrate that a single set of reflectron parameters served for both channels (cf.Fig. 7). The long-term stability of the glow discharge sources permitted the identification and eventual elimination of noise sources not related to the ionization source (e.g. interference noise pickup, turbomolecular pump electronic noise, and electronic crosstalk between the two channels) without a significant change in signal level.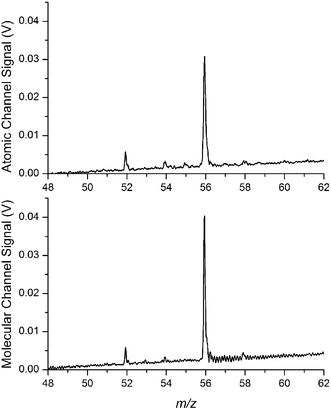 | ||
| Fig. 7 Simultaneous dual dc-glow discharge mass spectra of iron isotopes from a stainless steel sample. The top spectrum is taken from the atomic channel and the lower spectrum from the molecular channel. | ||
During the initial characterization of the ESI interface, a flowing atmospheric-pressure afterglow source65,66 was used for optical alignment and optimization. The benefit of this source over a low pressure glow discharge is that it operates at atmospheric pressure at temperatures of less than 500 K, so ion energies are similar to those in the ESI source but with greater ion flux.
Preliminary characterization of each source (ESI and ICP) was carried out with the other source off. Characterization of both sources during simultaneous operation will be the focus of the next publication in this series. The objective for characterizing the ESI source in this part of the work was to confirm that a variety of intact molecular ions could be produced over a wide mass range. Alkyl-ammonia salts have been demonstrated to serve as effective mass standards for positive-ion ESI.67 Alkyl-ammonia salts are available over a wide range of masses and typically retain only a single charge, which makes them convenient for optimization and mass calibration. However, the high sensitivity of ESI for these compounds produces substantial memory effects; the residual signal often lingers for months.
Fig. 8a shows an ESI mass spectrum containing hexadecyltrimethylammonium (HDTMA, 285 Da), tetrahexylammonium (THA, 355 Da), and tetraoctylammonium (TOA, 467 Da) (Sigma Aldrich, St. Louis, MO, USA). All of these compounds produced singly charged states along with a series of expected adducts (i.e. Na, H2O). Fig. 8b shows an ESI mass spectrum of leucine enkephalin (YGGFL, 556 Da) (Sigma Aldrich, St. Louis, MO, USA), principally containing the singly charged molecular ion. A small fraction of the doubly charged molecular ion is also observed, along with matrix adducts.
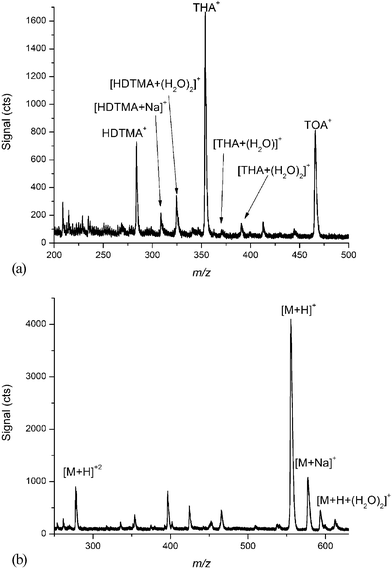 | ||
| Fig. 8 (a) Characteristic ESI mass spectrum of hexadecyltrimethylammonium (HDTMA, 285 Da), tetrahexylammonium (THA, 355 Da), and tetraoctylammonium (TOA, 467 Da) (b) Characteristic ESI mass spectrum of Leucine Enkephalin (556 Da) showing mainly the singly charged molecular ion. | ||
An ICP mass spectrum of a multi-elemental solution, shown in Fig. 9, covers full elemental mass coverage from 6Li+ to 238U+. This particular multi-elemental solution contains most of the common metals discussed in the speciation literature (i.e. Cr, Se, and As)
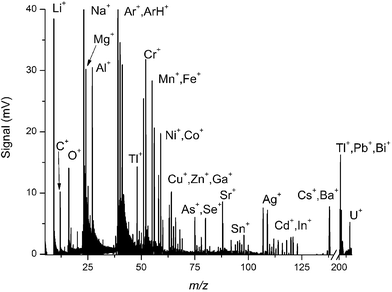 | ||
| Fig. 9 Characteristic ICP mass spectrum of a multi-elemental solution, illustrating the full mass range capabilities from 6Li+ to 238U+. | ||
Resolution as high as 1500 FWHM was obtained for 208Pb+ on the ICP channel. As shown in Fig. 10 such resolving power is sufficient to provide baseline separation of the individual isotopes of lead. Further work is necessary, though, to reach the target resolving power of 2000.
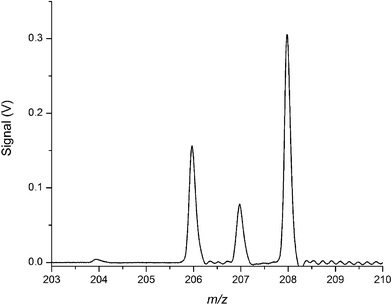 | ||
| Fig. 10 Characteristic ICP mass spectrum illustrating resolving power of 1500 (FWHM) for 208Pb+, which is sufficient for baseline resolution of the major lead isotopes. | ||
When both channels use identical electric fields from extraction to detection, a single set of reflectron potentials can be used for both of them. In Fig. 11 dc-glow discharge sources were used in place of the ESI and ICP sources. The resolution was determined for 208Pb+ in a solder sample divided between the two GD sources. Both channels used −2200 V post acceleration and no energy discrimination. As Fig. 11 demonstrates, the resolution for both channels is optimal under the same set of reflectron conditions. One should not infer from Fig. 11 that the ESI channel has a lower absolute resolving power than the ICP channel. Rather, the difference arises from the use of different focusing ion optics for the two channels, which were developed for different energy ranges and ion flux.
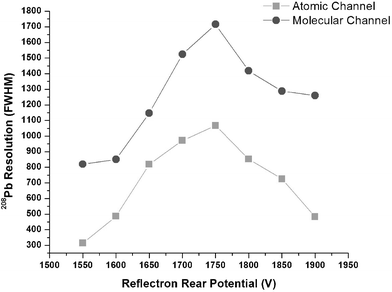 | ||
| Fig. 11 Optimization of the reflection potential in the single reflectron used for both channels while operating dc-glow discharge sources. The figure of merit is the resolution (FWHM) of 208Pb+ produced from a solder sample. Both channels optimize under similar conditions showing that a single reflectron can be used for both channels. | ||
While it was found that the use of energy discrimination and detector post acceleration did affect resolution, the difference in optimal reflectron potentials between the two channels was less than 15 V. Hence, a compromise between the optimized conditions for the two channels could be established without significant degradation of resolution on either channel.
Conclusions
A dual-source time-of-flight mass spectrometer was designed and constructed to simultaneously produce elemental, isotopic, quantitative, and molecular information on metal-containing species. The described work focuses on the instrument design and on experimental considerations in operating two sources simultaneously. Initial characterization of the instrument was carried out to optimize an electrospray source and an ICP source independently. Subsequent work will focus on characterization when the two sources are operated simultaneously. The flexible design of the instrument simplifies modification to optimize the energy bandpass of extracted ions and to produce fragment ions from the ESI source. Work is continuing to that end.Acknowledgements
The authors would like to acknowledge the scientific contributions of Ms Amy Rosen who performed a preliminary study on the feasibilty of dual-source time-of-flight mass spectrometry.This work was supported in part by the National Science Foundation through grant number CHE-0520777 and by the US Department of Energy through grant DE-FG02-98ER14890
References
- Anon, Nat. Chem. Biol., 2008, 4, 143 CrossRef.
- C. M. Moore, A. Gaballa, M. Hui, R. W. Ye and J. D. Helmann, Mol. Microbiol., 2005, 57, 27–40 CrossRef CAS.
- D. J. Thiele and J. D. Gitlin, Nat. Chem. Biol., 2008, 4, 145–147 CrossRef CAS.
- D. W. Koppenaal and G. M. Hieftje, J. Anal. At. Spectrom., 2007, 22, 855 RSC.
- J. Szpunar, Anal. Bioanal. Chem., 2004, 378, 54–56 CAS.
- N. Jakubowski, R. Lobinski and L. Moens, J. Anal. At. Spectrom., 2004, 19, 1–4 RSC.
- S. Langard, e, Department of Occupational Medicine, Telemark Central Hospital, Porsgrunn, Norway, United States, 1990.
- H. M. Shen and Q. F. Zhang, Environ. Health Persp. Suppl., 1994, 102, 275–282 Search PubMed.
- D. R. Williams, Chem. Rev. (Washington, D. C.), 1972, 72, 203–213 CrossRef CAS.
- M. Yaman, Curr. Med. Chem., 2006, 13, 2513–2525 CrossRef CAS.
- D. P. Perl and A. R. Brody, Science (Washington, D. C.), 1980, 208, 297–299 CAS.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- C. Hock, G. Drasch, S. Golombowski, F. Muller-Spahn, B. Willershausen-Zonnchen, P. Schwarz, U. Hock, J. H. Growdon and R. M. Nitsch, J. Neural Transm., 1998, 105, 59–68 CrossRef CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS.
- D. T. Dexter, F. R. Wells, A. J. Lees, F. Agid, Y. Agid, P. Jenner and C. D. Marsden, J. Neurochem., 1989, 52, 1830–1836 CrossRef CAS.
- J. M. Gorell, C. C. Johnson, B. A. Rybicki, E. L. Peterson, G. X. Kortsha, G. G. Brown and R. J. Richardson, Neurology, 1997, 48, 650–658 CAS.
- J. M. Gorell, C. C. Johnson, B. A. Rybicki, E. L. Peterson, G. X. Kortsha, G. G. Brown and R. J. Richardson, Neurotoxicology, 1999, 20, 239–248 CAS.
- G. A. Day, A. Dufresne, A. B. Stefaniak, C. R. Schuler, M. L. Stanton, W. E. Miller, M. S. Kent, D. C. Deubner, K. Kreiss and M. D. Hoover, Ann. Occup. Hyg., 2007, 51, 67–80 CAS.
- A. K. Madl, K. Unice, J. L. Brown, M. E. Kolanz and M. S. Kent, J. Occup. Environ. Hyg., 2007, 4, 448–466 CrossRef CAS.
- B. Nemery, Eur. Respir. J., 1990, 3, 202–219 CAS.
- K. Yoshizawa, B. Rimm Eric, J. S. Morris, L. Spate Vickie, C.-c. Hsieh, D. Spiegelman, J. Stampfer Meir and C. Willett Walter, N. Engl. J. Med., 2002, 347, 1755–1760 CrossRef CAS.
- J.-L. Lin, D.-T. Lin-Tan, K.-H. Hsu and C.-C. Yu, N. Engl. J. Med., 2003, 348, 277–286 CrossRef CAS.
- J. Chelly, Z. Tuemer, T. Toennesen, A. Petterson, Y. Ishikawa-Brush, N. Tommerup, N. Horn and A. P. Monaco, Nat. Genet., 1993, 3, 14–19 CrossRef CAS.
- D. M. Templeton, F. Ariese, R. Cornelis, L.-G. Danielsson, H. Muntau, H. P. Van Leeuwen and R. Lobinski, Pure. Appl. Chem., 2000, 72, 1453–1470 CrossRef CAS.
- P. A. Gallagher, X. Wei, J. A. Shoemaker, C. A. Brockhoff and J. T. Creed, J. Anal. At. Spectrom., 1999, 14, 1829–1834 RSC.
- V. Nischwitz and S. A. Pergantis, J. Anal. At. Spectrom., 2006, 21, 1277–1286 RSC.
- V. Nischwitz and S. A. Pergantis, Rapid Commun. Mass Spectrom., 2006, 20, 3579–3585 CrossRef CAS.
- V. Nischwitz and S. A. Pergantis, Environ. Chem., 2007, 4, 187–196 CrossRef CAS.
- A. P. Vonderheide, S. Mounicou, J. Meija, H. F. Henry, J. A. Caruso and J. R. Shann, Analyst (Cambridge, U. K.), 2006, 131, 33–40 RSC.
- J. L. Ellis, S. D. Conklin, C. M. Gallawa, K. M. Kubachka, A. R. Young, P. A. Creed, J. A. Caruso and J. T. Creed, Anal. Bioanal. Chem., 2008, 390, 1731–1737 CrossRef CAS.
- W. Buchberger, B. Czizsek, S. Hann and G. Stingeder, J. Anal. At. Spectrom., 2003, 18, 512–514 RSC.
- A. B. Gwizdala, S. K. Johnson, S. Mollah and R. S. Houk, J. Anal. At. Spectrom., 1997, 12, 503–506 RSC.
- J. L. Sterner, M. V. Johnston, G. R. Nicol and D. P. Ridge, J. Mass Spectrom., 2000, 35, 385–391 CrossRef CAS.
- P. P. Mahoney, J. P. Guzowski, Jr., S. J. Ray and G. M. Hieftje, Appl. Spectrosc., 1997, 51, 1464–1470 CAS.
- R. K. Marcus, Abstracts of Papers, ACS National Meeting, Washington DC, USA, August 20–24, 2000, ANYL-155 Search PubMed.
- J. Castro and R. Kenneth, GIT Lab. J., Eur., 2007, 11, 58–59 Search PubMed.
- E. M. Krupp, B. F. Milne, A. Mestrot, A. A. Meharg and J. Feldmann, Anal. Bioanal. Chem., 2008, 390, 1753–1764 CrossRef CAS.
- J. Mattusch, D. Moeller, M. P. E. Gonzalez and R. Wennrich, Anal. Bioanal. Chem., 2008, 390, 1707–1715 CrossRef CAS.
- D. P. Myers, G. Li, P. Yang and G. M. Hieftje, J. Am. Soc. Mass Spectrom., 1994, 5, 1008–1016 CrossRef CAS.
- D. P. Meyers, G. Li, P. P. Mahoney and G. M. Heiftje, J. Am. Soc. Mass Spectrom., 1995, 6, 400–410 CrossRef.
- J. H. Barnes, IV, G. D. Schilling, M. B. Denton, D. W. Koppenaal and G. M. Hieftje, J. Anal. At. Spectrom., 2003, 18, 1015–1018 RSC.
- R. J. Cotter, Time-of-flight mass spectrometry, American Chemical Society, Washington, DC. 1994 Search PubMed.
- H. Wollnik, Optics of Charged Particles, Academic Press, Orlando, FL. 1987 Search PubMed.
- M. Guilhaus, J. Am. Soc. Mass Spectrom., 1994, 5, 588–595 CrossRef CAS.
- B. Spengler, D. Kirsch and R. Kaufmann, J. Phys. Chem., 1992, 96, 9678–9684 CrossRef CAS.
- R. B. Marcus, K. K. Chin, Y. Yuan, H. Wang and W. N. Carr, IEEE Trans. Electron Devices, 1990, 37, 1545–1550 CrossRef.
- C. Marinach, A. Brunot, C. Beaugrand, G. Bolbach and J. C. Tabet, Int. J. Mass Spectrom., 2002, 213, 45–62 Search PubMed.
- S. D. Tanner, J. Anal. At. Spectrom., 1993, 8, 891–897 RSC.
- N. Jakubowski, B. J. Raeymaekers, J. A. C. Broekaert and D. Stuewer, Spectrochim. Acta, Part B, 1989, 44, 219–228 CrossRef.
- A. L. Gray and J. G. Williams, J. Anal. At. Spectrom., 1987, 2, 599–606 RSC.
- D. M. Chambers and G. M. Hieftje, Spectrochim. Acta, Part B, 1991, 46, 761–784 CrossRef.
- W. C. Wiley and I. H. McLaren, Rev. Sci. Instrum., 1955, 26, 1150–1157 CAS.
- D. P. Myers, PhD Thesis, Indiana University, 1994, pp. 238–258 Search PubMed.
- U. Boesl, R. Weinkauf and E. W. Schlag, Int. J. Mass Spectrom. Ion Processes, 1992, 112, 121–166 CrossRef CAS.
- J. H. D. Eland, Meas. Sci. Technol., 1993, 4, 1522–1524 CrossRef CAS.
- G. Sanzone, Rev. Sci. Instrum., 1970, 41, 741–742 CrossRef CAS.
- A. M. Cravath, Phys. Rev., 1929, 33, 605–613 CrossRef CAS.
- N. E. Bradbury and R. A. Nielsen, Phys. Rev., 1936, 49, 388–393 CrossRef CAS.
- P. R. B. Vlasak, Douglas J. Davenport, Michael R. Enke and G. Christie, Rev. Sci. Instrum., 1996, 67, 68–72 CrossRef CAS.
- C. W. Stoermer, S. Gilb, J. Friedrich, D. Schooss and M. M. Kappes, Rev. Sci. Instrum., 1998, 69, 1161–1164 CrossRef.
- J. R. Kimmel, F. Engelke and R. N. Zare, Rev. Sci. Instrum., 2001, 72, 4354–4357 CrossRef CAS.
- P. P. Mahoney, S. J. Ray, G. M. Hieftje and G. Li, J. Am. Soc. Mass Spectrom., 1997, 8, 125–131 CrossRef CAS.
- I. S. Gilmore and M. P. Seah, Int. J. Mass Spectrom., 2000, 202, 217–229 Search PubMed.
- D. M. McClenathan and G. M. Hieftje, J. Anal. At. Spectrom., 2005, 20, 1326–1331 RSC.
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2654–2663 CrossRef CAS.
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Analytical Chemistry (Washington, DC, United States), 2008, 80, 2646–2653 Search PubMed.
- J. Viidanoja, A. Sysoev, A. Adamov and T. Kotiaho, Rapid Commun. Mass Spectrom., 2005, 19, 3051–3055 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |