Asparagine β-hydroxylation stabilizes the ankyrin repeat domain fold†
Leanne
Kelly
a,
Michael A.
McDonough‡
a,
Mathew L.
Coleman‡
b,
Peter J.
Ratcliffe‡
b and
Christopher J.
Schofield‡
*b
aThe Chemistry Research Laboratory and Oxford Centre for Integrative Systems Biology, University of Oxford, Oxford, UK OX1 3TA
bThe Henry Wellcome Building for Molecular Physiology, University of Oxford, Oxford, UK OX3 7BN
First published on 29th October 2008
Abstract
Ankyrin repeats (ARs) are one of the most common structural motifs among eukaryotic proteins. Recent analyses have shown that factor inhibiting hypoxia-inducible factor (FIH) catalyses the hydroxylation of highly conserved Asn-residueswithin ankyrin repeat domains (ARDs). However, the effect of Asn-hydroxylation on ARD structure is unknown. Supporting the proposal that FIH-mediated ARD hydroxylation is ubiquitous we report that consensus ARD proteins are FIH substrates both in vitro and in vivo. X-ray diffraction analyses revealed that hydroxylation does not alter the archetypical ARD conformation in the crystalline state. However, other biophysical analyses revealed that hydroxylation significantly stabilizes the ARD fold in solution. We propose that intracellularprotein hydroxylation is much more common than previously thought and that one of its roles is stabilization of localized regions of ARD folds.
Introduction
Factor-inhibiting hypoxia-inducible factor (FIH) catalyzes the hydroxylation of Asn-803 of the C-terminal transcriptional activation domain (CAD) of human hypoxia-inducible factor HIF-1/2α. HIF-α CAD hydroxylation impairs HIF mediated transcription by blocking the HIF-p300 interaction.1–3 FIH is an Fe(II) and 2-oxoglutarate-dependent (2OG) dioxygenase and is one of a sub-family that is distinct from the HIFα prolyl hydroxylases, the activities of which regulate HIF-α stability. The oxygen dependence of FIH and the prolyl HIF hydroxylases enables their role as hypoxia sensors (for review see ref. 4 and 5).Following the finding that FIH also catalyzes Asn-hydroxylation of ARDs in NFκβ/IκBα family proteins6 FIH was reported to catalyse ARD hydroxylation of Notch receptors7,8 and the SOCS (suppressors of cytokine signaling) box protein 4 (ASB4).9 Since ARDs are ubiquitous (>250 present in human proteins and predicted to be part of >5% of all eukaryotic proteins) the observation that multiple peptide fragments of other ARD containing proteins are FIH substrates6 has increased the possibility that ARD hydroxylation, and more generally cytoplasmic post-translational hydroxylation, in human cells is common.
FIH-mediated ARD Asn-hydroxylation has not yet been shown to play a direct role in a signalling pathway. However, the Notch ARD was shown to compete with HIF-1α for FIH-dependent hydroxylation and to increase HIFα-CAD activity. Because the hydroxylated ARD products bound FIH less tightly than the ARD substrates, the hydroxylation status of the cytoplasmic ‘ankyrin pool’ was proposed to regulate the efficiency of FIH catalyzed HIF-α hydroxylation.7
The HIFα-CAD region is disordered when uncomplexed but adopts distinct and different conformations when bound to p30010,11 or FIH.12 In contrast, extensive structural studies have revealed a very well-conserved ARD fold comprised of ∼33 residue repeats forming a hairpin-α-helix-loop-α-helix motif;13 this fold likely must unravel to enable FIH catalyzed hydroxylation.7
The observations of ARD Asn-hydroxylation have raised questions as to the effect of hydroxylation on the ARD fold and as to the generality of FIH mediated Asn hydroxylation in this extensive protein family. The effect of individual Asn-hydroxylation events on natural ARDs are difficult to dissect because the ARDs are often located within larger proteins, are part of protein complexes, and more than one hydroxylation site may be present. We reasoned that if ARD hydroxylation is indeed ubiquitous then FIH should accept consensus ARD substrates and that such generic substrates may also enable us to identify the biophysical effects of Asn-hydroxylation. We report that consensus ARD proteins are FIH substrates and describe biophysical analyses that demonstrate significant stabilization of the archetypical ARD fold. Together with the previous structure–function data on ARD proteins our findings suggest a role for intracellular ARD protein hydroxylation.
Experimental procedures
Gene design
The designed sequence for the 4CA1A2A3Nprotein was based on the consensus ARD protein designed by Binz et al.14 and Mosavi et al.15 The capping repeats utilized in the consensus ARD proteins of Binz et al.14 were used in the design of the terminal repeats. The sequence for the internal consensus repeats were adapted from the consensus sequence derived by Mosavi et al.15 The 4CA1A2A3N sequence designed for this study, contained a single potential asparaginyl hydroxylation site. The Arg in the non-conserved 5th position of the 33-residue internal consensus sequence derived on Mosavi et al.15 was replaced with a Ser residue in the 4CA1A2A3N sequence to incorporate BamHI restriction endonuclease sites for the conversion of the 4 repeat ARD to a 3 repeat ARD. The 4CA1A2A3NDNA sequence was synthesized by Genescript (Piscataway, NJ, USA) and ligated into a pET-28a(+) (Novagen) vector using NdeI and HindIII restriction sites. Mutants were generated by standard techniques.Recombinant protein expression and purification
All recombinant proteins were transformed into E. coli BL21 (DE3) and induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) at 37 °C. His6-tagged proteins were purified using nickel affinity chromatography. Fractions containing the pure protein were concentrated before over night cleavage of the His6-tag with thrombin, the cleaved untagged protein was purified using size exclusion chromatography (Superdex S75). 3CA1A2N-OH was prepared by co-expression of pET-28a-3CA1A2N and pMAL-FIH in E. coli BL21 (DE3), cells were grown for 3.5 h at 37 °C after induction with IPTG. Purification was as for the unhydroxylated material.2OG decarboxylation and MS analyses
Purified proteins were tested for their ability to stimulate FIH-dependent decarboxylation of [14C]-labeled 2OG. Incubations contained 160 μM 2OG, 80 μM Fe(II), 5.75 μM FIH, and 57.5 μM substrate in 50 mM Tris/HCl, pH 7.4 and were carried out at 37 °C for 30 min. Material reacted with FIH for MS analysis was prepared under the same conditions without the radiolabeled 2OG. Time course reactions were quenched with an equal volume of SDS-PAGE loading buffer containing 10% SDS and 1% β-mercaptoethanol. For MS analysis proteins were resolved by SDS-PAGE, and excized gel fragments were digested with trypsin. After separation on an Agilent technologies 1100 capillary LC system, the peptides were analysed by positive ion electrospray with a QTof micro mass spectrometer (Waters, Milford, USA).CD and DSC Analyses
CD experiments were performed using a Chirascan Circular Dichroism Spectrometer (Applied Photophysics, UK) equipped with a peltier temperature controller. The temperature inside the cuvette was recorded during the scan and used in the subsequent analyses. A cuvette with a path length 0.1 cm was used. Protein samples were buffered in 100 mM sodium phosphatebuffer (NaH2PO4/NaOH) pH 6.1 at a typical concentration of 10 μM. Thermal scans were performed at a rate of 2 °C min−1, and the signal was averaged over 12 s. A buffer baseline was subtracted and the data smoothed prior to analysis using a 2 state model: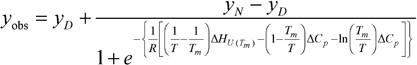
| yN = aN + bNT |
| yD = aD + bDT |
Transfections and immunoprecipitations
RNAi used Oligofectamine (Invitrogen). Human FIH and control Drosophila HIF siRNAoligonucleotides were as reported.16 Plasmid transfections used Fugene (Roche). 4CA1A2A3N and 3CA1A2N were PCR-cloned into pEF1/V5-His Invitrogen) using standard procedures. pEF FIH was prepared as described previously.7 Monoclonal anti-FIH was raised against full length recombinant FIH and used to detect human FIH.16 Anti-PK antibody was from Serotec and anti-β-tubulin was from Sigma. For immunoprecipitation analyses cells were lysed in IP+ buffer6 and PK-tagged 3CA1A2N precipitated with agarose beads conjugated with anti-PK antibody (Sigma).Crystallography
Crystallisation conditions were sought using high throughput robotic screening methods at the Oxford Protein Production Facility.17 Diffraction quality crystals were obtained from the initial screen for the His-tagged 3CA1A2N-OH within one week under conditions of the Hampton Research (Aliso Viejo, CA, USA) Crystal Screen II number 12 (30% (v/v) polyethylene glycol 400, 100 mM sodium acetate pH 4.6, 100 mM cadmium chloride) using a protein concentration of 29 mg ml−1. The His-tag cleaved 3CA1A2N crystallized within one week under conditions of the Hampton Research Index Screen number 64 which consists of 100 mM HEPES pH 7.5, 5 mM magnesium chloride, 5 mM cobalt(II) chloride, 5 mM cadmium chloride using a protein concentration of 13 mg ml−1 Single crystals were transferred from the crystallisation drop directly into well solution diluted with 30% glycerol and frozen in liquid nitrogen prior to mounting in a cryo-stream at 100 K for data collection on Beamline 14.2 of the SRS, Daresbury, UK tuned to a wavelength of 0.976 Å and equipped with a Quantum 4 CCD detector. The data were collected using DNA ∼1.0.0 and processed using HKL200018 (see Table 1 supporting information for data processing and refinement statistics† ). The structures of His-tagged 3CA1A2N-OH and 3CA1A2N were solved by molecular replacement using a prior 3 repeat consensus ankyrin structure (PDB entry 1N0Q)15 as the search model in PHASER19 v.1.3.1. The initial models for the individual structures were built using COOT v.0.3.120 and refined using CNS v1.1.21 Iterative cycles of model fitting and slowcool annealing maximum likelihood refinement with bulk solvent modelling and isotropic B-factor refinement continued until Rfree no longer improved at which point water molecules were added to the model where |2Fo-Fc| electron density peaks and distance and geometry criteria were met. Crystallographic and B-factor weighting factors were optimized before final refinement.Coordinates for the 3CA1A2N-OH and 3CA1A2N structures have been deposited with the PDB (2ZGD and 2ZGG).
Results
A consensus ankyrin repeat peptide is an FIH substrate. Two studies have independently reported the use of multiple sequence analyses to identify similar consensus AR sequences.14,15 Engineered consensus ARD proteins were shown to adopt the archetypical fold observed in the crystalline form of wildtype ARD. The consensus AR sequences are also similar to the natural AR peptides shown to be FIH substrates6,7,9 including the conservation of a Leu/hydrophobic residue at the −8 position relative to the hydroxylated Asn-residuewhich could be important for binding to FIH.7 Initially, we synthesized a 20 residue synthetic ankyrin peptide (1CA) based on the consensus sequence derived by Mosavi et al.15 (Fig. 1) and analysed it as an FIH substrate. In the presence of FIH the 1CA peptide stimulated 2OG turnover to a greater extent than that observed for HIF-1α CAD and an AR fragment peptide derived from IκBα under standard conditions (Supplemental data Fig. S1). Liquid chromatography (LC)-mass spectrometry (MS) analyses demonstrated that the 2OG turnover was coupled to hydroxylation and MS/MS analyses identified the sole site of modification as the single Asn-residue (Supplemental data Fig. S2† ).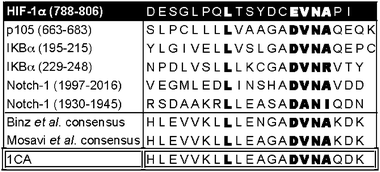 | ||
| Fig. 1 Peptide sequences of HIF-1α and ARD6,7 FIH substrates aligned with 1CA and previously reported AR consensus sequence.14,15 | ||
The observation that FIH can catalyse the hydroxylation of a consensus AR peptide is consistent with the proposal that FIH may have evolved to catalyze hydroxylation of multiple natural ARD proteins. Because significant conformational changes are likely required in order for the archetypical ARD fold to partially unfold and bind to FIH in a catalytically productive manner,7 the observation that conformationally flexible AR peptides act as FIH substrates does not necessarily imply that a longer natively-folded ARD protein will be a substrate. We addressed this question by testing consensus ARD proteins as FIH substrates.
Design of consensus ARD proteins
We designed a gene that encodes a consensus ARD protein with 4 ARs, using elements from previously reported strategies. As in the work of Binz et al.14 we employed N- and C-terminal modified capping domains to minimize the exposure of hydrophobic regions and enhance production of soluble protein. The sequences of the two central ARs were near identical to the consensus sequence identified by Mosavi et al.15 Considerations in the sequence design included predicted optimal protein expression in E. coli, solubility under the conditions of FIH catalysis, ready purification via affinity chromatography, ability to modify the number of ARs, amenability to trypsin digestion to quantify Asn-hydroxylation by MS, resistance to aggregation, and the potential for crystallisation. Because the extent of FIH-catalyzed hydroxylation varies at different sites within natural ARD substrates,6,7 we initially incorporated a single potential Asn-containing FIH substrate sequence ‘DVNA motif’ to simplify quantitative analyses. The DVNA motif was positioned in hairpin loop 3 (Fig. 2A), i.e. the loop linking the third and fourth ARs, because in both Notch 1 (N1) and IκBα, the Asn-residue closer to a terminus is hydroxylated to a greater extent than that further away from a terminus.6,7Ala-residues were incorporated at the equivalent position to the Asn of the DVNA motif creating DVAA sequences in the other two loops.![Some but not all consensus ARD proteins are FIH substrates. (A) Sequences of the 3CA1A2N and 4CA1A2A3N repeat consensus ARDs. The single potential hydroxylation site in these proteins is underlined. Numbers in the right hand boxes correspond to the residue numbers and the ankyrin repeat (AR) number. Excision of the DNA encoding for the second AR from the vector used for 4CA1A2A3N expression, by digestion with BamHI followed by religation, gave a vector encoding for a 3AR consensus protein with a single potential Asn-hydroxylation site in the loop linking the central and C-terminal ARs (3CA1A2N). (B) 3CA1A2N but not 4CA1A2A3N stimulates FIH mediated 2OG decarboxylation, as measured by 14CO2 formation. (C) Tryptic digestion followed by LC-MS analyses of 3CA1A2N incubated under standard assay conditions in the absence (top) and presence (bottom) of FIH. The expected monisotopic masses for the [M+2H]2+ unhydroxylated and hydroxylated tryptic peptides are m/z = 728.88 and 736.88. No mass change was observed for 4CA1A2A3N incubated under standard assay conditions with FIH. (D) MS/MS analysis of the tryptic digest peptide containing the Asn of interest incubated in the absence (top) and presence (bottom) of FIH. A +16 Da shift is observed in the y ion series appearing at y5, corresponding to fragments containing Asn-84 of 3CA1A2N. Some hydrolysis of the hydroxyasparagine amide is likely as both +16 Da and +17 Da mass shifts relative to the unhydroxylated sample were observed.](/image/article/2009/MB/b815271c/b815271c-f2.gif) | ||
| Fig. 2 Some but not all consensus ARD proteins are FIH substrates. (A) Sequences of the 3CA1A2N and 4CA1A2A3N repeat consensus ARDs. The single potential hydroxylation site in these proteins is underlined. Numbers in the right hand boxes correspond to the residue numbers and the ankyrin repeat (AR) number. Excision of the DNA encoding for the second AR from the vector used for 4CA1A2A3N expression, by digestion with BamHI followed by religation, gave a vector encoding for a 3AR consensus protein with a single potential Asn-hydroxylation site in the loop linking the central and C-terminal ARs (3CA1A2N). (B) 3CA1A2N but not 4CA1A2A3N stimulates FIH mediated 2OG decarboxylation, as measured by 14CO2 formation. (C) Tryptic digestion followed by LC-MS analyses of 3CA1A2N incubated under standard assay conditions in the absence (top) and presence (bottom) of FIH. The expected monisotopic masses for the [M+2H]2+ unhydroxylated and hydroxylated tryptic peptides are m/z = 728.88 and 736.88. No mass change was observed for 4CA1A2A3N incubated under standard assay conditions with FIH. (D) MS/MS analysis of the tryptic digest peptide containing the Asn of interest incubated in the absence (top) and presence (bottom) of FIH. A +16 Da shift is observed in the y ion series appearing at y5, corresponding to fragments containing Asn-84 of 3CA1A2N. Some hydrolysis of the hydroxyasparagine amide is likely as both +16 Da and +17 Da mass shifts relative to the unhydroxylated sample were observed. | ||
Some but not all consensus ARDs are FIH substrates
The N-terminally His-tagged consensus 4AR consensus protein with a single Asn-hydroxylation site on the third loop (4CA1A2A3Ni.e. with an Ala residue in the first and second hairpin loops, and with an Asn residue on loop 3), was produced in a highly soluble form in E. coli using a pET28a vector. Purification by nickel-affinity chromatography, cleavage of the His-tag, followed by gel -filtration chromatography produced >90% pure 4CA1A2A3N by SDS-PAGE and MS analyses. In contrast to the 1CA peptide, incubation of 4CA1A2A3N with FIH did not lead to the stimulation of 2OG turnover by FIH, nor hydroxylation as determined by trypsin digestion followed by MS analysis (Fig. 2B).Prior biophysical analyses have revealed that the conformational stability of consensus ARD-proteins increases with the number of ARs.15,22 We considered it possible that a 4AR consensus protein might not be an FIH substrate due to its high stability.15,22 Therefore we next made a 3AR consensus protein (3CA), predicted to be less stable than 4CA1A2A3N.
In contrast to 4CA1A2A3N, 3CA1A2N significantly stimulated 2OG turnover by FIH (Fig. 2B); The level of activity was ∼50% of the level observed with the natural ARD FIH substrate IκBα (Supplemental data Fig. S3). This observation is consistent with the presence of two hydroxylation sites in IκBα but only one in 3CA1A2N. Intact protein MS analyses following incubation with FIH revealed a +16 Da mass increment relative to non-incubated 3CA1A2N (data not shown). Amino acid analyses were consistent with the presence of 3-hydroxy-Asp after FIH incubation at a level consistent with complete hydroxylation of a single Asn-residue (data not shown); the Asn residues are hydrolysed to Asp under the acidic amino acid analysis conditions. Trypsin digestion followed by LC-MS and MS/MS sequencing analyses revealed the Asn-residue in the loop linking the second and third ARs as the only hydroxylation site (Fig. 2C and D). No other Asn-residue was hydroxylated indicating that 3CA1A2N possesses the same FIH specificity requirement as natural ARD proteins. A variant in which the target Asn was mutated to an Ala-residue(3CA1A2A) was not an FIH substrate (data not shown).
MS time-course analyses were then used to compare the efficiency of the 3CA1A2N as an FIH substrate relative to N1 ARD and IκBα. Both N1 and IκBα have two hydroxylation sites, one of which is favoured for hydroxylation6,7 (Supplemental data Fig. S4). These analyses indicated that 3CA1A2N has a similar efficiency as a substrate to the favoured sites in the intact N1 and IκBα ARDs. Kinetic analyses, using an oxygen electrode assay23 with 3CA1A2N gave a Km of 3.2 ± 1.3 μM (kcat 5.1 min−1), significantly lower than that reported for HIF-α CAD fragments.23,24 The kcat/km value of 2.7 × 104 s−1M−1, is also significantly higher than the reported values for HIF-α CAD fragments,23 consistent with the suggestion that some natural ARDs are better FIH substrates than HIF-α CAD.6
A consensus ARD protein is a substrate for FIH in human cells
To test whether a consensus ARD protein is a substrate for FIH in cells, we transfected 293T cells with PK-3CA1A2N or 4CA1A2A3N prior to immunopurification and MS/MS analyses. Consistent with the in vitro data, both unhydroxylated and Asn-hydroxylated DVNA peptides were detected by MS following trypsin digestion of 3CA1A2N, demonstrating that consensus ARD proteins can support FIH activity in vivo. To investigate the FIH-dependence of 3CA1A2NAsn hydroxylation we transfected 293T cells that either under- (siRNA) or over- (plasmid transfection) expressed FIH with PK-3CA1A2N followed by immunopurification and LC-MS quantification of the unhydroxylated:hydroxylated 3CA1A2N DVNA peptides . In cells expressing endogenous levels of FIH the 3CA1A2N DVNA peptide was between 8–12% hydroxylated (Fig. 3). PK-3CA1A2N hydroxylation was dependent on FIH since FIH siRNA treatment reduced Asn-hydroxylation to<5%. Conversely, FIH over-expression increased PK-3CA1A2N DVNA hydroxylation to ∼75%. FIH is therefore both necessary and sufficient for PK-3CA1A2N Asn hydroxylation in human cells. Consistent with the in vitro data PK-4CA1A2A3N was not hydroxylated under any of the conditions tested. The concordance between in vivo and in vitro data provide validation for the use of engineered consensus ARD proteins for the in vitro biophysical analyses on the effect of Asn-hydroxylation.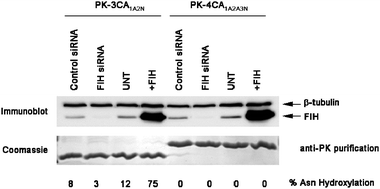 | ||
| Fig. 3 3CA1A2N is an FIH substrate in human cells. FIH is necessary and sufficient for 3CA1A2N Asn hydroxylation in vivo. 293T cells were transfected with PK-3CA1A2N and/or FIH or empty vector (EV) control for 48 h prior to immunoprecipitation and MS. For FIH-targeted RNA interference, cells were treated twice over 48 h with 10 nM siRNA prior to PK-3CA1A2N transfection. | ||
Biophysical analyses
To investigate the effect of Asn-hydroxylation on ARD structure, we then crystallized the 3CA1A2Nprotein in both the non-hydroxylated and Asn-hydroxylated (3CA1A2N-OH) forms. The 3CA1A2N-OHprotein was prepared to > 95% hydroxylation by co-expression of vectors encoding for MBP-tagged FIH and His-tagged 3CA1A2N. A refined crystal structure of His-tagged 3CA1A2N confirmed that our consensus sequences can adopt the archetypical ARD fold (Fig. 4A), in particular the backbone of the complete 3CA1A2Nprotein was superimposable on those of the complete ARs present in N1 and IκBα.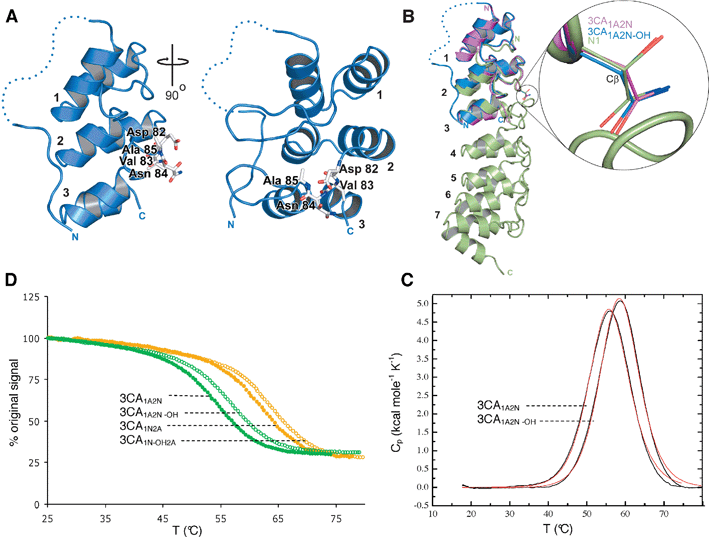 | ||
| Fig. 4 Hydroxylation increases the thermal stability of 3CA without altering the conformation adopted in the crystalline state. (a) Ribbons diagram views of the overall structure of the His-tagged 3CA1A2N-OH (blue) showing the DVNA motif in stick representation (white) and the hydroxylated Asn-84 on loop 2 between the second and third repeats (ARs are labeled 1,2,3). The dashed line indicates the disordered region of the N-terminal His-tag not visible in the electron density maps. (B) Superimposition of the structures of 3CA1A2N-OH (blue) and 3CA1A2N (magenta) and hydroxylated mouse N1 (green). Encircled is an expanded view of the FIH target asparagine in ARD proteins illustrating the lack of conformational change in its three-dimensional structure between, not only consensus ankyrins 3CA1A2N-OH and 3CA1A2N, but also when compared to the hydroxylated mouse N1 protein structure (PDB ID 2QC9). (C) Temperature induced equilibrium denaturation monitored by CD spectroscopy at 222 nm for unhydroxylated (closed circles) and hydroxylated (open circles) 3CA1A2N (green) and 3CA1N2A (orange), represented as percentage of the original signal at 25 °C. The midpoint of the transitions (Tm) are 54.1 °C for 3CA1A2N, 57.1 °C for 3CA1A2N-OH, 61.7 °C for 3CA1N2A and 63.5 °C for 3CA1N-OH2A. (D) DSC data showing the thermal unfolding of 3CA1A2N and 3CA1A2N-OH after baseline correction and normalization (black line), and curve fit using a 2-state model (red line). The average melt temperatures calculated by DSC were 55.4 °C for 3CA1A2N and 58.1 °C and 3CA1A2N-OH. | ||
The electron density maps of 3CA1A2N-OH clearly implied a single hydroxylation event occurring at the pro-S position of Asn-84 as observed in the N1 ARD7 and HIF-α CAD12 (Supplemental data Fig. S5† ). Overall the hydroxylated and non-hydroxylated CA structures were very similar (Fig. 4B), including the side chain conformation of Asn-84, where there were minor side chain differences mainly due to differences in metal ion interactions. Hydroxylation at the pro-S site of Asn-84 positions the alcohol to hydrogen bond with the side chain of the Asp-residue that forms part of the conserved DXNZ motif present in many natural ARs. In many natural ARD proteins where an Asp-residue is not present in this motif it is replaced with a residue (e.g.Asp, Gln, Glu) also capable of forming a hydrogen bond to the hydroxyl-group of a hydroxy-Asn residue. As in the hydroxylated N1 structure,7 this interaction is predicted, at least in part, to stabilize the ARD and/or AR.
Stability assays
To investigate the effect of Asn-hydroxylation on the thermal stability of 3CA1A2N we used circular dichroism (CD ) to monitor temperature induced changes in ellipticity at 222 nm (with concentrations used for the analyses the proteins were monomeric by non-denaturing MS analyses). Thermal unfolding was reversible (to >95% at temperatures up to 80 °C); the shapes of the curves for both 3CA1A2N and 3CA1A2N-OH were sigmoidal with no evidence for detectable folding intermediates. These analyses revealed a clear stabilizing effect for Asn-hydroxylation (Fig. 4C and D). To test whether the stabilizing effect of hydroxylation was limited to the C-terminal of the two potential sites we then made a 3CA1A2N variant (3CA1N2A) in which the hydroxylation site was in the N-terminal rather then the C-terminal of the 2 loops. The 3CA1N2A was also hydroxylated by FIH (data not shown) and a similar stabilizing effect of hydroxylation was observed for the 3CA1N2A/3CA1N-OH2A pair as for the 3CA1A2N/3CA1A2N-OH pair (Fig. 4C). The results also indicated that the non-hydroxylated 3CA1N2A was more stable than the non-hydroxylated 3CA1A2N. To further test the apparent stabilizing effect observed by the CD analyses, the thermal stability and unfolding of the hydroxylated (3CA1N-OH2A and 3CA1A2N-OH) and the non-hydroxylated (3CA1A2N and 3CA1N2A) proteins were then investigated by differential scanning calorimetry (DSC ). Again a clear stabilizing effect was observed for both proteins upon hydroxylation (Tm = 55.4 °C for 3CA1A2N, 58.1 °C for 3CA1A2N-OH, 63.4 °C for 3CA1N2A, and 67.6 °C for 3CA1N-OH2A) (Fig. 4D). The difference in the absolute Tm values calculated for the DSC and CD analyses coupled to differences in calculated ΔHu(cal) and ΔHu(VH) values (data not shown) implies that folding is more complex than a simple 2-state model observed for some ARDs under equilibrium conditions.Discussion
The observation that consensus ARD proteins are FIH substrates supports the proposal that intracellularAsn-hydroxylation of ARDs is common.6 As with studies on N17 3S-Asn-hydroxylation of a consensus ARD protein with 3ARs (3CA1A2N) did not alter the well-defined ARD fold in the crystalline state. CD and DSC analyses revealed that hydroxylation of Asn-residues in either of the two loops increased the thermal stability of the ARD by similar amounts. The consensus ARD proteins described here will be useful tools to investigate in detail the effects of Asn-hydroxylation on the dynamics of ARD structure and the chemical origin of the stabilizing effects described.In contrast to the 3AR proteins the 4AR protein was not an FIH substrate. It seems likely that this difference is related to the stability of the fold rather than the number of ARs. The stability of the studied consensus ARD proteins increases with the number of ARs,22 but destabilizing elements within natural ARD proteins means that the number of ARs does not necessarily correlate with stability either of the overall protein or within localized regions. Notch and IκBα, known substrates of FIH in human cells, contain ∼6 and ∼7ARs, respectively, thus, the number of ARs is unlikely by itself to be limiting in FIH catalysis.
Stabilization of the ARD fold by Asn hydroxylation has similarities with the role of 2OG-oxygenase-dependent prolyl-4-hydroxylation in stabilising the collagen triple helix fold.25 There are, however, important differences between collagenPro-hydroxylation and ARD Asn-hydroxylation that argue the role of the latter is not solely to optimize conformational stability of the archetypical ARD fold. Collagen has an optimized repeating Pro-Pro-Gly motif that places a glycine at every third residue for favourable packing and ubiquitous Pro-hydroxylation for optimal stabilization of the global triple helix fold. In contrast, FIH-mediated Asn-hydroxylation does not occur at every AR of natural ARDs, and hydroxylation is not always complete.6,7 Furthermore, in contrast to collagen, studies with consensus and natural ARD proteins reveal that their sequences are not optimized for conformational stability.26,27 Such observations suggest that hydroxylation of ARDs is unlikely to have evolved to optimize the global ARD fold of individual proteins, and that some degree of localized or global instability relative to the archetypical ARD fold may be required for the biological functions of ARD proteins.
Hydroxylation may ‘directly’ enable or ablate protein–protein interactions involving ARDs-as occurs for non-ARD proteins in the HIF system (e.g. HIFα CAD and p300). Although Asn hydroxylation was not observed to profoundly affect the interaction of known binding partners with the N1 ARD,7 some differences were noted for IκBα in vitro.6 Binding interactions, perhaps involving specific regions of an ARD could also be less directly modulated by hydroxylation. For instance, local as opposed to global effects of hydroxylation on the stability of the hydroxylated AR may regulate many specific conformations involved in regulating binding partner interactions. Modulating the stability of the archetypical ARD fold is known to regulate the affinity for binding partners.28 Local changes in ARD stability induced by sequence variations in natural ARDs alter their ‘folding energy landscape’.29–31 We propose that following Asn hydroxylation, changes in the local stability of regions within ARDs regulate protein–protein interactions, thereby providing a mechanism by which ARD hydroxylation can regulate signal transduction. It is also possible that stabilization of the ARD fold by hydroxylation only exerts a detectable effect through perturbation of the folding energy landscape of specific ARD proteins under specific (e.g. stressed) conditions when certain folding intermediates or conformations reach threshold levels.
Given that it seems likely that multiple ARD-proteins are hydroxylated; it is possible that the overall effect of many changes, in ARD-protein stability/interactions is to enable cells, and possibly tissues, to adapt robustly to different oxygen tensions.
Details of the mechanism by which Asn hydroxylation stabilizes the ARD fold are still being investigated. Our crystallographic analyses of hydroxylated mouse N17 and 3CA1A2N indicate that the introduced hydroxyl group is positioned to hydrogen bond to the side chain of a conserved Asp-residue -2 relative to the target asparaginyl-residue.
Although this additional intramolecular bond may contribute to stabilization of the ARD, studies on collagenPro-hydroxylation mean care should be taken in assigning the stabilizing effect solely this interaction. Rather than involving hydrogen bonding and a network of water molecules as initially proposed, it now appears that stabilization of the collagen triple helix by Pro-hydroxylation is primarily mediated via stereoelectronic effects and, biasing of conformation.25,32
It may be that post-translational hydroxylation also modulates the stability of structural domains in other systems. For example, hydroxylation of Asn-residues also occurs in epidermal growth factor (EGF) domains. Both Asp- and Asn-hydroxylation of EGF domains is carried out by a single 2OG dependent EGF domain Asp/Asn-hydroxylase (β-Asp/Asn hydroxylase, BAH.33 Sequence analyses reveal that FIH and BAH belong to different sub-families of 2OG oxygenases. The ability of BAH to catalyse Asp and Asnhydroxylation contrasts with FIH that only catalyses hydroxylation of Asn-residues. However, both FIH and BAH appear to accept multiple substrates with common motifs, ARDs for FIH and EGF domains for BAH. Similar to the FIH/ARD system, hydroxylation does not change the overall EGF fold.33,34 The work described here suggests that testing the effect of Asp/Asn-hydroxylation on EGF domain stability/folding will be of interest.
List of abbreviations
| AR | ankyrin repeat |
| ARD | ankyrin repeat domain |
| HIF | hypoxia-inducible factor |
| FIH | factor inhibiting HIF |
| CAD | C-terminal transcriptional activation domain |
| 2-OG | 2-oxoglutarate |
| LC/MS | liquid chromatography/mass spectrometry |
| MSMS | tandem mass spectrometry |
| N1 | notch 1 |
| siRNA | short interfering RNA |
| CD | circular dichroism |
| DSC | differential scanning calorimetry |
| EGF | epidermal growth factor |
Acknowledgements
This work was funded by the Biotechnology and Biological Research Council and the Wellcome Trust. We are grateful to Nicolas Mathioudakis for oxygen electrode analyses, Charlotte Coles and David Lancaster for their contributions with Notch and IκBα, Margaret Nutley for contributions to the DSC experiments and Mr A.C. Willis for amino acid analyses.References
- D. Lando, D. J. Peet, J. J. Gorman, D. A. Whelan, M. L. Whitelaw and R. K. Bruick, Genes Dev., 2002, 16(12), 1466–1471 CrossRef CAS.
- D. Lando, D. J. Peet, D. A. Whelan, J. J. Gorman and M. L. Whitelaw, Science, 2002, 295(5556), 858–861 CrossRef CAS.
- K. S. Hewitson, L. A. McNeill, M. V. Riordan, Y.-M. Tian, A. N. Bullock, R. W. Welford, J. M. Elkins, N. J. Oldham, S. Bhattacharya, J. M. Gleadle, P. J. Ratcliffe, C. W. Pugh and C. J. Schofield, J. Biol. Chem., 2002, 277(29), 26351–26355 CrossRef CAS.
- C. J. Schofield and P. J. Ratcliffe, Nat. Rev. Mol. Cell Biol., 2004, 5(5), 343–354 CrossRef CAS.
- W. G. Kaelin, Annu. Rev. Biochem., 2005, 74(1), 115–128 CrossRef CAS.
- M. E. Cockman, D. E. Lancaster, I. P. Stolze, K. S. Hewitson, M. A. McDonough, M. L. Coleman, C. H. Coles, X. Yu, R. T. Hay, S. C. Ley, C. W. Pugh, N. J. Oldham, N. Masson, C. J. Schofield and P. J. Ratcliffe, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(40), 14767–14772 CrossRef CAS.
- M. L. Coleman, M. A. McDonough, K. S. Hewitson, C. Coles, J. Mecinovic, M. Edelmann, K. M. Cook, M. E. Cockman, D. E. Lancaster, B. M. Kessler, N. J. Oldham, P. J. Ratcliffe and C. J. Schofield, J. Biol. Chem., 2007, 282(33), 24027–24038 CrossRef CAS.
- X. Zheng, S. Linke, J. M. Dias, X. Zheng, K. Gradin, T. P. Wallis, B. R. Hamilton, M. Gustafsson, J. L. Ruas, S. Wilkins, R. L. Bilton, K. Brismar, M. L. Whitelaw, T. Pereira and J. J. Gorman, et al., Proc. Natl. Acad. Sci. U. S. A., 2008, 105(9), 3368–3373 CrossRef CAS.
- J. E. Ferguson, III, Y. Wu, K. Smith, P. Charles, K. Powers, H. Wang and C. Patterson, Mol. Cell. Biol., 2007, 27(18), 6407–6419 CrossRef.
- S. A. Dames, M. Martinez-Yamout, R. N. De Guzman, H. J. Dyson and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(8), 5271–5276 CrossRef CAS.
- S. J. Freedman, Z.-Y. J. Sun, F. Poy, A. L. Kung, D. M. Livingston, G. Wagner and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(8), 5367–5372 CrossRef CAS.
- J. M. Elkins, K. S. Hewitson, L. A. McNeill, J. F. Seibel, I. Schlemminger, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, J. Biol. Chem., 2003, 278(3), 1802–1806 CrossRef CAS.
- L. K. Mosavi, T. J. Cammett, D. C. Desrosiers and Z.-Y. Peng, Protein Sci., 2004, 13(6), 1435–1448 CrossRef CAS.
- H. K. Binz, M. T. Stumpp, P. Forrer, P. Amstutz and A. Pluckthun, J. Mol. Biol., 2003, 332(2), 489–503 CrossRef CAS.
- L. K. Mosavi, D. L. Minor and Z.-Y. Peng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(25), 16029–16034 CrossRef CAS.
- I. P. Stolze, Y.-M. Tian, R. J. Appelhoff, H. Turley, C. C. Wykoff, J. M. Gleadle and P. J. Ratcliffe, J. Biol. Chem., 2004, 279(41), 42719–42725 CrossRef CAS.
- T. S. Walter, J. Diprose, J. Brown, M. Pickford, D. I. Stuart and K. Harlos, J. Appl. Crystallogr., 2003, 36, 308–314 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- A. T. Brünger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson and G. L. Warren, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 905–921 CrossRef.
- S. K. Wetzel, G. Settanni, M. Kenig, H. K. Binz and A. Pluckthun, J. Mol. Biol., 2008, 376(1), 241–257 CrossRef CAS.
- D. Ehrismann, E. Flashman, D. N. Genn, N. Mathioudakis, K. S. Hewitson, P. J. Ratcliffe and C. J. Schofield, Biochem. J., 2007, 401(1), 227–234 CrossRef CAS.
- P. Koivunen, M. Hirsila, V. Gunzler, K. I. Kivirikko and J. Myllyharju, J. Biol. Chem., 2004, 279(11), 9899–9904 CAS.
- C. L. Jenkins and R. T. Raines, Nat. Prod. Rep., 2002, 19, 49–59 RSC.
- T. J. Cammett, L. Luo and Z.-Y. Peng, J. Mol. Biol., 2003, 327(1), 285–297 CrossRef CAS.
- D. U. Ferreiro, C. F. Cervantes, S. M. E. Truhlar, S. S. Cho, P. G. Wolynes and E. A. Komives, J. Mol. Biol., 2007, 365(4), 1201–1216 CrossRef CAS.
- C. Zahnd, E. Wyler, J. M. Schwenk, D. Steiner, M. C. Lawrence, N. M. McKern, F. Pecorari, C. W. Ward, T. O. Joos and A. Pluckthun, J. Mol. Biol., 2007, 369(4), 1015–1028 CrossRef CAS.
- T. O. Street, C. M. Bradley and D. Barrick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(12), 4907–4912 CrossRef.
- A. R. Lowe and L. S. Itzhaki, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(8), 2679–2684 CrossRef CAS.
- N. D. Werbeck and L. S. Itzhaki, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(19), 7863–7868 CrossRef CAS.
- L. E. Bretscher, C. L. Jenkins, K. M. Taylor, M. L. DeRider and R. T. Raines, J. Am. Chem. Soc., 2001, 123(4), 777–778 CrossRef CAS.
- J. E. Dinchuk, R. J. Focht, J. A. Kelley, N. L. Henderson, N. I. Zolotarjova, R. Wynn, N. T. Neff, J. Link, R. M. Huber, T. C. Burn, M. J. Rupar, M. R. Cunningham, B. H. Selling, J. Ma and A. A. Stern, et al., J. Biol. Chem., 2002, 277(15), 12970–12977 CrossRef CAS.
- M. S. Sunnerhagen, E. Persson, I. Dahlqvist, T. Drakenberg, J. Stenflo, M. Mayhew, M. Robin, P. Handford, J. W. Tilley and I. D. Campbell, J. Biol. Chem., 1993, 268(31), 23339–23344 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data, further assay results and links to 3D visualisations in Jmol. See DOI: 10.1039/b815271c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2009 |