An integrated comparative phosphoproteomic and bioinformatic approach reveals a novel class of MPM-2 motifs upregulated in EGFRvIII-expressing glioblastoma cells†
Brian A.
Joughin‡
a,
Kristen M.
Naegle‡
b,
Paul H.
Huang‡
b,
Michael B.
Yaffe
ab,
Douglas A.
Lauffenburger
*ab and
Forest M.
White
*ab
aThe David H. Koch Institute for Integrative Cancer Research at MIT, Cambridge, MA, USA
bDept. of Biological Engineering, MIT, Cambridge, MA, USA. E-mail: fwhite@mit.edu; lauffen@mit.edu
First published on 30th October 2008
Abstract
Glioblastoma (GBM, WHO grade IV) is an aggressively proliferative and invasive brain tumor that carries a poor clinical prognosis with a median survival of 9 to 12 months. In a prior phosphoproteomic study performed in the U87MG glioblastoma cell line , we identified tyrosinephosphorylation events that are regulated as a result of titrating EGFRvIII, a constitutively active mutant of the epidermal growth factor receptor (EGFR) associated with poor prognosis in GBM patients. In the present study, we have used the phosphoserine/phosphothreonine-specific antibody MPM-2 (mitotic protein monoclonal #2) to quantify serine/threoninephosphorylation events in the same cell lines . By employing a bioinformatic tool to identify amino acid sequence motifs regulated in response to increasing oncogene levels, a set of previously undescribed MPM-2 epitope sequence motifs orthogonal to the canonical “pS/pT-P” motif was identified. These motifs contain acidic amino acids in combinations of the −5, −2, +1, +3, and +5 positions relative to the phosphorylated amino acid. Phosphopeptides containing these motifs are upregulated in cells expressing EGFRvIII, raising the possibility of a general role for a previously unrecognized acidophilic kinase (e.g.casein kinase II (CK2)) in cell proliferation downstream of EGFR signaling.
Introduction
Glioblastoma (GBM, WHO grade IV) is a complex disease driven by a number of genetic aberrations that dysregulate normal cellular processes such as proliferation, apoptosis and cell cycle control.1 In particular, expression of EGFRvIII, a constitutively active mutant of the epidermal growth factor receptor (EGFR), promotes GBM cell proliferation and survival by preventing cell cycle arrest upon serum withdrawal.2 This loss in serum dependency has been attributed to a downregulation of the cyclin-dependent kinase (CDK) inhibitor p27 as a result of phosphatidylinositol 3-kinase (PI3K) activation by EGFRvIII.2 Improved characterization of the regulatory network by which EGFRvIII alters mitotic processes in GBM would not only provide further insight into its mitogenic signaling networks but also generate a broader inventory of candidate target genes that may serve as points of therapeutic intervention.While proximal signals downstream of receptor tyrosine kinases (RTKs) such as EGFR are largely propagated by tyrosinephosphorylation, distal cellular processes are often the consequence of serine/threoninephosphorylation events, which comprise more than 99% of the phosphoproteome. This large background makes the enrichment of interesting phosphoproteomic subsets, such as mitogenic signaling proteins, particularly challenging.3 This problem is highlighted by a recent global phosphoproteomic study of EGF-mediated signaling in HeLa cells where fewer than 10% of the identified phosphorylation sites were found to be responsive to EGF stimulation.4 In order to overcome this limitation in global phosphoproteomic analysis, we have devised a sequential immunoprecipitation (IP) strategy coupled to mass spectrometry (MS ) that builds on a previously described phosphotyrosine-enrichment approach to quantify the mitotic phosphoproteome downstream of EGFRvIII (Fig. 1).5
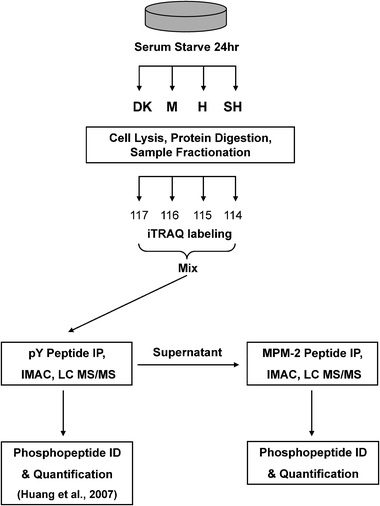 | ||
| Fig. 1 Outline of experimental strategy. U87MG sublines (U87-M, 1.5 × 106 copies/cell; U87-H, 2.0 × 106 copies/cell; U87-SH, 3.0 × 106 copies/cell; U87-DK, 2.0 × 106 inactive copies/cell) were serum starved for 24 h prior to cell lysis and protein digestion. Digested peptides were stable-isotope labeled with the isobaric iTRAQ reagent, mixed and subjected to phosphotyrosine immunoprecipitation (IP) using a pan-specific phosphotyrosine antibody .5 Mitotic phosphopeptides were then immunoprecipitated from the supernatant with the MPM-2 antibody . Eluted phosphopeptides were further enriched with immobilized metal affinity chromatography (IMAC) prior to liquid chromatography tandem mass spectrometry analysis (LC-MS/MS). Phosphopeptide identification (ID) and quantification was performed as described in the methods. | ||
To access the subset of phosphoserine and phosphothreonine modifications in the mitotic compartment, we have employed MPM-2, a monoclonal antibody derived from mitotic HeLa cell lysates that recognizes a wide variety of mitotic phosphorylated antigens.6 Despite its widespread use in the literature as a marker of serine/threoninephosphorylation in mitotic cells, only a small number of the substrates recognized by MPM-2 have been identified.7,8 Furthermore, only limited characterization of the in vivophosphorylation sites of these substrate proteins has been performed. However, in vitro peptide library screens have shown that the binding specificity of MPM-2 is dominated by the “pS/pT-P” motif commonly propagated by the cyclin-dependent kinases (CDKs) and mitogen-activated protein kinases (MAPKs).9,10
Quantitative phosphoproteomic mass spectrometry offers the ability to analyze the effects of different conditions, treatments, and cell lines on the global phosphorylation-mediated state of intracellular signaling.11,12 In order to obtain mechanistic insight into how changes in phosphorylation affect cell phenotype it is necessary to combine the data from quantitative phosphoproteomics with additional information, including protein sequence surrounding the phosphorylation site. Kinases that generate phosphosites, phosphopeptide-binding domains that use phosphosites as signals to prompt a response, and phosphatases that remove phosphosites are all regulated in part by the amino acid sequence surrounding the phosphorylated residue.13–15 There is a great deal of literature and a number of online resources linking linear amino acid sequence motifs to associated kinases and binding domains.16,17 Here we describe a bioinformatics tool to identify amino acid sequence motifs significantly enriched among the phosphopeptides associated most strongly with various expression levels of EGFRvIII. We anticipate that this new motif information will lead to enhanced mechanistic biological insight by connecting the probed processes to sequence motifs associated with known molecules and molecular functions and by revealing motifs of unknown biological function that can be explored further. We also expect that our new method will prove useful in many other problems of interest in basic cellular biochemistry and in therapeutics discovery applications.
Results and discussion
To characterize the effect of EGFRvIII on the mitotic cellular signaling networks, we have utilized the MPM-2 antibody to enrich for peptides containing sites of serine and threoninephosphorylation from U87MG glioblastoma cell lines with titrated levels of the EGFRvIII. A previous phosphoproteomic study of EGFRvIII receptor-mediated signaling has determined the effect of titrating EGFRvIII receptor levels on phosphotyrosine-driven networks.5 We now build on those foundational findings by investigating a key subset of serine/threonine substrate phosphorylation sites upregulated by EGFRvIII expression in this same battery of cell lines . Since this study is focused on signaling downstream of the EGFRvIII receptor, cells were subjected to serum starvation prior to analysis to minimize any confounding signaling events that may arise from components in serum and cell culture media. After depleting phosphotyrosine-containing peptides using the pan-specific phosphotyrosine antibody PY100, the iTRAQ-labeled supernatant was subjected to a subsequent immunoprecipitation using the MPM-2 monoclonal antibody (Fig. 1). Peptides eluted from the MPM-2 IP were further enriched for phosphopeptides using immobilized metal affinity chromatography (IMAC) prior to liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. Two biological replicates were performed, resulting in the identification and quantification of 87 unique sites of phosphorylation on 68 phosphopeptides (58 proteins). Of these sites, 11 were found to be novel (Supplementary Table S1† ) with respect to the resources Phospho.ELM,16 PHOSIDA,18 PhosphoSitePlus (www.phosphosite.org), and a recent study of mitotic phosphoproteins.19 Three of the sites have not been detected previously in humans, but only in homologousproteins.This phosphoproteomic analysis is, to our knowledge, the most extensive characterization of MPM-2 substrates to date. Our present study is also distinct from previous MPM-2 proteomic analyses in that our MS analysis provides quantitative information on in vivo MPM-2 substrates with site-specific resolution. A previous IVEC screen to identify MPM-2 substrates in Xenopus embryo extracts was performed by Stukenberg et al. and identified 20 candidate proteins that underwent mitotic phosphorylation.7 More recently, a proteomic study of MPM-2 substrates performed using 2D gel electrophoresis identified 22 MPM-2 candidate substrate proteins.8 Strikingly, there is no overlap between the proteins identified in these two studies and our current analysis. The lack of similarity in the datasets is likely due to context-dependent variation, including the use of different cell lines and chemically-induced cell cycle synchronization or mitotic activation in previous studies,7,8 compared to asynchronously cycling EGFRvIII-expressing cells in the current study. Additionally, we have performed substrate isolation using solution-based peptide IP coupled to mass spectrometry, an approach that may yield different substrates from the cDNA screens and 2D gel electrophoresis analysis carried out in the two prior studies.
Ectopic expression of EGFRvIII in U87MG cells results in an increased proliferation rate and a larger G2-M cell population under serum depravation conditions.2,20 Consistent with the well-recognized binding affinity of MPM-2 to phosphoproteins in mitotic cells, we observe that phosphorylation of the established proliferation markers Ki-67 and MCM3 (minichromosome maintenance protein 3) were upregulated 1.4-fold and 2.7-fold respectively in the U87-H subline, which expresses a high level of EGFRvIII, compared to the U87-DK kinase-dead control cells.21,22 It has previously been demonstrated that EGFRvIII downregulates p27 expression via activation of the PI3K pathway, resulting in an increase in CDK2-cyclin activity and Rb (retinoblastoma protein) hyperphosphorylation, allowing cells to enter the cell cycle.2 In line with this result, we also observe that phosphorylation of Rb (S608 & S612) and the Rb family member p107 (T385) increase more than 2-fold in the U87-H cells compared to the control U87-DK cells. These phosphorylation sites directly precede proline residues, a characteristic motif recognized by proline-directed kinases such as the CDKs.23
Of the 58 proteins identified in this study, only 8 are annotated in the Gene Ontology database24 as having a role in the cell cycle. It is surprising that only 15% of the phosphopeptides immunoprecipitated by MPM-2 have a previous association with cell cycle, especially given that MPM-2 is considered to specifically recognize substrates in proliferating cells and mitotic cell lysates.6 Nonetheless, those proteins that are labeled as having the GO process annotation term ‘cell cycle’ are enriched (p-value of 0.01) in the subgroup of peptides whose phosphorylation level is upregulated is in the top quartile in the U87-H cell line as compared to the control U87-DK cells.
Intriguingly, only 59 of the 87 phosphorylation events identified in this study were on a serine or threonine residue followed by a proline (Supplementary Table S1,†Fig. 2A). Of the 28 remaining sites, 20 had an aspartic or glutamic acid in the ‘+1’ position, directly to the C-terminal side of the phosphorylated residue. Moreover, 16 of the 68 phosphopeptides identified in the MS study contained at least one “pS/pT-D/E” site, and no “pS/pT-P” site, demonstrating that a large fraction of the acid-directed sites were specifically recognized by the MPM-2 antibody , and were not merely neighbors to proline-directed sites on the same peptide. To ensure that this surprising departure from the canonical MPM-2 epitope was not a byproduct of non-specific binding, a degenerate peptide library experiment was performed to determine whether non-“pS-P” containing motifs could be directly recognized by the MPM-2 antibody . Peptide libraries were synthesized on a cellulose membrane and immunoblotted with MPM-2 to discover the in vitro affinity of MPM-2 for positional dependence and amino acid composition of favorable motifs (Fig. 2). Importantly, due to the prevalence of the “pS-P” motif in MPM-2 literature, all libraries degenerate at the +1 position excluded proline in the +1 position, in an attempt to minimize the effect of what might be a dominant interaction. The results show that MPM-2 binds directly to peptides containing acidic residues in the +1 position, as well as to peptides containing proline, at levels significantly above the background. In fact, the in vitro binding of MPM-2 to the “pS-E” and “pS-D” libraries is higher than to “pS-P”.
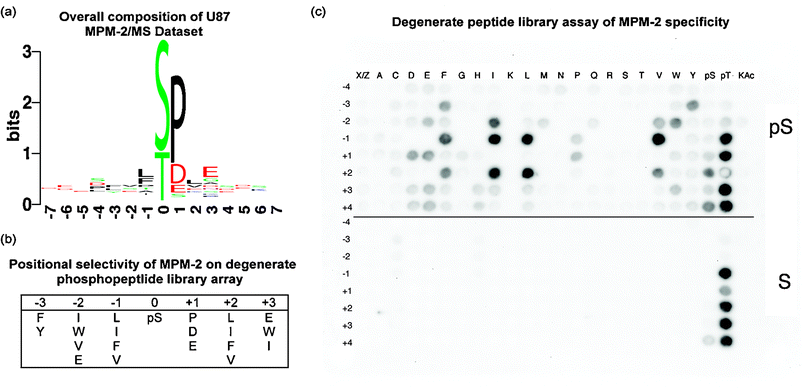 | ||
| Fig. 2 Specificity of the MPM-2 antibody . (a) Motif logo of mass spec dataset.47 The height of each amino acid represents its frequency at that position, and the total stack height of a position represents total conservation. (b) Positional selectivity of two-fold or greater (see Methods) of MPM-2 for residues surrounding a phosphorylated residue according to the results of a degenerate peptide library screen. (c) A degenerate library screen of MPM-2 selectivity. The blot is composed of two sections, the top-half contains a phosphoserine-oriented library and the bottom half is a serine-oriented control library. On each spot is an entire degenerate library oriented on the central serine residue, i.e. X-X-X-X-pS/S-Z-X-X-X, where X represents all naturally occurring amino acids except cysteine and Z additionally excludes proline. A second position, indicated by the row numbering position with respect to the orienting pSer or Ser residue, is fixed to a particular residue, indicated by column-wise position. All natural amino acids, as well as phosphoserine, phosphothreonine, and acetylated lysine, were tested for their contribution to MPM-2 recognition. Phosphoblot quantitation can be found in Supplementary Table S4.† | ||
The largest positional variance occurs in the −1 and +2 positions, where the aliphatic and aromatic amino acids, I, L, F, and V increase the affinity for MPM-2 significantly over pS alone. These results are in very good agreement with two previous degenerate peptide studies that also found aliphatic and aromatic dependence in the −1 and + 2 positions.9,10 In comparison with these previous studies, the most significant difference seen in this study is the preference for acidic residues in the +1 position in addition to the canonical pS-P. This acidic motif was obscured in one of the previous studies because Yaffe et al. chose to fix both the pS and a +1 proline in response to an initial screen that showed heavy +1 proline selectivity,10 although significant acidic residue preference was also detected in this initial screen (data not shown). The experiment by Rodriguez et al. did allow for degeneracy in the +1 position,9 yet only detected a preference for glycine and proline in this position. It is possible that the discrepancy between this study and our current results may be due to a mixture of pS and pT in the oriented position in the Rodriguez et al. screen, especially given that, as shown in Fig. 2C, phosphothreonine, but not phosphoserine, alone in many positions is sufficient to bind to MPM-2 in a negative control library with a fixed, oriented non-phosphorylated serine residue.
The selectivity of MPM-2 for pT is also evident in our MS phosphoproteomics data set, where 33% of the phosphorylated sites discovered in this study are phosphothreonines, a 3-fold increase over the previously reported pT:pS ratio in the phosphoproteome.3 This data also demonstrates that MPM-2 strongly favors pT followed by proline in the +1 position, as 87% of the pT sites enriched by MPM-2 match this “pT-P” motif. To gauge the specificity of MPM-2 for pT and surrounding amino acids, we compared our results to the general composition of the human phosphoproteome to date, as represented by the Phospho.ELM database25 (version 6.0) of identified proteinphosphorylation sites. By comparison, pT represents approximately 14% of the known human phosphoproteome, and 44% of these sites are have a proline in the +1 position. Since the phosphoproteome is still largely uncharacterized, it is difficult to predict whether this percentage is reflective of the true biological composition of pT sites, or whether it is the result of study bias. However, it is clear that there is enrichment for “pT-P” phosphorylation sites in our data set beyond that which can be accounted for in the known human phosphoproteome, indicating good agreement for phosphothreonine with the canonical “pS/pT-P” MPM-2 epitope.
To highlight the effect of EGFRvIII on mitotic regulatory networks, we performed a motif enrichment analysis (see Methods) on the sequence surrounding the mapped phosphorylation sites in the peptides captured by the MPM-2 antibody and upregulated in the top quartile of all detected phosphosites in cells expressing either a medium, high, or super-high level of EGFRvIII relative to the kinase-dead negative control. Surprisingly, the motifs enriched in the EGFR expressing cells did not contain the C-terminal proline corresponding to the generally accepted specificity of the MPM-2 antibody .7,10,26 Though the motif “pS/pT-P” was present in about half of the phosphopeptides in the top quartile for each of the U87-M, U87-H, and U87-SH cell lines , it was present in a higher fraction (over two-thirds) of the total data, and was therefore not enriched among sites upregulated downstream of EGFRvIII signaling and captured by MPM-2. Instead, a number of acid-directed motifs were found significantly enriched among EGFRvIII-regulated phosphosites (Table 1 for U87-H cells, Supplementary Tables S2 and S3 for U87-M and U87-SH cells† ). Motifs were found containing aspartic acid, glutamic acid, or both, at positions −5, −2, +1, +3, and +5 relative to the phosphorylated residue.
| Motifa | Motif in foreground | Motif in background | Foreground size | Background size | Statistical significance |
|---|---|---|---|---|---|
| a “s” = pS; “x” = pS/pT, “.” = Any amino acid, “–” = D/E, “O” = M/I/L/V. | |||||
| D.x | 8 | 10 | 25 | 95 | 2.73 × 10−4 |
| –.x | 12 | 20 | 25 | 95 | 3.27 × 10−4 |
| –.s | 10 | 15 | 25 | 95 | 3.98 × 10−4 |
| –.x– | 8 | 11 | 25 | 95 | 8.24 × 10−4 |
| –.s....E | 5 | 5 | 25 | 95 | 9.17 × 10−4 |
| –..D.x | 5 | 5 | 25 | 95 | 9.17 × 10−4 |
| –.s–.– | 7 | 9 | 25 | 95 | 1.05 × 10−3 |
| –.s....– | 6 | 7 | 25 | 95 | 1.17 × 10−3 |
| D.x– | 6 | 7 | 25 | 95 | 1.17 × 10−3 |
| x– | 12 | 23 | 25 | 95 | 2.09 × 10−3 |
| D.s | 6 | 8 | 25 | 95 | 3.80 × 10−3 |
| –.s–.E | 6 | 8 | 25 | 95 | 3.80 × 10−3 |
| –..–.x | 6 | 8 | 25 | 95 | 3.80 × 10−3 |
| –.xD | 6 | 8 | 25 | 95 | 3.80 × 10−3 |
| –.sD.E.E | 4 | 4 | 25 | 95 | 3.97 × 10−3 |
| –.s.L..– | 4 | 4 | 25 | 95 | 3.97 × 10−3 |
| sD.–.–O | 4 | 4 | 25 | 95 | 3.97 × 10−3 |
| –..D.x– | 4 | 4 | 25 | 95 | 3.97 × 10−3 |
| D.xD | 4 | 4 | 25 | 95 | 3.97 × 10−3 |
| –...xP..S | 4 | 4 | 25 | 95 | 3.97 × 10−3 |
| D.s–.E | 5 | 6 | 25 | 95 | 4.48 × 10−3 |
| –.sD.–.– | 5 | 6 | 25 | 95 | 4.48 × 10−3 |
| –.s–L– | 5 | 6 | 25 | 95 | 4.48 × 10−3 |
| xD.E.E | 5 | 6 | 25 | 95 | 4.48 × 10−3 |
| s– | 10 | 19 | 25 | 95 | 5.81 × 10−3 |
| xD.–.– | 7 | 11 | 25 | 95 | 6.47 × 10−3 |
| –.s.L | 6 | 9 | 25 | 95 | 9.30 × 10−3 |
| s–.– | 9 | 17 | 25 | 95 | 9.30 × 10−3 |
| sD.–.– | 6 | 9 | 25 | 95 | 9.30 × 10−3 |
| –..x | 6 | 9 | 25 | 95 | 9.30 × 10−3 |
| x–.– | 10 | 20 | 25 | 95 | 9.60 × 10−3 |
| xP | 13 | 64 | 25 | 95 | 0.983 |
The rate of false positive motif discovery must be considered because the statistical significance of a large number of amino acid sequence motifs has been calculated in this study. Since the statistical significances of sequence motifs are interdependent, the traditional straightforward Bonferroni false positive correction is unfortunately inappropriate. Therefore, to approach the question of whether the sequence motif enrichments we observed might be spurious false positives, we took an empirical approach. We generated 1000 random foregrounds of 25 phosphosites (corresponding in size to our foregrounds of interest), and tabulated the number of detected enriched motifs in each, as well as the statistical significance of the most significant discovered motif. Only 6.8%, 1%, and 2%, respectively, of random foregrounds have as many statistically significant (p < 0.01) motifs identified as the foregrounds built from the top quartile of sites in U87-M, U87-H, or U87-SH cells relative to the U87-DK control (Fig. 3A). Moreover, only 5.6%, 6%, and 6% of random foregrounds have a motif with a statistical significance as significant as the most significant motif found among the top quartile of phosphosites in U87-M, U87-H, or U87-SH cells, respectively, as compared to U87-DK controls (Fig. 3B). Finally, 2.6%, 0.8%, and 1.5% of random foreground datasets have both as many motifs and as significant a strongest motif as foregrounds generated from each of the three EGFRvIII-expressing cell lines—U87-M, U87H, and U87-SH. Taken together, these empirical metrics indicate that our motif analyses are identifying a biologically relevant phenomenon.
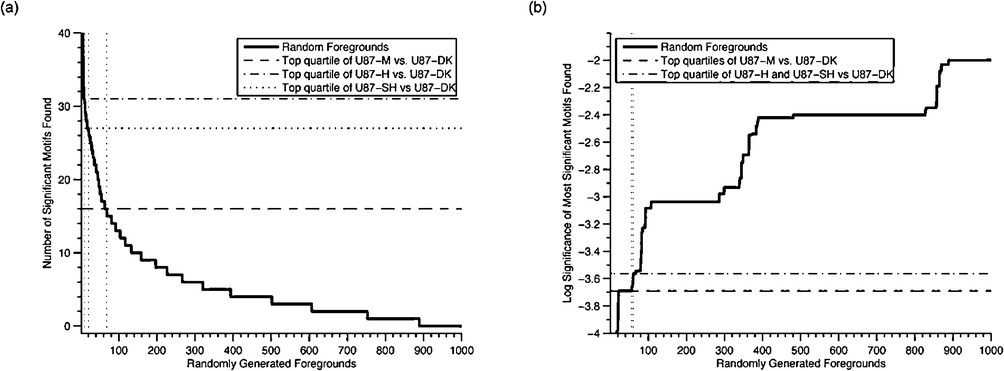 | ||
| Fig. 3 Comparison of motif enrichment analyses of regulated data with motif enrichment analyses of randomly selected phosphosites. Motif enrichment analyses of the top quartile of upregulated phosphosites in cells expressing medium, high, and super-high amounts of EGFRvIII versus a kinase dead control were compared to 1000 analyses of a randomly selected quarter of phosphosites. (a) Comparison of the statistical significance of the most significant motif found in each analysis. (b) Comparison of the number of motifs found in each analysis. Vertical lines indicate the number of random foregrounds with a metric value the same as, or more extreme than, the values for the MS-motivated foregrounds. | ||
Conclusions
Discovery of an antibody epitope through MS measurement revealed biologically relevant substrates and non-canonical selectivity for a well-studied biological probe. The previously-established belief that MPM-2 binds only to “pS/pT-P” sites was most likely derived from inadequate substrate-detection sensitivity, with earlier methods capable of only picking up the most abundant of MPM-2’s substrates: “pS-P” sites. With contemporary state-of-art peptide IP and MS peptide identification, we have now been able to complement the traditional in vitro affinity assay with in vivo biological substrates for MPM-2, thereby providing positional selectivity for cellular substrates. For example, of the singly phosphorylated peptides , 98% conform to selectivity for P/E/D in the +1 position. On average, each of these phosphopeptides conformed to three positions of selectivity defined in Fig. 2B, and all conformed to at least one. The range of conformity among the sequence positions varied greatly, with the −1 positional selectivity ranking second to the +1 position, whereas the −2 position contributed to only about 30% of the peptides identified. Of the peptides that conform to the −1 positional selectivity of MPM-2 for isoleucine, leucine, phenylalanine, and valine, 90% of the MS-identified singly-phosphorylated substrates contain either a leucine or a phenylalanine, while no peptides contain valine, although there are known “V-pS/pT” and “V-pS/pT-P” sites listed in the Phospho.ELM25 database. This observation demonstrates the difference between favorable in vitro degenerate library interactions and actual biological substrates. The most likely explanation for this difference is that proteinsin vivo must be optimized for both kinase-dependent phosphorylation and for MPM-2 binding, while in vitro MPM-2 binding is not restricted by kinase-dependent phosphorylation.The set of peptides identified by an MS experiment arises from a convolution of many factors. In order to appear in an IP/MS experiment, a peptide must be present in the cell, have sufficient affinity for the antibody , be compatible with any further purification, and be efficiently measured and sequenced viaMS . This convolution of effects makes it difficult to perform motif-enrichment analysis of MS data. To address these issues, we have developed a tool to identify amino acid sequence motifs enriched in a regulated subset of a larger dataset. By comparing a biologically interesting subset of an MS dataset to the entire MS dataset, many of the confounding elements are cancelled. Most existing motif identification tools, such TEIRESIAS,27EMOTIF,28 and PRATT,29 solve a fundamentally different problem: motif identification without regard to a biologically relevant background. In contrast, our method compares a list of phosphopeptides of interest to a proteomic background, and is therefore similar to the MOTIF-X method of Schwartz and Gygi.30 Although similar, these methods differ in several respects important to the goals of this study. First, and most importantly, our method uses the full cohort of peptides identified in the MS study as a comparative background. This dataset provides the most specific set of peptides possible, thereby effectively minimizing all influences except the biological regulation of interest. Second, our method allows each peptide to be associated with any number of enriched motifs by searching over individual and pairs of amino acid positions, expanding the space of motifs that may be found at a manageable computational expense. Third, statistical significance of enrichment is calculated by using the exact hypergeometric probability function, which is more appropriate to the relatively small background data sets of current interest than the binomial approximation. Finally, an empirical analysis of randomly selected data provides some indication that foregrounds generated from upregulated phosphosites within this dataset have meaningful arrays of enriched amino acid sequence motifs.
A very recent study by Dephoure et al. provides a great number of novel phosphosites upregulated in mitotic cells relative to an asynchronous control.19MOTIF-X30 was used to compare phosphoserine sites upregulated in mitotic cells to all serines in the human proteome. This comparison revealed four motifs that the authors associated with CDKs, two that the authors associated with the kinases Aurora A and Plk1, and two that the authors felt indicated uncharacterized mitotic kinase function. It is worth noting that the motifs we have discovered here do not correspond well to any of the motifs described by Dephoure et al. This disparity in findings is likely due to the targeted nature of our study: we have examined only phosphopeptides captured by the antibody MPM-2, and focused on those motifs that correspond explicitly to increased levels of EGFRvIII expression.
Although the accepted “pS/pT-P” epitope of MPM-2 is most often generated by the cell-cycle dependent kinases31 and the MAP kinases,32 a number of other kinases including Plk1, NIMA, and MEK are known to generate MPM-2 epitopes in at least one substrate,33 but not necessarily in a manner that matches the motifs found in this study. The motifs we identify here as being associated with EGFRvIII upregulation in U87 glioblastoma cells are most reminiscent of the specificity of casein kinase II, with acidic amino acids prevalent in the −2, +1, +3 and +5 positions, corresponding well to the substrate specificity of CK2 for serine and threonine residues with nearby negative amino acids, with the +1, +2, and particularly the +3 residues most often acidic.34,35 The presence of acidic residues in the + 1 position, in particular, seems to indicate that the known acidophilic mitotic kinase, Plk1, is not wholly responsible, as Plk1 substrates most often have an aliphatic residue in the +1 position.36 In fact, one of the phosphorylated sites identified in this study, serine 366 on the RNA-binding protein La, is a known CK2 substrate.37 The idea of a role for CK2 in mitosis is not a new one. Though thought to be constitutively active, CK2 is known to be required in the G2/M portion of the S. cerevisiaecell cycle,38 as well as to promote the meiosis of Xenopusoocytes.39 Specific roles have been found for CK2 in the activation of the cell-cycle regulatory phosphatase CDC25B,40 and at a mitosis-specific site on DNA topoisomerase IIα; in fact, a pair of studies has identified casein kinase II as being capable of generating an MPM-2 epitope on DNA topoisomerase IIα.41,42 The amino acid sequence surrounding the phosphorylated residue, “NRRKRKPpSTSDDSDS” contains the common +3 acid aspect of motifs found enriched in MPM-2-captured, EGFRvIII-regulated sites in our data, although it does not match directly to any of the motifs we have identified. CK2 activity has also been found to be upregulated in a large number of cancers (reviewed in ref. 43 and 44). While the similarity of the motifs we have identified to the known substrate specificity of CK2 is striking, it is similarly possible that there is another acidophilic kinase with an undescribed motif specificity and uncharacterized role in mitosis. In fact, the artificial neural network analysis available through the recently published NetPhorest algorithm provides the following consensus CK2 motif: E-E-E-(E/D/S)-(E/D)-S/T-(D/E/S/G)-(D/E/S)-(E/D)-(E/D)-(E/D)-(E/D)-E, with weak selectivity in italics, moderate selectivity in normal text, and strong selectivity in bold.45. Although this motif does not exactly match to any of the motifs found in this study, the panel of motifs identified here is a combination of kinasephosphorylation specificity, antibody binding specificity, and regulation downstream of EGFRvIII. The motifs described here, therefore, likely represent only a subset of the kinase recognition motif. Regardless of the kinase responsible, we have identified a set of motifs that are upregulated in a manner coordinated with the expression level of EGFRvIII in glioblastoma cell lines . EGFRvIII expression itself correlates with poor prognosis in GBM patients,1 therefore understanding the generation and regulation of these motifs may lead to an improved understanding of glioblastoma and to improvements in its treatment.
Similar analyses can be applied to other diseases and disorders. We hypothesize that identified motifs will be a signature for kinases, phosphopeptide-binding domains, and perhaps phosphatases, associated with the particular regulation being analyzed. Properly interpreted, these motifs may provide mechanistic insight into the origin and phenotype of the samples studied. This information may aid in determining systematic signaling differences among existing cell lines , thereby enabling mechanistic hypotheses with respect to treatment. Ultimately, it should be possible to apply these methods to characterize individual patient tumors, as differentiated from adjacent healthy tissue, with the goal of discovering hidden dysregulated signaling modules that could be effective therapeutic targets.
Experimental
Cell culture and retrovirus infection
For this study, U87MG glioblastoma cells lines were transfected with EGFRvIII and sorted to generate sublines that express the following mean receptor levels, U87-M (1.5 × 106 copies/cell), U87-H (2.0 × 106 copies/cell) and U87-SH (3.0 × 106 copies/cell). In addition, we have included the U87-DK subline that expresses 2.0 × 106 copies/cell of a kinase-dead version of the EGFRvIII receptor as a negative control for its activity.5,20Cell lines were cultured in DMEM with 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, and 100 mg ml−1streptomycin in 95% air/5% CO2 atmosphere at 37 °C. U87MG cells expressing EGFRvIII or DK receptors were selected in 400 μg ml−1 G418. For U87MG cells expressing titrated levels of EGFRvIII, a bulk population of cells was prepared by retroviral transduction with pLERNL and stained as previously described5 with an anti-EGFR monoclonal antibody Ab-1 (clone 528; Oncogene Science, Cambridge, MA), followed by fluorescein isothiocyanate-conjugated goat anti-mouse Ig antibody (PharMingen, Minneapolis, MN) and sorted for medium (1.5 × 106 receptors, U87-M), high (2.0 × 106 receptors, U87-H), and superhigh (3.0 × 106 receptors, U87-SH) receptor amounts. For this procedure, U87-EGFRvIII cells engineered previously and determined to express 2 × 106 receptors per cell were used as a gating control. The sorted cells were then maintained in culture and receptor levels were analyzed again by flow cytometry prior to experimental use.Cell lysis, protein digestion and peptide fractionation
U87MG cells were maintained in DMEM medium supplemented with 10% FBS. 1.5 × 106 cells per 10 cm plate were seeded for 24 h, then washed with PBS and incubated for 24 h in serum-free media. Cells were lysed in 1 ml of 8 M urea. For each of the two biological replicates performed, lysate from three 10 cm plates were pooled together. Cells were reduced with 10 mM DTT for 1 h at 56 °C, alkylated with 55 mM iodoacetamide for 45 min at room temperature, and diluted to 12 ml with 100 mM ammonium acetate, pH 8.9, prior to digestion with 40 μg of trypsin (Promega). The lysates were digested overnight at room temperature. Digested lysate were acidified to pH 3 with acetic acid and loaded onto a C18 Sep-Pak Plus cartridge (Waters). The peptides were desalted (10 ml 0.1% acetic acid) and eluted with 10 ml of a solution of 25% acetonitrile and 0.1% acetic acid. Each sample was divided into 5 aliquots and lyophilized to dryness.iTRAQ labeling of peptides and immunoprecipitation
Lyophilized peptides were subjected to labeling with the iTRAQ 4-plex reagent (Applied Biosystems). Each aliquot of peptides was dissolved in 30 μl of 0.5 M triethylammonium bicarbonate, pH 8.5 and reacted with two tubes of iTRAQ reagent (dissolved in 70 μl of ethanol each). The reagents for each of the conditions used were, iTRAQ-114 (U87-DK), iTRAQ-115 (U87-M), iTRAQ-116 (U87-H) and iTRAQ-117 (U87-SH). The mixture was incubated at room temperature for 50 min and then concentrated to 30 μl. The four different isotopically labeled samples were combined and acidified with 360 μl of 0.1% acetic acid and then reduced to dryness.The combined sample was reconstituted with 150 μl of IP buffer (100 mM Tris, 100 mM NaCl, 1% NP-40, pH 7.4), 300 μl of water and the pH was adjusted to 7.4. After immunoprecipitation with pTyr100 (Cell Signaling Technology, Beverly, MA) for the phosphotyrosine-containing peptides , which were used in a prior study,5 the supernatant was incubated with 10 μg of protein G Plus-agarose beads (Calbiochem) and 12 μg of MPM-2 antibody (Upstate) for 8 h at 4 °C. Phosphopeptides were washed and eluted as previously described.5.
Immobilized metal affinity chromatography (IMAC) and mass spectrometry
Immobilized metal affinity chromatography (IMAC) was performed to enrich for phosphorylated peptides and remove non-specifically retained non-phosphorylated peptides . Eluted peptides were loaded onto a 10 cm self-packed IMAC (20MC, Applied Biosystems) capillary column (200 μm ID, 360 μm OD), and rinsed with organic rinse solution (25% MeCN, 1% HOAc, 100 mM NaCl) for 10 min at 10 μl min−1. The column was then equilibrated with 0.1% HOAc for 10 min at 10 μl min−1 and then eluted onto a 10 cm self-packed C18 (YMC-Waters 10 × 5 μm) precolumn (100 μm ID, 360 μm OD) with 50 μl of 250 mM Na2HPO4, pH 8.0. After a 10 min rinse with 0.1% HOAc, the precolumn was connected to a 10 cm self-packed C18 (YMC-Waters 5 μm ODS-AQ) analytical capillary column (50 μm ID, 360 μm OD) with an integrated electrospray tip (1 μm orifice). Peptides were eluted with a 125 min gradient with solvents A (1% HOAC) and B (70% MeCN in 1% OHAc): 10 min from 0% to 13%, 95 min from 13% to 42%, 10 min from 42% to 60% and 10 min from 60% to 100%. Eluted peptides were directly electrosprayed into a QqTof mass spectrometer (QSTAR XL Pro, Applied Biosystems). MS/MS spectra of the five most intense peaks with 2–5 charge states in the full MS scan were automatically acquired in information-dependent acquisition mode with previously selected peaks excluded for 40 s.Phosphopeptide sequencing, quantification and clustering
MS/MS spectra were extracted and searched using MASCOT (Matrix Science). For MASCOT, data was searched against the human non-redundant protein database with trypsin specificity, 2 missed cleavages, precursor mass tolerance of 2.2 amu for the precursor ion and 0.15 for the fragment ion tolerance. Phosphorylation sites and peptide sequence assignments were validated and quantified by manual confirmation of raw MS/MS data (raw MS/MS data available at http://web.mit.edu/fwhitelab/data/index.html). Peak areas of iTRAQ marker ions (m/z 114, 115, 116 and 117) were obtained and corrected according to manufacturer’s instructions to account for isotopic overlap. The quantified data was then normalized with values from the iTRAQ marker ion peak areas of non-phosphorylated peptides in the supernatant of the immunoprecipitation (used as a loading control to account for possible variation in the starting amount of sample for each condition). Each condition was normalized against the U87-H cell line to obtain fold changes across all 4 conditions.Phosphopeptide library array
Methods used here are similar to Elia et al.46 An ABIMED peptide arrayer was used to synthesize degenerate libraries on an amino-PEG cellulose membrane. The libraries consisted of four degenerate positions each on the N- and C-terminal sides of the central phosphoserine or serine positions, i.e. X-X-X-X-pS/S-Z-X-X-X, where X represents all naturally occurring amino acids except cysteine and Z additionally excludes proline. On most spots, one of the degenerate positions was fixed as a specific amino acid in addition to the orienting phosphoserine or serine. The cellulose membrane was blocked for 1.5 h at room temperature in 3% milk and 1% TBS-T at pH 7.4. It was then incubated with the primary antibody , MPM-2 (Upstate), at room temperature for 1.5 h at 0.5 μg/mL in 1% TBS-T. The membrane was washed, blocked, and then probed with a secondary anti-mouse HRP-conjugated antibody (GE Healthcare) overnight at 4 °C. MPM-2 library binding was detected using enhanced chemiluminescence , imaged and quantified using a Kodak Image Station.Determination of MPM-2 selectivity
The quantified phosphoserine-oriented peptide library was normalized to the serine-oriented control library. Each fixed position was then normalized to the average of the completely degenerate column (column 1) to determine total selectivity of that fixed position over pS alone. Quantification and normalization are provided in Supplementary Table S4.†Preparation of data for motif enrichment analysis
Of the 68 phosphopeptides identified in this study, 44 are singly phosphorylated, 21 are doubly phosphorylated, and 3 are phosphorylated on three amino acids. We therefore expanded the dataset to include the full complement of individual phosphosites, each centered on a single phosphorylation. For sites quantified more than once in the context of different phosphopeptides with different partner residues simultaneously phosphorylated, we included each instance of the site, for a total of 95. In three instances, the exact residue of phosphorylation could not be determined, and a choice between two possible sites was made arbitrarily (see Supplementary Table S1† ). Analysis was repeated with all 8 possible selections of the identities of these three sites, with no significant qualitative effect on the results (data not shown). We expanded each site to include the 7 amino acids N-terminal and 7 amino acids C-terminal of the phosphorylated residue using the Entrez Protein database. For each of the three cell lines expressing medium, high, or super-high levels of EGFRvIII, we identified the top quartile of phosphosites upregulated relative to kinase-dead U87-DK control cells. For U87-M, U87-H, and U87-SH cells, this corresponded to 1.67-fold, 2.10-fold, and 2.20-fold enrichment, respectively, and 25 sites. The 24th- and 25th- most upregulated sites in each cell line came from the same phosphopeptide and had identical quantification.Enriched motif search
The space of possible motifs in a 15-mer peptide containing a central fixed phosphoresidue is enormous—over 2.1 × 1019, if a few combinations of chemically similar amino acids are allowed for. A strategy must therefore be used for restricting the search to those motifs most likely to be significantly enriched in the regulated data of interest. For each of phosphoserine, phosphothreonine, and the combination of the two, the significance of enrichment in the regulated data relative to the full background was calculated (see Motif significance calculation, below) for every motif that can be created by fixing any one or two of the seven positions on either side of the phosphorylated residue as any of the twenty amino acids, as the combination of the basic amino acidsarginine and lysine, as the combination of the acidic amino acidsglutamate and aspartate, or as the combination of the hydrophobic amino acidsleucine, isoleucine, valine and methionine. For each motif identified with an enrichment significance of 0.01 or less that appeared 3 or more times in the EGFRvIII-regulated foreground data, the significance of all motifs that can be created by fixing the identities of any further one or two amino acids was calculated. This procedure was recursed until no further significant motifs were found.Motif significance calculation
For each motif of potential interest, the statistical significance of enrichment of that motif in the EGFRvIII-regulated foreground subset of the total background data was calculated using the hypergeometric distribution:where N is the number of phosphosites in the full dataset, n is the number of sites in the EGFRvIII-regulated subset, m is the number of motif sites in the full data, and k is the number of motif sites in the regulated data subset. This corresponds exactly to the probability of seeing as many instances or more of the motif as are seen in the EGFRvIII-regulated dataset by chance if drawing a dataset the same size as the regulated data randomly from the full dataset.
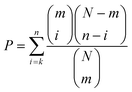
Empirical analysis of false positive rate
To characterize the rate of false positive motif discovery, 1000 foreground sets of 25 phosphosites (corresponding in size to the top-quartile datasets studied) were randomly generated, and motif enrichment analysis was performed on each. The number of motifs found and the significance of the most significantly enriched motif for each were tabulated and compared to the same statistics for the foreground datasets of interest.| GBM | Glioblastoma multiforme |
| EGFR | epidermal growth factor receptor |
| MPM-2 | mitotic protein monoclonal #2 |
| CK2 | casein kinase II |
| CDK | cyclin-dependent kinase |
| PI3K | phosphatidylinositol 3-kinase |
| RTK | receptor tyrosine kinase |
| IP | immunoprecipitation |
| MS | mass spectrometry |
Acknowledgements
The authors thank members of the White, Yaffe, and Lauffenburger groups for helpful discussion, and Webster Cavenee and Frank Furnari (UCSD) for providing U87MG cells and sublines. This work was partially supported by NCI Integrative Cancer Biology Program grant U54-CA112967, NCI Bioengineering Research Partnership grant R01-CA96504 and by NIH grant R01-GM60594.References
- F. B. Furnari, T. Fenton, R. M. Bachoo, A. Mukasa, J. M. Stommel, A. Stegh, W. C. Hahn, K. L. Ligon, D. N. Louis, C. Brennan, L. Chin, R. A. DePinho and W. K. Cavenee, Genes Dev., 2007, 21, 2683–2710 CrossRef CAS.
- Y. Narita, M. Nagane, K. Mishima, H. J. Huang, F. B. Furnari and W. K. Cavenee, Cancer Res., 2002, 62, 6764–6769 CAS.
- T. Hunter and B. M. Sefton, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 1311–1315 CrossRef CAS.
- J. V. Olsen, B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen and M. Mann, Cell, 2006, 127, 635–648 CrossRef CAS.
- P. H. Huang, A. Mukasa, R. Bonavia, R. A. Flynn, Z. E. Brewer, W. K. Cavenee, F. B. Furnari and F. M. White, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12867–12872 CrossRef CAS.
- F. M. Davis, T. Y. Tsao, S. K. Fowler and P. N. Rao, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 2926–2930 CrossRef CAS.
- P. T. Stukenberg, K. D. Lustig, T. J. McGarry, R. W. King, J. Kuang and M. W. Kirschner, Curr. Biol., 1997, 7, 338–348 CrossRef CAS.
- M. Xiang, C. Xue, L. Huicai, L. Jin, L. Hong and H. Dacheng, Biochim. Biophys. Acta, 2008, 1784, 882–890.
- M. Rodriguez, S. S. Li, J. W. Harper and Z. Songyang, J. Biol. Chem., 2004, 279, 8802–8807 CrossRef CAS.
- M. B. Yaffe, M. Schutkowski, M. Shen, X. Z. Zhou, P. T. Stukenberg, J. U. Rahfeld, J. Xu, J. Kuang, M. W. Kirschner, G. Fischer, L. C. Cantley and K. P. Lu, Science, 1997, 278, 1957–1960 CrossRef CAS.
- K. Schmelzle and F. M. White, Curr. Opin. Biotechnol., 2006, 17, 406–414 CrossRef CAS.
- Y. Zhang, A. Wolf-Yadlin, P. L. Ross, D. J. Pappin, J. Rush, D. A. Lauffenburger and F. M. White, Mol. Cell. Proteomics, 2005, 4, 1240–1250 CrossRef CAS.
- P. J. Kennelly and E. G. Krebs, J. Biol. Chem., 1991, 266, 15555–15558 CAS.
- M. B. Yaffe and A. E. Elia, Curr. Opin. Cell Biol., 2001, 13, 131–138 CrossRef CAS.
- M. B. Yaffe, Nat. Rev. Mol. Cell Biol., 2002, 3, 177–186 CrossRef CAS.
- R. Amanchy, B. Periaswamy, S. Mathivanan, R. Reddy, S. G. Tattikota and A. Pandey, Nat. Biotechnol., 2007, 25, 285–286 CrossRef CAS.
- J. C. Obenauer, L. C. Cantley and M. B. Yaffe, Nucleic Acids Res., 2003, 31, 3635–3641 CrossRef CAS.
- F. Gnad, S. Ren, J. Cox, J. V. Olsen, B. Macek, M. Oroshi and M. Mann, Genome Biol., 2007, 8, R250 CrossRef.
- N. Dephoure, C. Zhou, J. Villen, S. A. Beausoleil, C. E. Bakalarski, S. J. Elledge and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2008 Search PubMed.
- H. S. Huang, M. Nagane, C. K. Klingbeil, H. Lin, R. Nishikawa, X. D. Ji, C. M. Huang, G. N. Gill, H. S. Wiley and W. K. Cavenee, J. Biol. Chem., 1997, 272, 2927–2935 CrossRef CAS.
- S. A. Ha, S. M. Shin, H. Namkoong, H. Lee, G. W. Cho, S. Y. Hur, T. E. Kim and J. W. Kim, Clin. Cancer Res., 2004, 10, 8386–8395 CrossRef CAS.
- A. Soling, M. Sackewitz, M. Volkmar, D. Schaarschmidt, R. Jacob, H. J. Holzhausen and N. G. Rainov, Clin. Cancer Res., 2005, 11, 249–258.
- R. Linding, L. J. Jensen, G. J. Ostheimer, M. A. van Vugt, C. Jorgensen, I. M. Miron, F. Diella, K. Colwill, L. Taylor, K. Elder, P. Metalnikov, V. Nguyen, A. Pasculescu, J. Jin, J. G. Park, L. D. Samson, J. R. Woodgett, R. B. Russell, P. Bork, M. B. Yaffe and T. Pawson, Cell, 2007, 129, 1415–1426 CrossRef CAS.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS.
- F. Diella, C. M. Gould, C. Chica, A. Via and T. J. Gibson, Nucleic Acids Res., 2008, 36, D240–244 CAS.
- J. M. Westendorf, P. N. Rao and L. Gerace, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 714–718 CrossRef CAS.
- I. Rigoutsos and A. Floratos, Bioinformatics, 1998, 14, 55–67 CrossRef CAS.
- I. Jonassen, J. F. Collins and D. G. Higgins, Protein Sci., 1995, 4, 1587–1595 CrossRef CAS.
- C. G. Nevill-Manning, T. D. Wu and D. L. Brutlag, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5865–5871 CrossRef CAS.
- D. Schwartz and S. P. Gygi, Nat. Biotechnol., 2005, 23, 1391–1398 CrossRef CAS.
- Z. Songyang, S. Blechner, N. Hoagland, M. F. Hoekstra, H. Piwnica-Worms and L. C. Cantley, Curr. Biol., 1994, 4, 973–982 CrossRef CAS.
- F. A. Gonzalez, D. L. Raden and R. J. Davis, J. Biol. Chem., 1991, 266, 22159–22163 CAS.
- A. Kumagai and W. G. Dunphy, Science, 1996, 273, 1377–1380 CrossRef CAS.
- Z. Songyang, K. P. Lu, Y. T. Kwon, L. H. Tsai, O. Filhol, C. Cochet, D. A. Brickey, T. R. Soderling, C. Bartleson, D. J. Graves, A. J. DeMaggio, M. F. Hoekstra, J. Blenis, T. Hunter and L. C. Cantley, Mol. Cell. Biol., 1996, 16, 6486–6493 CAS.
- F. Meggio and L. A. Pinna, FASEB J., 2003, 17, 349–368 CrossRef CAS.
- H. Nakajima, F. Toyoshima-Morimoto, E. Taniguchi and E. Nishida, J. Biol. Chem., 2003, 278, 25277–25280 CrossRef CAS.
- E. I. Schwartz, R. V. Intine and R. J. Maraia, Mol. Cell. Biol., 2004, 24, 9580–9591 CrossRef CAS.
- D. E. Hanna, A. Rethinaswamy and C. V. Glover, J. Biol. Chem., 1995, 270, 25905–25914 CrossRef CAS.
- O. Mulner-Lorillon, J. Marot, X. Cayla, R. Pouhle and R. Belle, Eur. J. Biochem., 1988, 171, 107–117 CrossRef CAS.
- N. Theis-Febvre, O. Filhol, C. Froment, M. Cazales, C. Cochet, B. Monsarrat, B. Ducommun and V. Baldin, Oncogene, 2003, 22, 220–232 CrossRef CAS.
- A. E. Escargueil, S. Y. Plisov, O. Filhol, C. Cochet and A. K. Larsen, J. Biol. Chem., 2000, 275, 34710–34718 CrossRef CAS.
- A. E. Escargueil and A. K. Larsen, Biochem. J., 2007, 403, 235–242 CrossRef CAS.
- K. Ahmed, D. A. Gerber and C. Cochet, Trends Cell Biol., 2002, 12, 226–230 CrossRef CAS.
- S. Sarno and L. A. Pinna, Mol. BioSyst., 2008, 4, 889–894 RSC.
- M. L. Miller, L. J. Jensen, F. Diella, C. Jorgensen, M. Tinti, L. Li, M. Hsiung, S. A. Parker, J. Bordeaux, T. Sicheritz-Ponten, M. Olhovsky, A. Pasculescu, J. Alexander, S. Knapp, N. Blom, P. Bork, S. Li, G. Cesareni, T. Pawson, B. E. Turk, M. B. Yaffe, S. Brunak and R. Linding, Sci. Signal., 2008, 1, ra2 Search PubMed.
- A. E. Elia, P. Rellos, L. F. Haire, J. W. Chao, F. J. Ivins, K. Hoepker, D. Mohammad, L. C. Cantley, S. J. Smerdon and M. B. Yaffe, Cell, 2003, 115, 83–95 CrossRef CAS.
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary tables and supplemental figure S1. See DOI: 10.1039/b815075c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2009 |