Mathematical model of the Lux luminescence system in the terrestrial bacterium Photorhabdus luminescens†
Patricia A.
Welham
* and
Dov J.
Stekel
University of Birmingham, Birmingham, UK B15 2TT. E-mail: PAW631@bham.ac.uk
First published on 4th November 2008
Abstract
A mathematical model of the Lux luminescence system, governed by the operonluxCDABE in the terrestrial bacterium Photorhabdus luminescens, was constructed using a set of coupled ordinary differential equations. This model will have value in the interpretation of Lux data when used as a reporter in time-course gene expression experiments. The system was tested on time series and stationary data from published papers and the model is in good agreement with the published data. Metabolic control analysis demonstrates that control of the system lies mainly with the aldehyde recycling pathway (LuxE and LuxC). The rate at which light is produced in the steady state model shows a low sensitivity to changes in kinetic parameter values to those measured in other species of luminescent bacteria, demonstrating the robustness of the Lux system.
Introduction
Bioluminescent species are widely distributed in nature. It seems that the light-emitting systems employed by the different phylogenetic groups evolved independently—the reactions and structures of the enzymes (luciferases) and substrates (luciferins) involved are very varied. The only common feature is that oxygen is required for the bioluminescent reaction.1Species of luminescent bacteria are found in terrestrial, freshwater and marine environments. Most species belong to the Aliivibrio, Photobacterium or Vibrio genera from the Vibrionaceae family (Gammaproteobacteria).2 The genes which encode the enzymes for the luminescent reactions in prokaryotes are known as luxgenes . Luxgenes are vertically inherited in the majority of these species, but those of Shewanella hanedai and Shewanella woodyi, two members of another Gammaproteobacteria family, are closely related to those of Aliivibrio, suggesting that horizontal gene transfer has occurred in these cases.
Marine species of luminescent bacteria include Vibrio fischeri, Vibrio harveyi and Photobacterium phosphoreum.3,4 The only light-emitting terrestrial bacterium found so far is Photorhabdus luminescens. It was once thought that there was also another terrestrial species, Xenorhabdus luminescens, but since 1996 it has been recognised that these are the same species.5Luxgenes have been cloned and sequenced for three strains of P. luminescens, and the luciferases produced from these genes have been characterized.6–9 The luciferase from P. luminescens has a very high thermal stability (a half life of over three hours at 45 °C), making the luxoperon of this organism a very good choice as a reporter system.7 It was for this reason that P. luminescens was chosen for study.
The operon responsible for the reactions involved in terrestrial bacterial luminescence is luxCDABE6 (Fig. 1). LuxA and luxB encode for the α and β subunits of the luciferase. There is 30% identity between luxA and luxB, indicating gene duplication. It has been suggested that the duplication may have arisen prior to the divergence of the lines leading to present-day luminescent bacteria.10 The active site is thought to be on the α subunit. The LuxAB enzyme catalyses the oxidation of FMNH2 (reduced flavin) and a long chain fatty aldehyde to oxidised flavin (FMN) and a long chain fatty acid respectively. This reaction results in the emission of blue-green light (wavelength 490 nm).
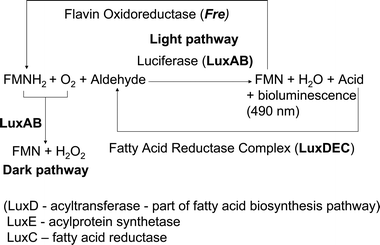 | ||
| Fig. 1 Reactions catalysed by the products of the LuxABCDE operon in Photorhabdus luminescens. In the light pathway the luciferase LuxAB catalyses the oxidation of reduced flavin and an aldehyde to flavin and a long chain fatty acid, resulting in emission of blue-green light. The acid is recycled to aldehyde by the action of the fatty acidreductase complex LuxCDE. Oxidised flavin is reduced by the enzyme Fre. If aldehyde concentration is low or zero the dark pathway is followed, leading to the production of flavin and hydrogen peroxide. | ||
There are two pathways for the luciferase reaction:
The light pathway, in the presence of aldehyde, leads to production of light by the following sequence of steps:
FMNH2 + LuxAB ⇌ LuxAB·FMNH2
LuxAB·FMNH2 + O2 → LuxAB·FMNH2·O2
LuxAB·FMNH2·O2 + RCHO → LuxAB·FMNH2·O2–RCHO
LuxAB·FMNH2·O2–RCHO → LuxAB + FMN + RCOOH + H2O + light
The dark pathway, in which aldehyde is not consumed, and light is not produced, consists of a sequence of three steps:
FMNH2 + LuxAB ⇌ LuxAB·FMNH2
LuxAB·FMNH2 + O2 → LuxAB·FMNH2.O2
LuxAB·FMNH2·O2 → LuxAB + FMN + H2O2
It can be seen that light is produced in the light pathway when the LuxAB·FMNH2O2–RCHO complex breaks down to FMN, RCOOH and H2O. Energy may also be released, however, by the breakdown of the previous intermediate, LuxAB·FMNH2·O2, to FMN and H2O2via the dark pathway. Light production does not occur in this pathway. The total decay rate (kT) is given by:
| kT = (kLA + kDKA)/(KA + A) |
The luxCDEgenes encode the fatty acidreductase complex required for the generation and recycling of fatty acid to aldehyde.12 The enzyme Fre (NAD(P)H: flavin oxidoreductase) supplies reduced flavin for the light emitting reaction.13 The fatty acidreductase complex consists of three components—a fatty acidreductase encoded by luxC, an acyltransferase encoded by luxD, and an acylprotein synthetase encoded by luxE. The proteins LuxE and LuxC are associated in an equal molar ratio in a multienzyme complex14 that is responsible for the recycling of fatty acid to aldehyde. LuxD catalyses a reaction leading to further production of fatty acid that appears to be decoupled from the main recycling pathway.15 The reactions catalysed by the products of luxCDABE are linked with those of the fatty acid biosynthesis pathway.
The motivation for constructing a mathematical model of the Lux system was the use of the luxoperon as a reporter system.16–19 The reporter is constructed by cloning the promoter region of interest into the plasmid, upstream of the luxCDABEoperon. The promoter controls the expression of the luxgenes and therefore controls the intensity of the light produced by the LuxAB reaction. The light intensity is therefore a measure of the activity level of the promoter.
The Lux mathematical model could be subjected to different conditions, such as varying protein concentrations, to see whether such changes could be used to optimise the light output from the reporter system. In particular, it could be useful to know which proteins had most control over the steady state concentrations of the system. The model is used to investigate the following: (i) whether such a model could be fitted to the existing experimental data published for the Lux system; (ii) which protein(s) control the light production; (iii) the effects of changing the concentrations of the Lux enzymes; (iv) how best to split the luxgenes between two plasmids to ensure maximum light production; (v) the sensitivity of the light production rate to changes in parameter values, and in particular to values measured in other bacterial species.
In order to develop the model, values were required for all the kinetic constants involved in the velocity equations. Most of the experimental work on the Lux system has been focused on three bacterial species. The LuxAB reaction has been investigated using Vibrio fischeri and Vibrio harveyi.13,20–23Photobacterium phosphoreum has been used for experiments on the LuxC, LuxD and LuxE reactions.24–28
Two independent sets of Km values were available for the LuxAB reaction of P. luminescens enzymes.29,30Km and Vmax values were available from measurements with V. fischeri and V. harveyi enzymes.20,23,31–34
The values for Fre were taken from the work of Inouye and Nakamura,31 in which H-NMR spectroscopy was used to determine the stereospecificity of the hydride transfer in the Fre reaction. This revealed the mechanism of the reaction and allowed the substrate specificity to be characterised.
The Km and Vmax values for the aldehyde substrate of the LuxAB reaction were taken from the work on the dependence of the intensity of the light output on the aldehyde chain length in Achromobacter fischeri (Vibrio fischeri) by Hastings, Spudich and Malnic.20 The values were calculated from measurements on the pentadecanal and decanal graphs (Fig. 3A and 3B in the paper).
The work of Meighen and Hastings,23 used kinetic data to determine that there is just one FMNH2-binding site in LuxAB. Both MAV (Vibrio harveyi) and Pf (Vibrio fischeri) luciferases were studied. The kinetic constants used in the model for FMNH2 were calculated from the two Lineweaver–Burk plots for Pf luciferase (Fig. 8 in the paper), which represented two ranges of FMNH2 concentrations. The time series for the light intensity in the MAV luciferase reaction was used in testing the model.
The kinetic constants for the LuxE reaction were obtained from a study of the fatty acidreductase complex in Photobacterium phosphoreum by Rodriguez, Nabi and Meighen.28 In this study a high pressure liquid chromatographic assay was used to show that the complex consisted of the three polypeptides now known as LuxD, LuxE and LuxC. The values of the kinetic constants were obtained from the Lineweaver-Burk plots for tetradecanoic acid and ATP (Fig. 1 in the paper). The time series in Fig. 3B was also used in testing the model.
LuxC was purified to homogeneity by Rodriguez, Riendeau and Meighen.27 It was found that in addition to catalysing the reaction of acyl-CoA and NADPH, the enzyme could transfer the acyl group to different thiol reagents in the absence of NADPH. The Lineweaver–Burk plot for the NADPH dependence of tetradecanoyl-CoA was used to obtain the kinetic constants for LuxE.
More detailed information about the graphs used in testing the model is included in the ESI (Lux Data Summary).†
Metabolic control analysis35,36 has been used successfully to investigate the dependence of the flux through a multi-enzyme system on changes in the concentrations of the component enzymes and substrates.37 The analysis identifies the extent to which the control of flux is spread in varying proportions between each of the enzymes and substrates in the system. A flux control coefficient is calculated for each enzyme. This is a measure of the rate at which the flux through the system changes as the enzyme activity is changed. It can be expressed in the non-dimensional form
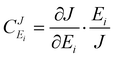
The effect on the flux of changing substrate concentrations can be measured for each substrate by the elasticity coefficient:
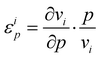
Methods
Model structure
The chemical equations for the reactions catalysed by the products of the luxoperon in Photorhabdus luminescens are:
In this paper these are modelled as a system of coupled ordinary differential equations. For this to be done, the set of equations for the steady state velocities of the reactions had to be obtained.
All of the reactions had two or three substrates, so the exact nature of the mechanism of each reaction had to be taken into account in the velocity equations, in particular the order in which substrates bind and products are released. The reactions are modelled as irreversible. The rate equations proposed by Alberty38 were used. In these equations kinetic parameters are represented by equilibrium constants, rather than rate constants for each step of the reaction as proposed by Dalziel.39
The mechanisms employed in each reaction were obtained from the literature.1,15,22,24,40–44 The relevant steady state equations were found in Segel,45 Copeland46 and Cornish-Bowden,47 where the methods of their derivation included the schematic approach of King and Altman.48 The method of grouping the rate constants was that of Cleland.49 For each reaction the version of the velocity equation used was that for the forward reaction in absence of products. The velocities are:
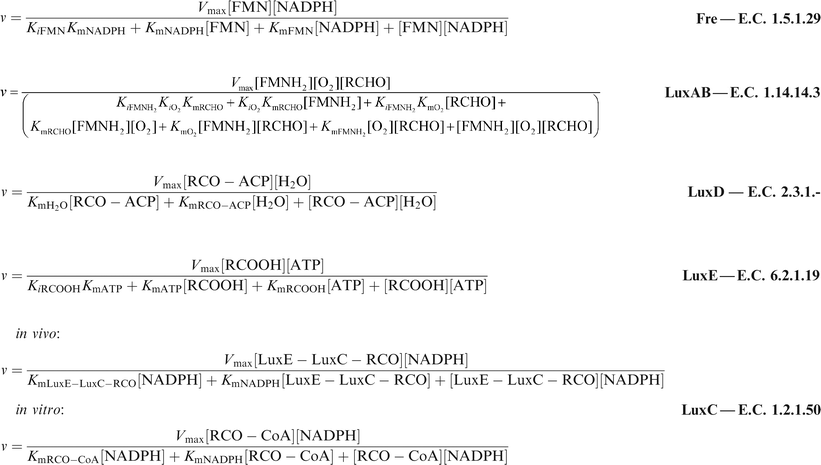
The velocity equations were combined to produce a set of differential equations to represent the flux through the system of each of the substrates of interest: flavin (FMN), reduced flavin (FMNH2), tetradecanoic acid (represented as RCOOH), tetradecanal (represented as RCHO), and either tetradecanoyl–LuxC–LuxE and tetradecanoyl–LuxE–LuxC (in vivo) or tetradecanoyl–CoA (in vitro), represented as LuxE–LuxC–RCO, LuxC–LuxE–RCO and RCO–CoA, respectively. The equations are:
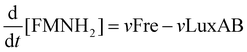
| [FMNH2] + [FMN] = F |
Preliminary work revealed that the inclusion of the LuxD reaction makes no difference to the output of the system since the reactions of LuxC and LuxE recycle all of the tetradecanoic acid from the LuxAB reaction. Leaving out LuxD gives the following:
in vivo:
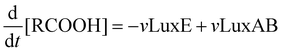
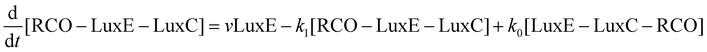
where k1 and k0 are the rate constants for the forward and back reactions, respectively in the reversible reaction:
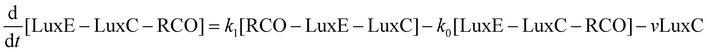
| RCO–LuxE–LuxC ⇌ LuxE–LuxC–RCO |
| [RCOOH] + [RCHO] + [RCO–LuxE–LuxC] + [LuxE–LuxC–RCO] = R |
in vitro:
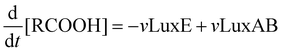
so
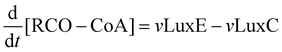
| [RCOOH] + [RCO–CoA] + [RCHO] = R |
Parameter values and testing the model
MAPLE‡ was used to model the sensitivity of the steady state to substrate and enzyme concentrations and kinetic parameter values. Estimates of the limits on the Km values used in the model were obtained from the enzyme database BRENDA50 and from the values given in the ESI (Sensitivity to Km Values).† The sensitivity was expressed as the ratio of the percentage change in substrate concentration to the percentage change in the Km value.Generally the velocity of the LuxAB reaction was measured by the intensity of the light generated in the reaction at each time point, so published Vmax values were expressed in units of quanta per second. For consistency these were converted to units of μMP min−1 μME−1 (P = product, E = enzyme) using a quantum yield value of 0.2. This was used as an approximate average of the three values of quantum yield for bacterial luciferase found in the literature (0.27,21 0.2151 and 0.16452).
For both LuxAB and Fre different values of Vmax were given for the different substrates in the reactions. These were ‘apparent’ values of Vmax since the concentration of one substrate was varied while the other was kept constant. The value appropriate for the substrate being varied in the experiment was used in the model.
The concentrations of the other reactants involved in the system also needed to be estimated. The other reactants estimated and tested were dissolved oxygen, ATP and NADPH. Since the LuxD reaction is omitted from the model, the concentrations of water and RCO–ACP, only associated with the LuxD reaction, were not needed. The environment of the Lux enzymes in typical experiments is that of an E. coli cell, so the concentrations of ATP, NADPH and FMN found in an E. coli cell were used.37 The values were 1310 μM for ATP, 560 μM for NADPH, and 88 μM for FMN. The value of the concentration of dissolved oxygen in the cell was not available, so its concentration in water at 37 °C at an atmospheric pressure of 101.3 kPa was calculated,§ giving a value of 214 μM. The values for ATP, NADPH and oxygen were taken as fixed within each simulation, and the sensitivity of the steady state light intensity to the values of these concentrations was analysed by varying each of them over five orders of magnitude. The results are included in the ESI (Sensitivity to Substrate Concentrations).†
The accuracy of the model was tested by comparing its output against the results from published experiments on the Lux system. One difficulty with this was that the LuxE and LuxC reactions in vivo are thought to involve the transfer of the tetradecanoyl group from LuxE to LuxC, although the process is not well understood.44 This is shown in the statement of the chemical equations for LuxE and LuxC. The in vitro experiments in the literature, however, use the fact that LuxC can also reduce other activated fatty acids such as acyl-CoA.26 In many papers tetradecanoyl-CoA is used in assays for LuxC,27 so the steady state velocity equation for LuxC would have to be in the in vitro form if the model was to be tested against these results.
The system of equations was solved in two ways, each employing the kinetic parameters and fixed concentrations listed in Table 1. Firstly, each of the in vivo differential equations were set to zero and the system was solved to obtain the steady state concentrations of the six products and the rate at which light was produced in this state. Secondly, the in vitro equations in velocity form were used to obtain a time-dependent solution for the concentration of each of the five products being investigated, and the intensity of the light being produced.
| Apparent values | Full model values | ||||
|---|---|---|---|---|---|
| K m/μM | V max/μMP min−1 μME−1 | K m/μM | V max/μMP min−1 μME−1 | ||
| LuxAB | LuxAB | ||||
| Decanal | 13.726 | 0.7441 | Decanal | 24.5233 | 1.3297 |
| FMNH2 | 1.1729 | 13.06 | FMNH2 | 1.9669 | 21.904 |
| O2 | 0.04 | — | O2 | 0.0198 | |
| LuxE | LuxE | ||||
| RCOOH | 0.4342 | 0.6117 | RCOOH | 0.4342 | 0.6117 |
| ATP | 0.02 | 0.692 | ATP | 0.02 | 0.7522 |
| LuxC | LuxC | ||||
| NADPH | 4.3691 | 2.2176 | NADPH | 5.0246 | 2.5503 |
| RCOCOA | 1 | RCOCOA | 1.0502 | ||
| Fixed Concentrations (μM): F = 88 R = 231 NADPH = 560 O2 = 214 ATP = 1310 | |||||
The model was tested by being used to reproduce time series data from papers using the published parameter values, and to reproduce stationary data from concentration curves.
To improve the accuracy of the steady state concentration values and the fit to the time series data, estimates for the Km and Vmax values for each set of stationary data were obtained by using the nonlinear least squares data fitting routine in MATLAB¶ on a series of data points measured from a suitable graph in the literature. Each data set and corresponding graph showed the output from the simulation of an experiment involving a single enzyme: LuxAB, LuxE or LuxC. These estimates were used as parameters in a Michaelis–Menten equation , which was plotted on the same graph as the relevant set of data points. The time series data were also plotted and compared with the curve obtained using the relevant Km and Vmax values.
The Km and Vmax values used in the model fitting are apparent values, and needed to be converted to values appropriate for the model using suitable sets of simultaneous equations. The flux control coefficient was calculated for each enzyme held at the concentration used in the relevant experiment. The variation in flux control coefficient values with enzyme concentration was examined. The kinetic parameters used in the model are shown in Table 1 above.
Results and Discussion
There is good agreement between the model and experimental data
Fig. 2 shows the results of fitting the model to the published experimental data.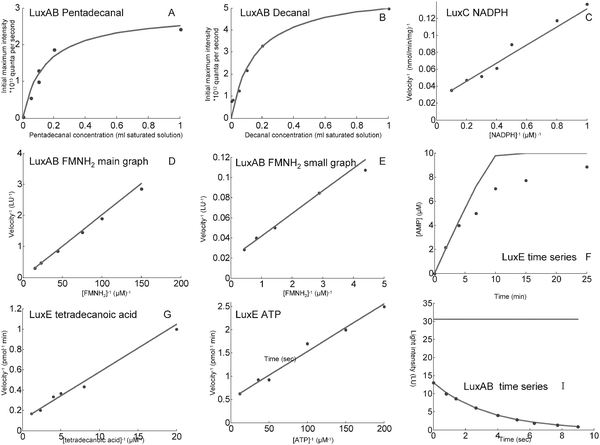 | ||
| Fig. 2 Comparisons between model predictions and experimental observations. Each graph displays the velocity of the reaction and the substrate concentrations in the units used in the relevant paper. A,B. Effect of aldehyde concentration on the velocity of the luminescence reaction. The velocity is measured by the initial intensity of the bioluminescence. The concentrations are expressed in millilitres of a saturated solution of aldehyde in a final volume of 2.0 ml. Both graphs show a very good fit of the model to the data. (Data obtained from Fig. 3, Hastings et al., 1963.20) C. Lineweaver–Burk plot for the NADPH dependence of LuxC activity. The velocity of the LuxC reaction is measured from the stimulation of luciferase activity by the aldehyde product of the reaction. It is expressed as nanomoles of aldehyde produced per minute per milligram of Lux C. Again the model fits well to the data. (Data from Fig. 2S, Rodriguez et al., 1983.27) D,E. Lineweaver–Burk plots for the dependence of the initial light intensity of the LuxAB reaction on the concentration of FMNH2. The light intensity is measured in light units (LU), where 1LU is equivalent to 2.2 × 1010 quanta per second. There is a close fit to the data over two separate ranges of concentration. (Data from Fig. 8, Meighen and Hastings, 1971.23) F. Time series of an assay for ATP hydrolysis using a mixture of LuxE and LuxC. The velocity is measured by the rate of formation of AMP, the product of the LuxE reaction, using a high performance liquid chromatography assay . It can be seen that the model has the same initial velocity and asymptotic value as the data. However, the data is more smoothly saturating than the model, which is based on Michaelis–Menten kinetics . This indicates that there is probably a more complex mechanism involved in the experiment. It is possible that the presence of LuxC has some influence on this, but the LuxC reaction which forms part of the luminescent recycling system requires the presence of NADPH. The data set used here was for a reaction mixture which did not contain NADPH. (Data from Fig. 3B, Rodriguez et al. 1985.43) G,H. Lineweaver–Burk plots for the tetradecanoic acid and ATP dependence of LuxE. The velocity is measured by including LuxC in the reaction mixture and coupling this to luciferase. The velocity is given as the rate in picomoles per minute at which aldehyde is produced from the luminescent reaction. As with the other sets of stationary data, the model shows a very good fit. (Data obtained from Fig. 1, Rodriguez et al. 1985.43) I. Time series for decay of the LuxAB reaction in Vibrio harveyi. The velocity is measured by the intensity of the luminescent reaction in light units as defined for Fig. D and E. The intensity initially rises sharply, then decays at a constant rate of 0.1 s−1 on a log scale. (Data from Fig. 2, Meighen and Hastings 1971.23) The rate for the model without the dark reaction is 3.04 × 10−5 s−1; this model does not fit the data. The green line is an exponential decay with a constant rate of 0.1 s−1 on a log scale, that would reflect the inclusion of a dark reaction (in which decay occurs without emission of light) with a kD of 0.0931 s−1. Therefore the inclusion of the dark reaction can explain these experimental time series data. | ||
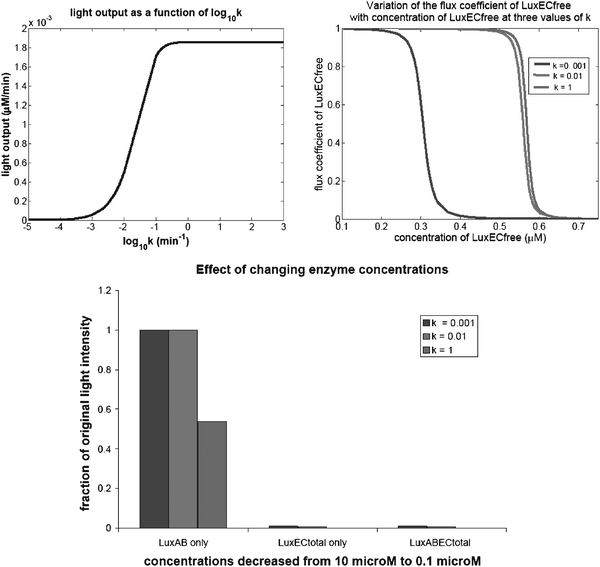 | ||
| Fig. 3 Change of control with enzyme concentration in the recycling pathway. A. The light output of the system as a function of the rate constant of the reaction in which an acyl group is transferred from LuxE to LuxC. It can be seen that the light output rises from close to zero to 1.8 × 10−3 μM min−1 over a range of k values of three orders of magnitude. The total concentration of the LuxEC dimer (denoted ‘LuxECtotal’) is divided between three forms in the system: free enzyme (LuxECfree), with an acyl group bound to LuxE, and with an acyl group bound to LuxC. B. The variation of the flux coefficient of LuxECfree with the concentration of LuxECfree. It can be seen that the control of LuxECfree over the system extends to higher concentrations as the value of k increases. The concentration of LuxECtotal used in the system is 0.0588 μM. This corresponds to a LuxECfree concentration of 8.5 × 10−5 μM. Even with k as low as 0.001 min−1, the control coefficient at this concentration is almost 1. C. A summary of the effects on the steady state light intensity of changing the concentrations of the Lux enzymes, in a way that might correspond to placing different groups of genes under the control of an inducible plasmid. Each set of bars corresponds to a different enzyme or combination of enzymes. The colour of the bar indicates the value of k used in that simulation. The height of the bar indicates the fraction to which the light intensity is reduced when the concentration of the enzyme(s) indicated decreases from 10 μM to 0.1 μM. The concentrations of the other enzymes remain at 10 μM. Part C shows that if the concentration of LuxAB only is reduced, the light intensity does not change if k is 0.001 or 0.01, but reduces to almost half if k is 1. If the total concentration of LuxEC is decreased, with or without a reduction in LuxAB, the light intensity reduces to less than 1% at all values of k, the reduction being greatest for k = 1. | ||
For LuxAB, the model is in good agreement with the graphs for both pentadecanal and decanal, with the fit for decanal being slightly better than that for pentadecanal. The main graph for FMNH2 spans a lower range of concentrations than the small graph, and shows slightly better fit.
The time series for rate of AMP formation with LuxE follows the Michaelis–Menten curve only approximately, with similar initial velocity and asymptotic value, but smoother shape. The reason for the discrepancy may be that in the experiment LuxC was also present.
There is, however, no correlation of the model with the LuxAB time series. On a log scale the rate of light production decays at a rate of 0.1 s−1. The model has a log decay rate of 3.04 × 10−5 s−1.
The most likely explanation for the lack of agreement of the model and the LuxAB time series data is that the fast decay in the experimental time series results from the dark pathway. To test this hypothesis, the equation for the total decay rate was applied to data from studies on Photobacterium phosphoreum.25
For these data,
| KA = 2.8 × 10−4 M, kL = 0.15 s−1 and kD = 0.25 s−1. |
The total decay rate is therefore
| kT = (0.15 × A + 0.25 × 2.8 × 10−4)/(2.8 × 10−4 + A) |
Control of the system lies mainly with LuxE and LuxC
Fig. 3A shows the light output of the system as a function of the rate constant of the reaction in which an acyl group is transferred from LuxE to LuxC. It can be seen that the light output rises from close to zero to 1.8 × 10−3 μM min−1 over a range of k values of three orders of magnitude. For values of k above 0.1, the light output is constant.The total concentration of the LuxEC dimer (denoted ‘LuxECtotal’) is divided between three forms in the system: free enzyme (LuxECfree), with an acyl group bound to LuxE, and with an acyl group bound to LuxC. Fig. 3B shows the variation of the flux coefficient of LuxECfree with the concentration of LuxECfree. It can be seen that the control of LuxECfree over the system extends to higher concentrations as the value of k increases.
The concentration of LuxECtotal used in the system is 0.0588 μM. This corresponds to a LuxECfree concentration of 8.5 × 10−5 μM. Even with k as low as 0.001 min−1, the control coefficient at this concentration is almost 1. This is further evidence that LuxEC has almost total control over the light output of the system.
Fig. 3C shows a summary of the effects on the steady state light intensity of changing the concentrations of the Lux enzymes, in a way that might correspond to placing different groups of genes under the control of an inducible plasmid. Each set of bars corresponds to a different enzyme or combination of enzymes. The colour of the bar indicates the value of k used in that simulation. The height of the bar indicates the fraction to which the light intensity is reduced when the concentration of the enzyme(s) indicated decreases from 10 μM to 0.1 μM. The concentrations of the other enzymes remain at 10 μM.
Fig. 3C shows that if the concentration of LuxAB only is reduced, the light intensity does not change if k is 0.001 or 0.01, but reduces to almost half if k is 1. If the total concentration of LuxEC is decreased, with or without a reduction in LuxAB, the light intensity reduces to less than 1% at all values of k, the reduction being greatest for k = 1. This is further evidence that control of light production lies within the recycling path rather than in the luminescent reaction, and suggests that LuxEC should be inducible, but LuxAB and Fre should be constitutively expressed (Changing the concentration of Fre has already been shown to have no effect on light intensity).
The steady state light production rate shows a low sensitivity to changes in parameter values
The data concerning the sensitivity of the steady state of the model to its Km values is included in the ESI.† It was found that most of the Km values for the four species covered by the available literature differ considerably from those used in the model, but the resulting light production rate only changed by at most 0.03%. Where a range of observed Km values were not available from the literature, we tested sensitivity using a range of Km over several orders of magnitude. The largest percentage change in light production rate using these additional values was −4%. This was for an increase in the Km value used in the model by a factor of 25.The analysis of the sensitivity of the steady state light intensity to the values of the concentrations of ATP, NADPH and oxygen showed that for seven combinations of concentrations varying over five orders of magnitude the light intensity was changed at most by 1.5%, demonstrating that the model is not particularly sensitive to the values of these parameters.
Concluding remarks
We have constructed a differential equation model for the Lux luminescence system. We have tested the model on published experimental data from several sources, and found good agreement. We have used metabolic control analysis to show that the control of the system lies mainly with the enzymes LuxE and LuxC. This conclusion is also supported by the results of using the model to show how changes in enzyme concentrations affect the steady state light intensity.A prediction from this study is that a reporter system constructed from the luxABCDEoperon of P. luminescens, expressing LuxAB constitutively, and with the promoter under study in front of LuxCDE, should act as an efficient reporter; this might have the advantage of speeding up the response time. The reverse configuration should not work. However, the utility of these ideas would need to be investigated through experiment.
The model we construct considers the dynamics of the luminescence system itself and does not incorporate the kinetics of protein synthesis and degradation. There are two reasons for this. First, we are interested in constructing a model that is explanatory of in vitro data, where these processes are not relevant. Second, in vivoprotein synthesis and turnover will depend on the promoter under study, the host organism and experimental conditions. The advantage of our approach is that we develop and analyze a generic chemical model that can be embedded into larger models for specific applications that also incorporate the protein dynamics.
Thus this study lays the foundation for future work that would allow powerful analysis of data from Lux reporter experiments. In particular, such work could include the reverse engineering of promoter activity from luminescent readout. This would require not only a sophisticated model in the form of the one developed here, but also additional parameters that would need to be experimentally defined, including the rates of synthesis of Lux proteins and their molecular stability.
Acknowledgements
We thank Neil Burton, Dafyd Jenkins, Jan Kreft, Peter Lund, Helen Parsons, and Chris Wharton for invaluable discussion and technical help. PAW is funded by a BBSRC Targeted Priority Research Studentship (BBS/S/E/2006/13186).References
- E. Meighen, Molecular biology of bacterial bioluminescence, Microbiol. Rev., 1991, 55(1), 123–142 CAS.
- H. Urbanczyk, J. C. Ast, A. J. Kaeding, J. D. Oliver and P. V. Dunlap, Phylogenetic analysis of the incidence of lux gene horizontal transfer in Vibrionaceae, J. Bacteriol., 2008, 190(10), 3494–3504 CrossRef CAS.
- S. Zenno and K. Saigo, Identification of the genes encoding NAD(P)H-flavin oxidoreductases that are similar in sequence to Escherichia coli Fre in four species of luminous bacteria: Photorhabdus luminescens, Vibrio fischeri, Vibrio harveyi, and Vibrio orientalis, J. Bacteriol., 1994, 176, 3544–3551 CAS.
- D. Byers and E. A. Meighen, Vibrio harveyi aldehyde dehydrogenase. Partial reversal of aldehyde oxidation and its possible role in the reduction of fatty acids for the bioluminescence reaction, J. Biol. Chem., 1984, 259(11), 7109–7114 CAS.
- L. F. Greer and A. A. Szalay, Imaging of light emission from the expression of luciferases in living cells and organisms: a review, Luminescence, 2002, 17, 43–74 CrossRef.
- S. Frackman, M. Anhalt and K. H. Nealson, Cloning, organization, and expression of the bioluminescence genes of Xenorhabdus luminescens, J. Bacteriol., 1990, 172(10), 5767–5773 CAS.
- R. Szittner and E. Meighen, Nucleotide sequence, expression, and properties of luciferase coded by lux genes from a terrestrial bacterium, J. Biol. Chem., 1990, 265(27), 16581–16587 CAS.
- L. Xi, K.-W. Cho and S.-C. Tu, Cloning and nucleotide sequences of lux genes and characterization of luciferase of Xenorhabdus luminescens from a human wound, J. Bacteriol., 1991, 173(4), 1399–1405 CAS.
- E. A. Meighen and R. B. Szittner, Multiple repetitive elements and organization of the lux operons of luminescent terrestrial bacteria, J. Bacteriol., 1992, 174(16), 5371–5381 CAS.
- T. O. Baldwin, M. M. Ziegler and D. A. Powers, Covalent structure of subunits of bacterial luciferase: NH2-terminal sequence demonstrates subunit homology, Proc. Natl. Acad. Sci. USA, 1979, 76(10), 4887–4889 CrossRef CAS.
- N. Valkova, R. Szittner and E. A. Meighen, Control of luminescent decay and flavin binding by the LuxA carboxyl-terminal regions in chimeric bacterial luciferases, Biochemistry, 1999, 38, 13820–13828 CrossRef CAS.
- D. Riendeau and E. Meighen, Co-induction of fatty acid reductase and luciferase during development of bacterial luminescence, J. Biol. Chem., 1989, 255, 12060–12065.
- E. Jablonski and M. DeLuca, Studies of the control of luminescence in Beneckea harveyi, Biochemistry, 1978, 17(4), 672–678 CrossRef CAS.
- L. Wall and E. A. Meighen, Subunit structure of the fatty acid reductase complex from Photobacterium phosphoreum, Biochemistry, 1986, 25, 4315–4321 CrossRef CAS.
- E. A. Meighen, Bacterial bioluminescence: organization, regulation, and application of the lux genes, FASEB. J., 1993, 7, 1016–1022 CAS.
- A. Zaslaver, A. E. Mayo, R. Rosenberg, P. Bashkin, H. Sberro, M. Tsalyuk, M. G. Surette and U. Alon, Just in time transcription program in metabolic pathways, Nat. Genet., 2004, 36, 486–491 CrossRef CAS.
- W. H. Tsui, G. Yim, H. H. Wang, J. E. McClure, M. G. Surette and J. Davies, Dual effects of MLS antibiotics: transcriptional modulation and interactions on the ribosome, Chem. Biol., 2004, 11(9), 1307–1316 CrossRef CAS.
- K. Duan, C. Dammel, J. Stein, H. Rabin and M. G. Surette, Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication, Mol. Microbiol., 2003, 50(5), 1477–91 CrossRef CAS.
- E. B. Goh, G. Yim, W. Tsui, J. Mc Clure, M. G. Surette and J. Davies, Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics, Proc. Natl. Acad. Sci. USA, 2002, 99(26), 17025–30 CrossRef CAS.
- J. W. Hastings, J. Spudich and G. Malnic, The influence of aldehyde chain length upon the relative quantum yield of the bioluminescent reaction of Achromobacter fischeri, J. Biol. Chem., 1963, 238(9), 3100–3105 CAS.
- J. W. Hastings, W. H. Riley and J. Massa, The purification, properties and chemiluminescent quantum yield of bacterial luciferase, J. Biol. Chem., 1965, 240(3), 1473–1481 CAS.
- J. W. Hastings and C. Balny, The oxygenated bacterial luciferase-flavin intermediate. Reaction products via the light and dark pathways, J. Biol. Chem., 1975, 250(18), 7288–7293 CAS.
- E. A. Meighen and J. W. Hastings, Binding site determination from kinetic data. Reduced flavin mononucleotide binding to bacterial luciferase, J. Biol. Chem., 1971, 246(24), 7666–7674 CAS.
- L. M. Carey, A. Rodriguez and E. Meighen, Generation of fatty acids by an acyl esterase in the bioluminescent system of Photobacterium phosphoreum, J. Biol. Chem., 1984, 259(16), 10216–10221 CAS.
- K. Yoshida, M. Takahashi and T. Nakamura, Studies on luciferase from Photobacterium phosphoreum. An enzyme-FMN intermediate complex in the bioluminescent reaction, J. Biochem., 1974, 75, 583–589 CAS.
- D. Riendeau, A. Rodriguez and E. Meighen, Resolution of the fatty acid reductase from Photobacterium phosphoreum into acyl protein synthetase and acyl-CoA reductase activities. Evidence for an enzyme complex, J. Biol. Chem., 1982, 257(12), 6908–6915 CAS.
- A. Rodriguez, D. Riendeau and E. Meighen, Purification of the acyl coenzyme A reductase component from a complex responsible for the reduction of fatty acids in bioluminescent bacteria, J. Biol. Chem., 1983, 258(8), 5233–5237 CAS.
- A. Rodriguez, I. R. Nabi and E. Meighen, ATP turnover by the fatty acid reductase complex of Photobacterium phosphoreum, Can. J. Biochem. Cell Biol., 1985, 63, 1106–1111 CAS.
- Z. Li and E. A. Meighen, The turnover of bacterial luciferase is limited by a slow decomposition of the ternary enzyme product complex of luciferase, FMN, and fatty acid, J. Biol. Chem., 1994, 269, 6640–6644 CAS.
- P. Colepicolo, K.-W. Cho, G. O. Poinar and J. W. Hastings, Growth and luminescence of the bacterium Xenorhabdus luminescens from a human wound, Appl. Environ. Microbiol., 1989, 55(10), 2601–2606 CAS.
- S. Inouye and H. Nakamura, Stereospecificity of hydride transfer and substrate specificity for FMN-containing NAD(P)H-flavin oxidoreductase from the luminescent bacterium, Vibrio fischeri ATCC 7744, Biochem. Biophys. Res. Commun., 1994, 205(1), 275–281 CrossRef CAS.
- W. Duane and J. W. Hastings, Flavin mononucleotide reductase of luminous bacteria, Mol. Cell. Biochem., 1975, 6(1), 53–64 CrossRef CAS.
- J. W. Hastings, K. Weber, J. Friedland, A. Eberhard, G. W. Mitchell and A. Gunsalas, Structurally distinct bacterial luciferases, Biochemistry, 1969, 8(12), 4681–4689 CrossRef CAS.
- J. Spudich and J. W. Hastings, Inhibition of the bioluminescent oxidation of reduced flavin mononucleotide by 2-decanal, J. Biol. Chem., 1963, 238(9), 3106–3108 CAS.
- H. Kacser and J. A. Burns, The control of flux, Symp. Soc. Exp. Biol., 1973, 27, 65–104 Search PubMed.
- D. Fell, Understanding the control of metabolism, Portland Press Ltd, London, 1997 Search PubMed.
- C. Chassagnole, D. A. Fell, B. Rais, B. Kudla and J.-P. Mazat, Control of the threonine-synthesis pathway in Escherichia coli: a theoretical and experimental approach, Biochem. J., 2001, 356, 433–444 CrossRef CAS.
- R. A. Alberty, The relationship between Michaelis constants, maximum velocities and the equilibrium constant for an enzyme-catalyzed reaction, J. Am. Chem. Soc., 1953, 75, 1928–1932 CrossRef CAS.
- K. Dalziel, Kinetics and mechanism of nicotinamide-dinucleotide-linked dehydrogenases, in The Enzymes, ed. P. D. Boyer, Academic Press, San Diego, CA, 3rd edn., 1975, pp. 1–60 Search PubMed.
- L. Xi, D.-C. Chow and S.-C. Tu, Thermodynamic analysis of the binding of oxidised and reduced FMN cofactor to Vibrio harveyi NADPH-FMN oxidoreductase FRP apoenzyme, Biochemistry, 2006, 45(49), 14781–14787 CrossRef.
- C. E. Jeffers and S. T. Tu, Differential transfers of reduced flavin cofactor and product by bacterial flavin reductase to luciferase, Biochemistry, 2001, 40(6), 1749–1754 CrossRef CAS.
- M. Kurfurst, S. Ghisla and J. W. Hastings, Characterization and postulated structure of the primary emitter in the bacterial luciferase reaction, Proc. Natl. Acad. Sci. USA, 1984, 81, 2990–2994 CrossRef.
- A. Rodriguez and E. Meighen, Fatty acyl-AMP as an intermediate in fatty acid reduction to aldehyde in luminescent bacteria, J. Biol. Chem., 1985, 260(2), 771–774 CAS.
- L. Wall, A. Rodriguez and E. Meighen, Intersubunit transfer of fatty acyl groups during fatty acid reduction, J. Biol. Chem., 1986, 261(34), 15981–15988 CAS.
- I. H. Segel, Enzyme Kinetics, Wiley, New York, 1975, pp. 505–883 Search PubMed.
- R. A. Copeland, Enzymes: a practical introduction to structure, mechanism, and data analysis, Wiley-VCH, 1996, pp. 263–278 Search PubMed.
- A. Cornish-Bowden, Fundamentals of enzyme kinetics, Butterworth and Co Ltd, London, 1979 Search PubMed.
- E. L. King and C. Altman, A schematic method of deriving the rate laws for enzyme-catalyzed reactions, J. Phys. Chem., 1956, 60, 1375–1378 CrossRef CAS.
- W. W. Cleland, The kinetics of enzyme-catalyzed reactions with two or more substrates or products. Prediction of initial velocity and inhibition patterns by inspection, Biochim. Biophys. Acta, 1963, 67, 188–196 CAS.
- I. Schomburg, O. Hofmann, C. Baensch, A. Chang and D. Schomburg, Enzyme data and metabolic information: BRENDA, a resource for research in biology, biochemistry, and medicine, Gene Funct. Dis., 2000, 3–4, 109–118 Search PubMed.
- I. V. Sokolova, G. S. Kalacheva and N. A. Tyulkova, Analysis of the ratio of quantum yield and fatty acid formation of Photobacterium leiognathi bioluminescence, Vestnik Moskovskogo Universiteta. Khimiya., 2000, 41(6 Supplement), 118–120 Search PubMed.
- O. Shimomura, F. H. Johnson and Y. Kohama, Reactions involved in bioluminescence systems of limpet (Latia neritoides) and luminous bacteria, Proc. Nat. Acad. Sci. USA, 1972, 69(8), 2086–2089 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary tables. See DOI: 10.1039/b812094c |
| ‡ http://www.maplesoft.com/ |
| § http://antoine.frostburg.edu/chem/senese/101/solutions/faq/predicting-DO.shtml |
| ¶ http://www.mathworks.com/ |
| This journal is © The Royal Society of Chemistry 2009 |