On-chip culturing of hepatocytes and monitoring their ammonia metabolism
Wataru
Satoh
,
Shintaro
Takahashi
,
Fumihiro
Sassa
,
Junji
Fukuda
and
Hiroaki
Suzuki
*
Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8573, Japan. E-mail: hsuzuki@ims.tsukuba.ac.jp; Fax: +81-29-853-4490; Tel: +81-29-853-5598
First published on 27th October 2008
Abstract
Ammonia metabolism of hepatocytes was monitored at nanolitre-scale volumes of a medium in a microsystem with microfluidic and sensing functions.
Recent progress in microfluidic technology has revolutionized methodologies in many basic research areas, particularly cell engineering.1 Since components of fabricated microdevices have dimensions comparable with those of cells, they provide unprecedented abilities to control cellular microenvironments in vitro and miniaturize assays.2 High-throughput processing achieved by microdevices is beneficial for stem cell research and drug screening, which require many trials to achieve the best conditions for stem-cell differentiation,3 and the identification of promising drug candidates from a library of compounds.4
We have developed novel devices for electrowetting-based precise handling of solutions on a nanolitre scale; this type of handling of solutions is dependent on the wettability change in a metal electrode that occurs in response to a change in its potential.5 As a practical application, we attempted to culture hepatocytes using the on-chip culture system and monitor their ammonia metabolism with a minimum amount of culture medium. Ammonia metabolism is an important hepatic differentiation marker and a critical function in cell sources employed in cell therapy for liver failure. Hepatocytes or hepatic lineage cells that derive from stem cells have been evaluated using conventional methods in terms of their ammonia metabolism and expressions of associated enzymes.6
In order to evaluate ammonia metabolism, ammonium ions are transformed into gaseous ammonia by raising the pH of the cell-culture medium. The ammonia dissipates into an air gap in the microfluidic channel and dissolves into the electrolyte solution. The ammonia concentration is determined by the ammonia sensor integrated to the microfluidic channel.
The internal structure of the microdevice was constructed from glass and polydimethylsiloxane (PDMS) substrates (Fig. 1). A culture chamber (volume, 1 µl), a reservoir for the electrolyte solution, a compartment for a liquid-junction reference electrode, and 2 flow channels were fabricated in the PDMS substrate (Fig. 1(b)). The flow channels comprised of segments (height, 40 µm; width, 500 µm) separated by valves. The flow channels were fabricated in close proximity in the area where a pH control unit and the air gap ammonia sensor were located. An air gap (length, 370 µm; width, 30 µm) was formed between the 2 flow channels in the sensing area (Fig. 1(d)). The ammonia sensor comprised of an iridium oxide pH-indicator electrode and a Ag/AgCl reference electrode situated in the upper channel on 1 side of the air gap.7 A gold working electrode and a liquid-junction Ag/AgCl electrode were located in the lower channel containing the culture medium and were used to control the pH in the culture medium by electrolysis. The cell culture medium and the electrolyte solution were pipetted into the corresponding reservoirs. The transport of solutions in the channels was by capillary action and controlled by electrowetting-based valves.8 In order to mobilize the electrolyte solution, an electric potential of −0.9 V was applied to the working electrode with respect to the Ag/AgCl reference electrode located at the exit of the reservoir (see Fig. 2(b)). However, for the culture medium, the driving force in the valve area was smaller for the same potential. Therefore, in the case of the culture medium, a potential of −1.2 V was applied against the Ag/AgCl reference electrode located at the exit of the culture chamber. The solutions passed each valve within 1 s, minimizing the accumulation of gas bubbles produced during electrolysis.
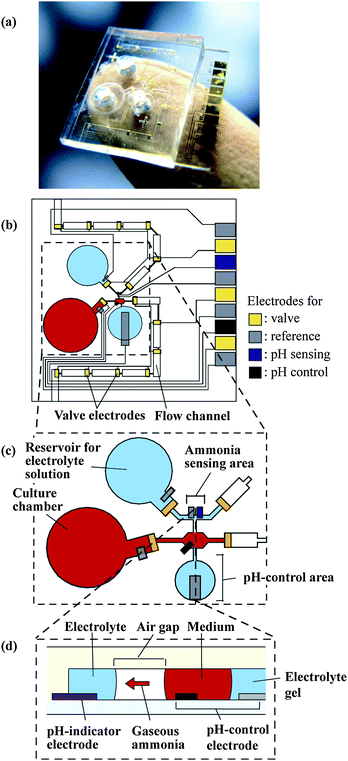 | ||
| Fig. 1 Microsystem for monitoring ammonia metabolism of hepatocytes. (a) The microsystem. (b) Detailed layout of the microfluidic components. (c) Magnified view of the ammonia sensing area. (d) Cross-section of the ammonia sensing area. | ||
 | ||
| Fig. 2 Ammonia measurement procedure. (a) The culture medium and electrolyte solution are filled into the culture chamber and the reservoir, respectively. (b) The electrolyte solution is filled into the ammonia sensing area; subsequently, the culture medium is extracted and transported to the pH control area. (c) The pH in the sampled medium is elevated and the gaseous ammonia diffuses into the electrolyte solution. (d) The used solutions are renewed by one-segment transport. W.E., working electrode; R.E., reference electrode. | ||
Fig. 2 shows the procedure for the extraction and transport of solutions and the determination of ammonia concentration. By opening the first valve located at the end of the reservoir for the electrolyte solution, the electrolyte solution (0.1 M KCl, 0.1 M NH4Cl) was introduced into the sensing area (Fig. 2(b)). The culture medium was also transported to the pH control area (Fig. 2(b)). The pH of the medium in the sensing area was increased from 7.4 to 10.8 by the application of −2.0 µA to the gold electrode in the pH control area for 2 min. (The pH had been checked previously by filling the air gap with the electrolyte solution and subsequently using the calibrated pH-indicator electrode for the ammonia sensor.) Elevating the pH in the medium transforms ammonium ions into gaseous ammonia, which is transported through the air-gap and subsequently dissolves into the electrolyte solution of the ammonia sensing area (Fig. 2(c)). As a result, the pH of the electrolyte solution is elevated. The relationship between the potential of the pH-sensing electrode and the concentration of ammonia is expressed by the Nernst equation (Fig. 3). Therefore, the ammonia concentration is obtained by measuring the potential change in the pH-indicator electrode with respect to the Ag/AgCl reference electrode. The pH change caused during the measurement may affect subsequent analyses of the medium in the neighboring segment. Furthermore, the pace of recovery of the potential of the ammonia sensor is generally slow. Therefore, after the measurement was determined, the pH of the medium in the pH control area was recovered to 7.4 by electrolysis to the acidic direction; this was accomplished by applying a current of +2.0 µA to the gold electrode in the pH control area for 2 min. After the measurement was determined, the electrolyte solution and the medium in the ammonia sensing area were replaced with fresh solutions by opening the next valves and were mobilized into the next segment (Fig. 2(d)), promoting the rapid recovery of the pH of the electrolyte solution. This procedure was repeated until the solutions reached the last segment.
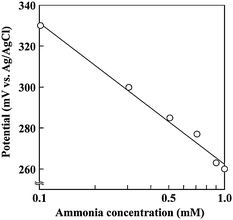 | ||
| Fig. 3 Dependence of the potential of the pH-indicator electrode on the ammonia concentration. | ||
We subsequently analyzed on-chip culturing of hepatocytes. Primary hepatocytes were isolated from the liver of an adult male Wistar rat (age, 8 weeks; weight, approximately 200 g) by liver perfusion using 0.05% collagenase. Cell viability was checked using the trypan blue dye exclusion assay, and cells with greater than 85% viability were used for culture. Hepatocytes were seeded in the culture chamber at 2 different densities (11 and 115 cells/mm2), and were cultivated in a humidified 95% air/5% CO2 incubator at 37 °C.
After 3 h of culture in the microsystem, hepatocytes attached and spread out on the surface of the culture chamber (Fig. 4(a)). Since the culture chamber was transparent, the cells could be analyzed by typical cytofluorometric assays. A live/dead fluorometric assay was performed after 1 day of culture after double staining the hepatocytes with fluorescein diacetate and ethidium bromide. When this assay is performed, the cytoplasm of live cells fluoresce green, whereas the nucleus of dead cells fluoresce red. The assay confirmed that almost all hepatocytes were alive (Fig. 4(b)). After 3 days of culture, we further conducted fluorometric assays to evaluate the metabolic activities, protein secretion, and drug metabolism of the hepatocytes. Fig. 4(c) shows an image of hepatocytes demonstrating immunofluorescence due to their immunoreactivity for albumin that has been overlapped with the corresponding phase-contrast image. Fig. 4(d) shows fluorescence in hepatocytes of resorufin that was converted from ethoxyresorufin during the enzymatic reactions of cytochrome P450. These results suggest that hepatocytes stably maintain their metabolic functions in microsystems.
 | ||
| Fig. 4 Hepatocytes cultured in the microsystem at a density of 11 cells/mm2. (a) Phase-contrast microscopic image. (b) Live/dead staining after 1 day of culture. (c), (d) Images showing albumin synthesis (c) and cytochrome P450 activity (d) after 3 days of culture. Scale bars correspond to 200 µm. | ||
After the hepatocytes were seeded and cultured for 3 h, a solution of NH4Cl was added to the medium (final concentration in the mixture, 1.0 mM). The change in the ammonia concentration was measured according to the previously mentioned procedure. Fig. 5 shows the change in the ammonia concentration measured using the microsystem and a conventional culture dish. The medium in the microchamber was sampled every 30 min. Ammonia metabolism commenced immediately after the addition of ammonia, and approximately half of the ammonia was metabolized during the first 30 min. Finally, the concentration decreased to the normal level in the human body (<0.1 mM) after 3 h at a density of 115 cells/mm2. For comparison, hepatocytes were seeded on a 35 mm type I collagen-coated dish at a density of 11 cells/mm2 and cultured for 3 h. In this case, the medium was replaced with a fresh culture medium supplemented with 1 mM NH4Cl. The medium in the dish was sampled at 0 and 3 h of culture after NH4Cl addition. The ammonia concentration was measured using a commercial measurement kit (Ammonia-Test Wako, Wako Pure Chemical Industries, Osaka, Japan) based on absorbance measurements. The data presented in Fig. 5 clearly shows the advantage of the microsystem employed in this study and the limitation of the conventional method. Measurements with shorter time intervals provided data that were difficult to acquire using the conventional method. This is crucial especially in situations when a rapid and accurate validation of hepatocytes is required for clinical use. Table 1 compares the corresponding dimensions of the culture and volumes of the medium. The sampled volume was 84 nl for 7 measurements in the microsystem, whereas 400 µl was necessary for the 2 measurements using the conventional method. It is important to note that the difference in the volumes utilized by the 2 methods is more than 3 orders of magnitude. Furthermore, obtaining a greater number of data points was difficult with the conventional method because even the 2 measurements consumed 20% of the medium and further consumption may have affected the experiment. With regard to this, only <10% of the total culture medium was consumed by the microsystem.
| Medium volume (µl) | Volume required for a single assay (µl) | Culture surface (mm2) | Seeding density (cells/mm2) | |
|---|---|---|---|---|
| Microsystem | 1 | 0.012 | 13 | 11 or 115 |
| Culture dish | 2000 | 200 | 962 | 11 |
 | ||
| Fig. 5 Monitoring of ammonia metabolism of hepatocytes. Hepatocytes were seeded at densities of 11 cells/mm2 (○) or 115 cells/mm2 (□) in the microsystem. Hepatocytes were also seeded in a conventional culture dish at a density of 11 cells/mm2, and the ammonia concentration was measured using a commercial kit (●). The dashed line that connects the black circles suggests the difficulty in obtaining data in this region. | ||
In conclusion, cytofluorometric assays and ammonia monitoring could be conducted using only a drop of reagents and cells. Minimizing the sample size employed in cell-based analysis systems will be beneficial to various studies that use valuable biological materials such as human stem cells, growth factors, and drug candidates.
References
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS; J. Atencia and D. J. Beebe, Nature, 2005, 437, 648–655 CrossRef CAS.
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS; L. Kang, B. G. Chung, R. Langer and A. Khademhosseini, Drug Discov. Today, 2008, 13, 1–13 CrossRef CAS; B. J. Kane, M. J. Zinner, M. L. Yarmush and M. Toner, Anal. Chem., 2006, 78, 4291–4298 CrossRef CAS.
- E. Figallo, C. Cannizzaro, S. Gerecht, J. A. Burdick, R. Langer, N. Elvassore and G. Vunjak-Novakovi, Lab Chip, 2007, 7, 710–719 RSC.
- J. Inglese, R. L. Johnson, A. Simeonov, M. Xia, W. Zheng, C. P. Austin and D. S. Auld, Nat. Chem. Biol., 2007, 3, 466–479 CrossRef CAS.
- W. Satoh, H. Hosono and H. Suzuki, Anal. Chem., 2005, 77, 6857–6863 CrossRef CAS; N. Nashida, W. Satoh, J. Fukuda and H. Suzuki, Biosens. Bioelectron., 2007, 22, 3167–3173 CrossRef CAS.
- T. Ishii, K. Yasuchika, H. Fujii, T. Hoppo, S. Baba, M. Naito, T. Machimoto, N. Kamo, H. Suemori, N. Nakatsuji and I. Ikai, Exp. Cell Res., 2005, 309, 68–77 CrossRef CAS; J. Fukuda, Y. Sakai and K. Nakazawa, Biomaterials, 2006, 27, 1061–1070 CrossRef CAS.
- H. Suzuki and Y. Matsugi, Sens. Actuators B, 2004, 98, 101–111 CrossRef.
- W. Satoh, H. Yokomaku, H. Hosono, N. Ohnishi and H. Suzuki, J. Appl. Phys., 2008, 103 Search PubMed 034903.
| This journal is © The Royal Society of Chemistry 2009 |