Microfluidic crystallization
Jacques
Leng
and
Jean-Baptiste
Salmon
*
Université Bordeaux-1, Laboratoire du Futur, 178 avenue du Docteur Schweitzer, F–33608, Pessac cedex, France. E-mail: jean-baptiste.salmon-exterieur@eu.rhodia.com
First published on 24th October 2008
Abstract
Microfluidics offers a wide range of new tools that permit one to revisit the formation of crystals in solution and yield insights into crystallization processes. We review such recent microfluidic devices and particularly emphasize lab-on-chips dedicated to the high-throughput screening of crystallization conditions of proteins with nanolitre consumption. We also thoroughly discuss the possibilities offered by the microfluidic tools to acquire thermodynamic and kinetic data that may improve industrial processes and shed a new light on nucleation and growth mechanisms.
I. Towards microfluidic crystallization
Nucleation and growth of crystals from a liquid phase is an experience of everyday life: production of salt by evaporation of seawater, formation of snowflakes at adequate temperature conditions… Understanding, predicting and optimizing crystallization mechanisms are also important needs in our industrialized world. The requested information specifically depends on the research field. For chemistry, crystallization is a key point of many processes (purification, food engineering, drugs synthesis…1–3) and the useful data are the solubilities, habits, existence of polymorphs, estimations of nucleation/growth rates etc. In structural biology, and due to an overwhelming genetic information, there is a huge demand for well-diffracting crystals of biological macromolecules. The determination of the tri-dimensional structures of these molecules from X-ray crystallography, is indeed essential to understand their functions, and for a rational design of specific drugs.In this work, we review how microfluidic technologies may assist the investigation of crystallization. The latter is a complex process involving nucleation and growth until at least one germ is visible, and thus couples kinetics to thermodynamics. Despite many experimental and theoretical developments, crystallization still remains a puzzling phenomenon,1,2 and there is no accurate theory to substitute for empirical approaches. We will show below how miniaturized fluidic tools permit a unique control of the kinetic pathways undergone in a phase diagram and also yield thorough insight into nucleation and growth processes for inorganic and biological molecules.
The crystallization process deals both with thermodynamic and kinetic features in multidimensional phase spaces as sketched in Fig. 1. Thermodynamic data are the solubility lines, the presence of metastable phases, polymorphs, liquid–liquid separation…, and they depend on multiple parameters such as the temperature, pH, solvent, impurities, etc. Additionally, kinetic trajectories in the phase diagram are relevant to control most of the final properties of the synthesized crystals. The path followed in the diagram controls the nucleation and growth of the crystals, and thus their number, size, and morphology. Major theoretical and computational work, and the emergence of model systems such as globular proteins and colloidal systems, recently brought insights into the general understanding of nucleation.4–7 Indeed, these soft matter systems display long relaxation times and tunable interactions that allow to partially decouple thermodynamics from kinetics, and thus to revisit the process of nucleation and growth under a microscope.
 | ||
| Fig. 1 Schematic view of a multidimensional diagram. T, c1 and c2 are, respectively, the temperature, the concentration in solute and in precipitating agent. Continuous lines are the solubilities and dashed lines indicate the presence of a metastable phase and the kinetic extent of the metastable zone. The arrow represents a complex and specific kinetic pathway. | ||
The benefits of microfluidics – studying crystallization sets specific objectives; we may want to collect thermodynamic or kinetic data, or grow good crystals for diffraction purposes. In the crystallization literature, there are essentially two strategies for getting such information: (i) adopt brute force as in combinatorial chemistry to multiply experiments; (ii) focus on a specific point to unveil fundamental mechanisms.
Microfluidics actually offers a range of tools that are helpful to revisit these two approaches of crystallization. The microfluidic technology is a toolbox to manipulate liquids in networks of microchannels with 1–100 µm length scales. Such networks mimic classical experiments performed in a laboratory, but with an unequalled control of the transport phenomena.8–10 In the specific context of crystallization,11,12 these fluidic tools essentially permit the manipulation of aqueous solutions around room temperature. The range of application was originally quite limited but constant progress in the microfluidic technology now yields original features that permits the researcher to:
1. Perform high-throughput data acquisition using crystallization assays down to 1 nL;
2. Design specific kinetic routes using the excellent control of the mass and heat transfers due to the reduction of the length scales and on-chip integration of sensors and actuators;
3. Bring new experimental conditions to investigate crystallization, with no turbulence, no or little gravity effect, confinement, and large surface/volume ratio. Additionally, the small volumes V of microfluidics are of special interest for nucleation. The mean nucleation time (∝ 1/V) may exceed the growth kinetics of the crystals13 and only one nucleation event is therefore statistically observed: this mononuclear mechanism is essential to estimate nucleation kinetics and investigate polymorphism.
In the following review, we describe the most recent microfluidic developments for studying crystallization. We focus first on the acquisition of fundamental data for chemical and pharmaceutical industries, with a detailed description of nucleation kinetics measurements. We then turn to the specific case of the protein crystallization for which microfluidics displays state of the art fluid manipulation at the nL scale.
II. Data acquisition for microfluidic crystallization
We start with the important and recurrent task of characterizing crystallization, as for drug discovery for instance. There is a need of high throughput and in situ analytical characterization (such as Raman spectroscopy, X-ray diffraction). The tools presented below challenge in efficiency, cost, and accuracy the most modern robotic platforms and are now nearly mature for an everyday laboratory use. We will detail how they provide original information on basic thermodynamic and kinetic features of crystallization. When required, we use classical nucleation theory as a guide for understanding crystallization. It relates the nucleation rate J, the number of critical nuclei produced per unit of time and volume to the supersaturation S (concentration/solubility), the actual driving force of nucleation.16,17 As a stochastic model, it defines the mean time of appearance of the first event of nucleation in a volume V: τn = 1/JV. While not perfectly adapted to nucleation in solution,16–18 this theoretical guide nevertheless points out the important control parameters offered by microfluidics, as for instance a very good control of V and thermal transfers upon cooling…We describe now several measurements that fully exploit these advantages.A. On-chip solubility measurement
The most important quantity when studying a solid compound is probably its solubility in various solvents. It indeed governs the crystallization process (temperature, solvent exchange, evaporation…), and plays an important role for the level of supersaturation that can be reached. Measurements of solubilities may be rather long and fastidious,1,2 whereas industrial researchers often need rapid screening of solubilities. Costly robotic workstations are thus often required.Laval et al. recently proposed a droplet-based microfluidic chip for rapid screening of solubility diagrams.14 This silicon/PDMS device permits the generation of aqueous nanolitre-sized droplets carried in an oil stream (see ref. 19,20 for reviews on two-phase flow microfluidics). Automated valves allow the storing of a bi-dimensional array of hundreds of aqueous droplets, at different compositions and temperatures (see Fig. 2a). By visual inspection of the droplets containing crystals, this array of ≈100 nL droplets permits a direct and quantitative reading of a solubility diagram concentration vs. temperature. In ref. 14, ten points of the solubility curve of a small organic molecule are estimated in only 1 h using ≈250 µL of solution. The future developments of such tools will concern the possibilities to deal with a large range of organic solvents, not always compatible with the standard PDMS technology.
 | ||
| Fig. 2 (a) On-chip solubility measurement: PDMS device to store hundreds of droplets of different concentrations and create two-dimensional array of droplets containing an aqueous solution at different concentrations and temperatures. Crystals appear as bright pixels using birefringence and solubility is directly read from such measurements (reproduced with permission from ref. 14). (b) Statistics: microfluidic device to store hundreds of nanolitre droplets and investigate polymorphism of aqueous solutions (reproduced with permission from ref. 15). This chip gives measurements of the solubilities of forms II and III of potassium nitrate in water, the corresponding habits in the droplets are shown (scale bars 100 µm). | ||
B. Probing polymorphism
For pharmaceutical and chemical industries, the control of polymorphism is essential since it governs, for instance, the biodisponibility of the active molecule, and more generally all the physical properties of the solid state (concerning polymorphism see ref. 21–24). Despite intense fundamental research, the following McCrone's statement “the number of polymorphs of a material is proportional to the time spent investigating” often holds.25 Again, high-throughput strategies involving robotics coupled with analytical measurements, is often necessary.26 Microfluidics, as a tool to perform multiple assays with small amounts of liquids, is thus promising.Recently, Shinohara et al. performed a microfluidic screening of C60crystallization that reveals various metastable phases.27 These authors exploit a very large interfacial area per unit of volume to identify metastable forms of C60 during its precipitation at an organic/alcohol liquid interface, that would not be observed in larger volumes. Laval et al. also developed recently a droplet-based device to investigate polymorphism15 (see Fig. 2b). First, hundreds of ≈100 nL droplets of an aqueous solution (potassium nitrate) are engineered in a silicone oil stream, and stored above its solubility temperature. Then, the temperature is decreased slowly until all the droplets contain crystals. In the range of investigated supersaturations, the volume of the droplets is small enough to induce mononuclear nucleation. Eventually, the temperature is slowly increased and dissolution temperatures of the crystals are measured. This thermal cycle reveals two different solubility temperatures. Raman microspectroscopy28 measurements performed directly in the droplets can confirm the vibrational signature of several species or polymorphs.
The mononuclear feature of the nucleation step is essential to explain these results. Indeed, polynuclear nucleation would certainly occur in larger volumes, resulting in the nucleation of the two different polymorphs in the same volume. Thus, polymorphic transitions would dissolve the metastable forms, and induce the growth of the stable ones.22 In the droplets, such polymorphic transformations cannot occur as nucleation is mononuclear most of the time. Moreover, other polymorphic transformations may be hindered because the mass transfers are governed by diffusion. Eventually, the small volume of the crystallizer allows one to reach large supersaturations that may unveil unexpected metastable forms.
C. Nucleation kinetics at the nL scale
Estimating the nucleation rate J in solution is a difficult task, and the available data remain confusing.16 First, it is a stochastic process and statistical measurements are required. The nature itself of the measured value must be questioned: the classical method consists in measuring the induction time τi between the application of a supersaturation S and the appearance of the first detectable crystals. But it is generally difficult to relate this induction time to the nucleation rate J: indeed, τi depends on the sensitivity of the detection device and many crystals may nucleate before the detection of the first one. Further, it implicitly includes a growth stage before the first germ is detectable.17 Additionally, a large sample of unavoidable impurities induce heterogeneous nucleation and homogeneous rates are thus often misestimated. Eventually, it is very difficult to apply sudden and homogeneous supersaturation in bulk crystallizers, particularly when dealing with thermal treatments.To overcome these difficulties, several authors proposed in the 50s the droplet method,29–31 that consists of investigating crystallization in small reactors. More precisely, an initial volume is divided into a large number N of small, identical and independent crystallizers of volume V. When N greatly exceeds the number of impurities present in the initial volume, some of them will not contain foreign nucleation sites and homogeneous nucleation may thus occur there. Moreover, the temporal evolution of the fraction of the reactors containing crystals can be related to the rate J when their volumes are small enough for mononuclear nucleation. In such a simplified case, the probability P(t) of a nucleation event is given by: P(t) = 1 − exp(− JVt), where t = 0 corresponds to the applied supersaturation. In a typical experiment using this method, the fraction ϕ(t) of reactors containing a crystal evolves at small time scales according to the heterogeneous mechanism since some reactors contain impurities. At long time scales, all these impurities induced the formation of crystals, and homogeneous nucleation occurs in the remaining reactors. Nucleation rate may then be estimated since ϕ evolves as log(1 − ϕ) ∼ − JVt.31
Since the pioneering work of Vonnegut, Turnbull, Pound and La Mer29–31 concerning the crystallization of supercooled droplets (mercury, tinetc.), this method has been used by many groups to estimate homogeneous rates of alkane crystallization,34,35 ice formation,36 and crystallization of fats.37 However, the droplet method raised many experimental difficulties, especially when dealing with solutions. Indeed, droplets are generally produced by emulsification of the solution in an inert phase, and are never perfectly monodisperse.38 Polydispersity therefore alters the effective nucleation frequency and makes the precise measurement of the nucleation rate J difficult.39Surfactants may also interfere with the bulk crystallization process, and surface nucleation may occur at the interface between the solution and the inert phase. Moreover, nucleation events in the different droplets may not be independent, especially in concentrated emulsions.34 Eventually, the detection of the fraction of droplets containing crystals is difficult to achieve experimentally and most of the time indirect.
Microfluidic technologies provide tools to overcome most of these experimental limitations. For instance, droplet-based systems provide a unique way to produce a large number of monodisperse reactors whose volume can be finely tuned. Thermal control is easily implemented on-chip, and analytical tools can be used to monitor the presence of the crystals in the droplets. White and Frost proposed in 1959 the first microfluidic droplet method for investigating nucleation kinetics in solution.32 In this innovative work, droplets of an aqueous solution are continuously formed in a mineral oil stream using a flow dropper similar to recent systems.40 The flow dropper is a microfluidic device consisting of a capillary tube nested in another one (see Fig. 3a). The volume of the droplets can be tuned between a few tens of nanolitres to one microlitre by varying the flow rates of the aqueous and oil streams. Droplets are then stored in a column of mineral oil, and supersaturation is created with a defined temperature quench. Microfluidics in such a case allows the production of perfectly monodisperse droplets acting as microreactors for crystallization, without the need of surfactants. This device has been used later in 1964 by Melia and Moffitt to investigate nucleation of various inorganic salts in aqueous solutions.41 Importantly, they demonstrated that nucleation is always heterogeneous for the investigated range of parameters.
 | ||
| Fig. 3 (a) Flow dropper developed by White and Frost in 1959 to produce monodisperse droplets. Right: evolution of the fraction of droplets that do not contain a crystal after a temperature quench below the solubility temperature of an aqueous solution (reproduced with permission from ref. 32). (b) A modern microfluidic device to produce droplets containing thermoresponsive colloids (PNIPAM). Right: polynuclear nucleation of colloidal crystals occurs in large droplets (500 µm size), whereas only one crystal nucleates in smaller droplets (100 µm size) (reproduced with permission from ref. 33). | ||
Using similar ideas and devices, Gong et al. recently measured the kinetics of nucleation of colloidal crystals33 (see Fig. 3b). Droplets of thermoresponsive colloids were engineered using a flow dropper device and stored for further analysis. Crystallization was induced by cooling, and the authors identified using microscopy a maximal droplet size for mononuclear nucleation (V < 1 nL). The evolution of the fraction of droplets containing colloidal crystals provided estimations of the nucleation rates that were different from those performed in bulk using light scattering. Bulk measurements indeed overestimate the kinetics due to heterogeneous and polynuclear nucleation. The same year, Dombrowski et al. developed a similar microfluidic device to study the crystallization of lactose.42 Droplets of an aqueous lactose solution were engineered using a T-junction and incubated in a PTFE (polytetrafluoroethylene) tube, at a given cooling temperature. For the investigated range of droplet volume and supersaturation, polynuclear nucleation occured. The measurement of the fraction of droplets containing zero, only one, or multiple crystals at a given time led to estimations of the nucleation kinetics. The authors also point out that the crystal size distribution of the particles produced using such a device was significantly decreased compared to the classical process in large crystallizers. Also the same year, Laval et al. developed a microfluidic PDMS device to perform a continuous droplet method on-chip.43 Monodisperse droplets (<100 nL) of an aqueous solution were continuously produced above the solubility temperature of the investigated solution, and carried by a silicone oil stream. With a specific channel network and thanks to a controlled thermal gradient, large temperature quenches were applied to the droplets as they flowed downstream a long microchannel (up to 50 °C in 10 s). The long residence time in the device permits the performance of continuous measurements of the fraction ϕ of droplets containing crystals, from 10 s up to a few minutes. The confrontation of data obtained on an aqueous potassium nitrate solution with the classical nucleation theory revealed heterogeneous mechanisms.
For proteins and organic molecules, there is to our knowledge, no microfluidic developments of the droplet method to measure nucleation kinetics. Several groups used robotic approaches44 or iterative measurements45 to estimate nucleation rates of model proteins. For these complex molecules, growth rates are intrinsically small and mononuclear mechanisms do not occur even if the volumes involved are small. Specific supersaturation profiles such as those used in ref. 44 are therefore needed to perform precise rate measurements. Droplet-based microfluidics permits the manipulation of microreactors on-chip, and will thus certainly prove useful in the near future to investigate nucleation kinetics of biological molecules.
D. Nucleation and growth in confined geometries
There is virtually no work devoted to crystalline growth in microfluidic cells. This might be due to the ubiquitous yet undesirable clogging of channels upon apparition of solid particles. While droplets prevent blocking and are unique for studying nucleation, they are not quite adequate for the investigation of growth. Indeed, they are of finite volume and are thus soon exhausted after nucleation. Instead, an open system such as a microevaporator46 is used to induce the nucleation and growth of electrolyte crystals in complex geometries (Fig. 4). | ||
| Fig. 4 Examples of directed growth of electrolyte crystals in confined microevaporation geometries:46 (a) linear growth of copper sulfate, here showing faceted crystals; (b) exploiting microfabrication for imparting a right-angle turn to a growing cubic lattice of a potassium chloride; (c) copper sulfate observed in between crossed polarizers and growing in a forest of PDMS poles. The thickness of the crystals is about 20 µm in the three pictures. | ||
These tools use a membrane to extract solvent from a microchannel and to concentrate virtually any solute: as the channel in contact with a membrane is connected to a reservoir, evaporation drains the solution from the reservoir and progressively concentrates the solute at the tip of the channel. The device has proved useful for inducing the crystallization of several species46 with access to quantitative data about nucleation.
Soon after nucleation, growth takes place as the crystal is continuously fed by fresh solute advected from the reservoir. Fig. 4a shows an example of long needle-like crystals of electrolyte that grew at a pace imparted by geometry, and which is in this case adequate for getting afaceted crystals. Interestingly, the habit of the crystals seems to change significantly when fed at an extremely low rate.46
Microevaporation is a natural tool for crystal growth, as by essence it feeds continuously a nucleated crystal. Note also that the absence of turbulent and gravity effects makes the technique prone to growing good quality crystals. Microfabrication may also add up an important contribution with the extreme control of fabricated geometry. The coupling of a neatly controlled growth and specific geometries leads to results of Fig. 4b and c which illustrate the emergence of experimental conditions which were not possible before. Fig. 4b shows a cubic atomic crystal growing at a controlled rate in a geometry compatible with its structure, a right-angle turn: the crystal changes facet to carry on the growth. Fundamental questions arise: what would happen during the growth in a more progressive turn? How does the symmetry of the crystal get coupled to the symmetry of the complex geometry? A more complex situation (Fig. 4c) couples two geometries: that of a non-cubic lattice growing in a channel pierced with a square lattice of poles. The two lattices are not expected to interfere too strongly; yet, the crystal nucleates a region of defects after crossing a pole which is of considerable extent and may alter the growth. The extension of this work to colloidal systems seems promising as it might be more straightforward to investigate the fault managing system (as compared to molecular systems) and how a material may heal during the growth process.
III. Microfluidic devices for protein crystallization
Since there is no way to predict a priori the crystallization conditions of a given protein (except for globular ones5), also because multi-dimensional phase diagrams have to be explored (pH, temperature, crystallizing agents…) and due to the minute amounts of bio-molecules available, high-throughput approaches consuming small amounts are required. The current strategies consist of:47,48 (i) performing random screening using sparse matrix reactants known to induce crystallization of other proteins; (ii) making optimizations (change the kinetics, finely tune the concentrations, control mass and heat transfers) around the crystallization hit to improve the diffraction qualities of the crystal; (iii) harvesting the crystal and perform X-ray measurements. Since high-throughput is required, and because proteins are often available in small quantities, robotic tools manipulating 50 nL–1 µL volumes of liquids are often used.47–51 Such small volumes are not a limitation since the minimal size of the crystals can be of the order of ≈50 µm thanks to the developments of X-ray optics.52Moreover, kinetic aspects always play an important role, and nucleation and growth of the crystals have to be finely tuned to obtain suitable diffraction features.53 Crystal growth conditions are optimal in the metastable zone but nucleation rarely occurs there, whereas nucleation is favored in the labile zone, but growth conditions lead there to crystals of poor diffraction qualities (see Fig. 5).
 | ||
| Fig. 5 Typical solubility diagram of a protein (adapted from ref. 53), and some crystallization conditions: vapor-diffusion (blue), batch (green), and free-interface diffusion FID (red). | ||
Therefore, optimized experiments rely on well-defined trajectories in the phase diagram. Microbatch crystallization only investigates a fixed supersaturation S, and thus does not exploit any kinetic path through the phase diagram. For fast nucleation kinetics however, mixing of the different compounds can influence the final crystalline state. Methods such as free-interface diffusion FID and counter-diffusion (gradient of S both in space and time), vapor-diffusion (increase of S until equilibrium is reached), and dialysis (increase of S at fixed concentration of protein) correspond all to different kinetic trajectories in the phase diagram (see Fig. 6a), that help to optimize crystallization. Performing high-throughput screening of both kinetic routes and crystallization conditions is highly necessary but difficult to implement with the classical means. Microfluidics, as shown below, can improve the high-throughput strategy (smaller consumption with more assays), and the control of the kinetic pathways to grow crystals with acceptable qualities.
 | ||
| Fig. 6 (a) Microfluidic FID device containing 144 crystallization chambers. Middle: details on three of these 25 nL chambers, and right: thaumatin crystal obtained using this method (reproduced with permission from ref. 54). (b) Left: screening of crystallization conditions of a membrane protein in ≈15 nL droplets (reproduced with permission from ref. 58). Middle: array of ≈40 nL wells in a PDMS chip containing different reactants, and right: crystals of an unknown protein obtained using this chip (reproduced with permission from ref. 63). | ||
A. Miniaturized free-interface diffusion devices for high-throughput crystallization down to several nL
The first microfluidic device for high-throughput screening of protein crystallization conditions is a miniaturized and parallelized version of the free-interface diffusion (FID) method by Hansen et al.54 This glass/PDMS device integrates pumps and valves (up to 480 valves in the present version, see Fig. 6a) and is automated to perform 144 crystallization trials in 25 nL reaction chambers on the same chip. Since PDMS is gas-permeable, priming of all the wells is possible with only a few microlitres of a protein sample, by pipetting manually different crystallizing agents. FID starts with well-defined interfaces in all the chambers when valves are opened. Miniaturization of the crystallizer is essential: mixing between crystallizing agents and proteins only occurs by molecular diffusion. There is no buoyancy-driven convection instabilities at such small length scales that would induce unwanted mixing and may disturb the growth of crystals. Moreover, the lithography permits the precise design of the geometries of the crystallization chambers, and therefore to play with the kinetics of mixing, without any changes on the final equilibrium states.B. Droplet-based microfluidics for high-throughput screening of crystallization conditions
Ismagilov et al. proposed to screen crystallization conditions of proteins using two-phase flow microfluidics. These devices exploit the possibility to generate aqueous nanolitre-sized droplets carried in an inert oil stream.55,56 Such droplets containing given amounts of crystallizing agents and proteins, act as small crystallizers whose volume can be precisely fixed (from about 0.1 to 100 nL).In ref. 55,56 the screening is performed by continuous changes of the flow rates of different aqueous streams (protein, crystallizing agents) before the formation of the droplets. Providing a low frequency droplet formation (≈2 Hz for 7.5 nL droplets in ref. 55), the authors manage to control the concentrations within a 15% level of confidence. An array of droplets with different crystallization conditions is therefore created, and can be incubated either in the PDMS chip or in an external glass capillary once flow rates are stopped. Using such techniques, many conditions can be screened with small amounts of consumed proteins (several µL).
A crucial issue in such experiments, is the nature of the oil. Perfluoroalkanes oils (PFP) prevent any transport of water from the droplets over long periods of time (up to a few weeks as mentioned in ref. 56), and the droplets thus act as perfect microbatch reactors. Using water-permeable oils, it is however possible to reproduce vapor-diffusion experiments. Indeed, the authors managed to store arrays of droplets with alternating compositions, and the chemical potential difference between the droplets concentrates or swells the plugs containing the proteins. Such a control of the kinetic pathway in the phase diagram will be discussed in section IIIE.
The same group proposed later a simpler approach using stored arrays of droplets.57 Basically, several ≈15 nL plugs containing different aqueous reactants in PFP are prepared manually and stored in a PTFE tube. To avoid coalescence and water permeation between these droplets, they are separated by gas plugs. This array can be stored for months when transferred in a glass capillary. To screen crystallization conditions, the array is first transferred again into a PTFE tube which is then inserted in a PDMS T-junction. This chip permits the merging of the array with a protein stream driven at a given flow rate. In the conditions studied by these authors, the protein stream merges within the different plugs of the array, and the resulting droplets are collected again in a glass capillary for incubation. These authors managed to screen 48 different conditions of crystallization provided by a commercial sparse matrix, using only 1 µL of a model protein.
Once hits of crystallization are found using random screening, one generally tries to optimize the crystallization by slight changes of the conditions. Liet al. proposed a hybrid method including the ideas of the works55,57 mentioned previously, to perform simultaneously the screening and the subsequent optimization in nanolitre droplets.58 The idea consists of storing an array of large drops (≈100–150 nL) containing different precipitants as in ref. 57. All these droplets are again separated by gas plugs to avoid coalescence and contamination. This array is then inserted in a PDMS chip, and flowed under pressure. The large plugs then form long segments that act as continuous streams and form, when combined with a protein and a buffer streams, smaller droplets (≈15 nL) whose composition can be varied by continuous changes of the flow rates. These plugs are again stored in a PTFE tube for incubation (see Fig. 6b). Using such a hybrid method, Liet al. performed up to 1300 crystallization trials in ≈10 nL plugs, using only 10 µL of sample, and showed successfully that this strategy is suitable for handling solutions of membrane proteins.58 This method, however, requires a high control of the microfluidic aspects of droplet flows. First, one needs to store arrays of drops and manipulate them as long streams in a PDMS chip. Second, one also needs to implement continuous changes of the flow rates to change the composition. Indexing the droplets is an important issue because thousands of different reactors can be formed in less than 20 min. In the work of Liet al., indexing the droplets is done using a continuous change of their volume by continuously changing the carrier fluid flow rate. Finally, the input array and the output droplets are stored in long PFTE tubes inserted in the PDMS chip to avoid evaporation for long term storage.
Several problems when dealing with droplets have to be mentioned. First, elasticity of PDMS devices and injection systems often induce long transients with pressure-driven flows, and large volumes of samples (several µL) may therefore be consumed before droplet flows are stable (this stability is required to know the concentrations). More importantly, difficulties often concern the mechanism of droplet formation itself. Indeed, it requires a good control of surface chemistry, and strongly depends on many parameters such as the viscosities, the geometries of the channels, the surface tensions etc. Even if recent works successfully captured the fundamental mechanisms,59,60 a complete understanding of droplet formation is still missing. It may thus be difficult for a group without a strong knowledge of two-phase flows and without any microfluidic know-how, to create droplets, to control their traffic and their merging, namely when dealing with membrane protein solutions or with solutions presenting very different properties (viscosity, surface tensions etc.).
Ismagilov et al. did major work concerning droplet microfluidics19 and concerning the chemistry of the surfactants and oils used to perform the above experiments.61 Indeed, in order to mimic perfect microbatch conditions, they identified fluorinated oils that were nearly impermeable to water, and synthesized oligoethylene glycol-capped perfluorinated surfactants that prevented protein adsorption at the interface of the droplets. To handle membrane proteins, they also used perfluoroamines that provided high surface tension.
C. Engineered microbatch experiments
Another strategy relies on the microfabrication of microbatch using lithography techniques. Early on, Juarez et al. manufactured a 10 × 10 array of 5 µL wells etched in a silicon wafer.62 The silicon presents a high thermal conductivity and therefore linear temperature gradients can be applied across the array of wells (typically from 15 to 35 °C in ref. 62). Such a microfabricated plate permits screening with only 250 µL of protein solution manually pipetted in the wells, different conditions of temperature that give different nucleation and growth mechanisms. There is however, no microfluidic handling of the fluids, and microfabrication is only used to manufacture specific geometries in given substrates.Recently, Zhou et al.63 proposed a microfluidic approach that does not require the use of valves or syringes for handling fluids. The authors microfabricate an array of wells (typically 150 wells of 20 nL), in various substrates such as PDMS, glass or poly(methyl methacrylate) (PMMA). For priming these wells, PDMS microchannels are fabricated and aligned above them. As proposed by Hansen et al.,54 aqueous solutions plugged at the inlets of the device naturally fill the chambers when PDMS is previously degassed. All the wells can thus be filled with only 5 µL of protein sample. Then by aligning chambers filled by the same method with different precipitants, an array of crystallization conditions is created without any syringes or valves (see Fig. 6b). Mixing between proteins and precipitants occurs through a FID route as in the device of Hansen et al., even if the valves in ref. 54 allow a better control of the kinetics of mixing. In the above chips, evaporation is a key point, since long incubation times are often required. To overcome these difficulties, layers of paraffin oil covering wells etched in glass slides prevent evaporation for months, as noted in ref. 63.
D. Microfluidic phase diagram determination to optimize crystallization conditions
Generally, hits of crystallization are randomly searched using reactants known to crystallize other proteins. Then conditions have to be optimized to improve the quality of the crystals. To rationalize such a strategy, Hansen et al. developed a microfluidic method consisting roughly in three steps.64 First, a vast screening of the solubility of a given protein is performed in a parameter space including different buffers and crystallizing agents, but also different protein concentrations. Promising conditions are found within this solubility fingerprint when instantaneous precipitation is observed. This screening does not measure the thermodynamic solubility, but the kinetic extent of the metastability limit, sometimes called the supersolubility.18 In a second step, precise phase diagrams are obtained around the promising conditions revealed previously, in order to estimate the limit of the supersolubility of the target protein. Conditions that reside just below the supersolubility are propitious for crystallizing the protein. Eventually, crystallization trials are performed at these conditions, using either microfluidic FID described above,54 or conventional microbatch or vapor-diffusion experiments.This ambitious strategy is made possible thanks to the development of a microfluidic formulator64 (see Fig. 7). This complex chip, based on the PDMS multilayer technique, integrates a microfluidic multiplexor and a rotary mixer.65,66 Mixtures from 32 reactants (salts, polymers, proteinsetc.) are generated using this chip at precise known concentrations in a 5 nL ring. The rotary device then mixes the reactants in a few seconds, and image analysis reveals whether precipitation occurs at this stage. About 4000 titration experiments can be performed in only one day, and using only 8 µL of protein sample. The formulator chip is used in the same way to screen phase diagrams around the most promising conditions found during the measurement of the solubility fingerprint. Each phase space consisting of 72 different mixtures, is performed using only ≈100 nL of protein sample, and helps to define the extent of the metastable region (see Fig. 7). This strategy has been applied successfully to crystallize diverse proteins known to be difficult to crystallize.67,68 Such a method also significantly improves the probability of obtaining crystals, compared to the classical random strategy. Moreover, it is important to mention the high-throughput features of this device, that consumes less sample (a few µL) for more tests (thousands of titrations per day).
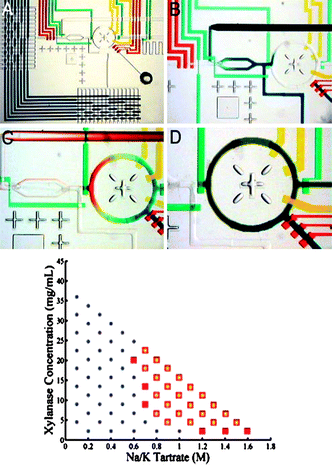 | ||
| Fig. 7 Microfluidic formulator to investigate protein phase diagram. The diameter of the mixing ring is 1.5 mm. This device allows an automated exploration of the protein supersolubility as shown below on a given example, squares indicate the precipitation region (reproduced with permission from ref. 64). | ||
E. Passive and active control with kinetics
As already mentioned, kinetics plays a highly relevant role to obtain good quality crystals for X-ray measurements. This is particularly relevant to the field of protein crystallization which is prone to metastability with several habits, polymorphs, and other out-of-equilibrium structures (e.g. gels, clusters69). Microfluidics, as demonstrated below, offers great opportunities to precisely follow defined kinetic pathways such as those displayed in Fig. 5.Often mentioned but rarely discussed, evaporation is a real problem for long term storage, especially in microfluidic devices (droplets, micro-wells). Volume loss and concentration evolution lead to the wrong determination of supersaturations that are involved during crystallization. If getting a crystal is often the only goal of the study, knowing the exact conditions is, however, important to reproduce the crystallization conditions in other systems, for instance vapor-batch diffusion. To bypass the difficulty due to evaporation, the chosen route is to promote it instead of preventing it. More precisely, by contacting microsystems with an osmotic bath, it is possible to regulate the exchanges between the microsystem and the bath so to control the chemical potential of a solution. Including an exchange membrane is thus a key issue for technological improvement of microsystems. While many microsystems incorporate polymeric membranes for dialysis, concentration, filtration, etc.,70 the role of a PDMS membrane is of special interest, it being the core material of many microfluidic chips. Indeed, PDMS is permeable to gas and liquids such as water, even though hydrophobic, and has been extensively used as such since the 60s in the industry for pervaporation.71 In PDMS microsystems dedicated to protein crystallization, evaporation through the PDMS can be a major disadvantage for long term storage72 and humidity around the chip has to be controlled.73 Furthermore, osmotic regulation across PDMS membranes has been implemented in several microsystems and the most advanced ones do combine several modules: formulation, addressing, and concentration/dissolution cycles. As shown below, such osmotic regulation also permits the tuning of the kinetic paths followed in the phase diagram during a crystallization experiment.
Using this approach, Fraden's group developed the so-called phase chip which is designed to efficiently screen the phase diagram of multi-component aqueous systems (see Fig. 8a). The two main innovations of this droplet-based microsystem are (i) to dock droplets (initially formulated) into wells where (ii) they are in contact through a thin layer of oil with a PDMS membrane and an osmotic bath underneath. In this osmotic bath either dry gas or water at fixed chemical potential, tuned with a dissolved salt (NaCl), is flowed. In a set of two papers,74,75 the authors carefully check the working conditions of the device: they are able to swell or concentrate droplets with excellent kinetic control (see Fig. 8a). A typical swelling/shrinkage time is ≈1 h, but depends on the ionic strength of the bath, as quantitatively described by a simple model. These osmotic conditions are used to study phase diagrams of aqueous solutions such as polymer and salt in water: the latter system is successfully studied and homogeneous phases along with liquid–liquid decomposition are observed.74 The chip is then used to study the crystallization of model proteins75 with a neat kinetic control: in particular, the pathway followed in the experiments permits the formation of many nuclei (high supersaturations), most of which are then dissolved by osmotic swelling of the drop to select only a few of them, that will eventually produce good quality crystals in the sense of X-ray scattering. It is therefore possible via an osmotic control to decouple nucleation and growth of protein crystals (see Fig. 8a).
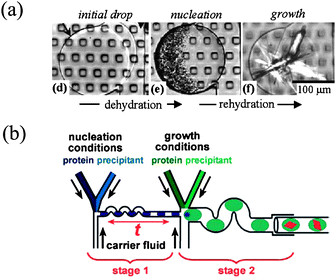 | ||
| Fig. 8 (a) Phase chip developed by Shim et al. to control the crystallization conditions in stored nanolitre droplets, using dialysis through a PDMS membrane (reproduced with permission from ref. 75). (b) Schematic time-controlled seeding droplets to separate nucleation and growth stages (reproduced with permission from ref. 78). | ||
Analogue results based on osmotic exchange were obtained in a large scale version of FID76 with valves that control the diffusion process and a membrane that offers an osmotic regulation. It is used to crystallize several proteins and to investigate what are the ideal conditions. Additionally, the authors observe that the crystallization habits are modified by the experimental conditions, but no explanation is given. As explained later in section IIIF, one of the main advantages of the chip is to directly allow the use of cryoprotectant to shield the crystal from X-ray damages.
A last example of membrane-controlled crystallization is given by Lau et al.77 This complex chip integrates a formulation stage, a droplet injector, and a two-phase storage module. The formulator, as described previously in section IIID, gives estimations of the limit of metastability by monitoring the occurrence of a precipitate in a 5 nL mixture of protein and reagents.64 Furthermore, this chip allows the formation of nanolitre droplets from the formulated mixtures, and their incubation in a storage module. The latter is associated to a membrane exchange zone which insures a constant chemical potential of the plugs, unlike preliminary versions of the same device where evaporation was problematic. Here, the real strength is the real-time feedback on precipitation, and the further kinetic optimization of crystallization, through an active control of the osmotic bath.
Droplet-based crystallization assays are thus interesting for controlling a kinetic pathway viamembrane exchange. Analogous yet less efficient is the exchange between droplets in a train across the oil separating them.56 As mentioned previously in section IIIB, it is indeed possible to alternate droplets of the solution to crystallize and droplets of brine that are used as a reservoir to concentrate the protein solution by diffusion across the oil spacer. The choice of the latter is important, along with the distance between droplets which control the (slow) maturation process. However, the evolution is unidirectional, there is no active control on the concentration process, unlike the case of membrane exchange. Besides evaporation, droplets are used for another kinetic pathway: the so-called seeding technique for crystal growth. It essentially decouples the nucleation of a seed from the growth of the latter into another bath were conditions are optimized. In general, high supersaturation is required for nucleation of seeds while low supersaturation is needed for ordered growth, and the two sets of conditions are often disjointed. Ismagilov's group uses this technique to induce the formation of seeds in highly saturated mixtures of proteins and precipitant (nucleation conditions) and then sends the droplets into larger ones where the solution gets diluted to reach ordered growth conditions78 (see Fig. 8b). The number of seeds can be optimized by tuning the incubation time prior to lowering the supersaturation. The two stages are thus decoupled and challenging proteins were crystallized.
Eventually, microfluidics allows passive action on the kinetics through a neat control of the mass transfers, since it involves optimized geometries (designed by lithography) with small length scales. The microfluidic FID described previously in section IIIA is a perfect example: through the design of the geometry of the connecting chambers (see Fig. 6a), it is possible to tune the pathway followed during the FID route to the final mixed state.54 In a recent work by Sauter et al.,12channel networks were manufactured to reproduce counter-diffusion crystallization experiments in a microfluidic format. Gradients of crystallization conditions depending on the channel geometries are obtained. In all these devices and due to the small length scales, mixing of the compounds only occurs through molecular diffusion, and the design of the channel network permits it to act on the mixing kinetics. Using similar ideas, Taljera et al. manufactured an evaporation-based crystallization platform to identify and optimize conditions for protein crystallization.79 In this device, microlitre-sized droplets containing known concentrations of protein and reactants evaporate through a channel of well-defined geometry. The design of the channel geometry permits the precise control of the rate of evaporation, and thus the kinetic pathway through the phase diagram. Using such a precise control of the evaporation, and thus on the drying rates, these authors demonstrate recently that kinetic parameters (nucleation, crystal growth) can be estimated from such experiments using kinetic models.80
F. Crystal harvesting vs. on-chip X-ray diffraction
Once a protein crystal is obtained, one has to harvest it and mount it for X-ray measurements. To reduce radiation damage due to the X-rays, and also to minimize thermal noise inducing unwanted background, crystals are often flash-frozen in liquid nitrogen. To avoid cracking of the crystals during this precarious step, crystals are generally soaked with a cryoprotectant solution before freezing. Harvesting and cryoprotecting steps are both difficult since diffraction qualities of the crystals may be altered.Standard crystal harvesting is often possible in the PDMS chips mentioned previously,54,63 since these microfluidic devices are not irreversibly sealed. However, X-ray diffraction measurements performed in-situ are promising to minimize harvesting perturbations. As shown recently, X-ray analysis can be performed directly on-chip in the PDMS devices developed by Hansen et al.76 Cryoprotectant solutions were introduced in the crystal wells by diffusion over a long period of time, and sections of the PDMS chip containing the crystals were frozen by immersion in liquid nitrogen. Issues concerning the effects of the PDMS membranes on the flash-freezing step, X-ray absorption, and scattered background are discussed in the supporting information of ref. 76. Strong X-ray absorption by the PDMS may be a limitation for thick devices, and other polymeric materials may thus be interesting leads.81,82 Sauter et al. already showed successful X-ray diffraction analysis of protein crystals grown by counter-diffusion in thick PMMA chips.12
In the droplet-based devices, crystal harvesting is a priori a highly precarious step, and in-situ data collection seems to be unavoidable. Zheng et al. demonstrate X-ray diffraction performed directly in the capillaries used to store the droplets,56 but they also mentioned strong X-ray damages due to the lack of cryocooling. More recently, the same group investigates the possibilities and limitations of in-situ data collection in such droplet-based systems.83 Critical issues such as the material of the capillaries (glass vs.PTFE), the absorption and diffraction background due to the oil phase, and also the radiation damages, are thoroughly discussed in this reference. These authors also point out that X-ray analysis performed on multiple droplets containing identical crystals may be convenient to overcome the above difficulties.
G. Heterogeneous nucleation and the role of the surfaces
One point rarely mentioned concerned the possibility of heterogeneous nucleation in microfluidic devices. Indeed, at these small length scales, the ratio surface/volume is large and surface effects are likely to play an active role. Recent work evidenced the role of the surface of the vessels on the nucleation kinetics of the crystals,84 and point out the important role of the surface chemistry of the microfluidic crystallizers. Sanjoh et al. used microfabrication techniques to engineer microfluidic devices with a patterned silicon wafer.85 The patterning of different doping sites (p-Si and n-Si) creates different surface charge conditions on the wafer that act as a spatial nucleation site, as shown on a model protein. This original work paves the way for many possible experiments to study the fundamental role surfaces play on the nucleation and growth mechanisms.IV. Conclusion
As shown all along this review, microfluidics offers unequalled experimental conditions to explore the complexity of crystallization in solution. These opportunities are mainly due to the possibility to decrease the investigated volumes and increase the number of assays (high-throughput screening), but also due to the excellent control of the transport phenomena at these small length scales. This active control on the kinetic pathway is essential to obtain reliable data and protein crystals with suitable diffraction qualities. Finally, microfluidics lead to uncommon conditions through the reduction of the volumes of the crystallizers: growth in confined geometries, mononuclear nucleation, etc., that bring a new understanding of the nucleation and growth mechanisms.The microfluidic technologies are quite recent, and the current developments of the lab-on-chip still intensify. Some of the works detailed above have already found some commercial applications, namely in the field of protein crystallization, and we believe that more companies and laboratories will implement these new techniques in the near future. We hope such promising technologies and uncommon experimental conditions will shed a new light on the fundamental mechanisms of nucleation and growth.
Acknowledgements
We acknowledge the precious help from P. Laval, and M. Joanicot.References
- S. Myerson, Handbook of industrial crystallization (Butterworth-Heinemann, 2002) Search PubMed.
- J. W. Mullin, Crystallization (Butterworth-Heinemann, Oxford, 2001) Search PubMed.
- N. Rodriguez-Hornedo and D. Murphy, Journal of Pharmaceutical Sciences, 1999, 88, 651 Search PubMed.
- P. R. ten Wolde and D. Frenkel, Science, 1997, 277, 1975 CrossRef.
- W. C. K. Poon, Phys. Rev. E, 1997, 55, 3762 Search PubMed.
- C. Desgranges and J. Delhommelle, Phys. Rev. Lett., 2007, 98, 235502 CrossRef.
- R. P. Sear, J. Phys.: Condens. Matter, 2007, 19 Search PubMed 033101.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977 CrossRef CAS.
- T. Vilkner, D. Janasek and A. Manz, Anal. Chem., 2004, 76, 3373 CrossRef CAS.
- H. A. Stone, A. D. Stroock and A. Ajdari, Annu. Rev. Fluid Mech, 2004, 36, 381 CrossRef.
- M. van der Woerd, D. Ferree and M. Pusey, J. Struct. Biol., 2003, 142, 180 CrossRef CAS.
- C. Sauter, K. Dhouib and B. Lorber, Cryst. Growth Des, 2007, 7, 2247 CrossRef CAS.
- D. Kashchiev, D. Verdoes and G. M. van Rosmalen, J. Crystal Growth, 1991, 110, 373 CrossRef.
- P. Laval, N. Lisai, J.-B. Salmon and M. Joanicot, Lab Chip, 2007, 7, 829 RSC.
- P. Laval, C. Giroux, J. Leng and J.-B. Salmon, J. Crystal Growth, 2008, 310, 3121 CrossRef CAS.
- A. C. Zettlemoyer, Nucleation, Marcel Dekker, New-York, 1969 Search PubMed.
- D. Kashchiev and G. M. van Rosmalen, Cryst. Res. Technol, 2003, 38, 555 CrossRef CAS.
- J. M. García-Ruiz, J. Struct. Biol., 2003, 142, 22 CrossRef.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem. Int. Ed, 2006, 45, 7336 CrossRef CAS.
- S.-Y. Teh, R. Lin, L. H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198 RSC.
- S. Veesler, F. Puel and G. Fevotte, STP Pharma Pratiques, 2003, 13, 1 Search PubMed.
- K. Sato, J. Phys. D: Appl. Phys., 1993, 26, 77.
- J. A. McCauley, R. J. Varsolona and D. A. Levorse, J. Phys. D: Appl. Phys., 1993, 26, 85.
- J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem. Int. Ed, 1999, 38, 3440 CrossRef.
- R. J. Davey, Chem. Commun, 2003, 13, 1463 Search PubMed.
- M. L. Peterson, S. L. Morissette, C. McNulty, A. Goldsweig, P. Shaw, M. LeQuesne, J. Monagle, N. Encina, J. Marchionna and A. Johnson, J. Am. Chem. Soc., 2002, 124, 10958 CrossRef CAS.
- K. Shinohara, T. Fukui, H. Abe, N. Sekimura and K. Okamoto, Langmuir, 2006, 22, 6477 CrossRef CAS.
- P. A. Anquetil, C. J. H. Brenan, C. Marcolli and I. W. Hunter, Journal of Pharmaceutical Sciences, 2003, 92, 149 Search PubMed.
- B. Vonnegut, J. Colloid Sci., 1948, 3, 563 CrossRef CAS.
- D. Turnbull, J. Chem. Phys., 1952, 20, 411 CrossRef CAS.
- G. M. Pound and V. K. LaMer, J. Am. Chem. Soc., 1952, 74, 2323 CrossRef CAS.
- M. L. White and A. A. Frost, J. Colloid Sci., 1959, 14 Search PubMed.
- T. Gong, J. Shen, Z. Hu, M. Marquez and Z. Cheng, Langmuir, 2007, 23, 2919 CrossRef CAS.
- A. B. Herhold, D. Ertas, A. J. Levine and J. H. E. King, Phys. Rev. E, 1999, 59, 6946 Search PubMed.
- H. Kraack, E. B. Sirota and M. Deutsch, J. Chem. Phys., 2000, 112, 6873 CrossRef CAS.
- G. R. Wood and A. G. Walton, J. Appl. Phys., 1970, 41, 3027 CrossRef CAS.
- J. N. Coupland, Current Opinion in Colloid and Interface Science, 2002, 7, 445 CrossRef CAS.
- J. B. Newkirk and D. Turnbull, J. Appl. Phys., 1955, 26, 579 CrossRef CAS.
- D. Kashchiev, N. Kaneko and K. Sato, J. Colloid Interface Sci., 1998, 208, 167 CrossRef CAS.
- A. S. Utada, E. Lorenceau, D. R. Link, P. D. Kaplan, H. A. Stone and D. A. Weitz, Science, 2005, 308, 537 CrossRef.
- T. P. Melia and W. P. Moffitt, J., Colloid Sci., 1964, 19 Search PubMed.
- R. D. Dombrowski, J. D. Litster, N. J. Wagner and Y. He, Chemical Engineering Science, 2007, 62, 4802 Search PubMed.
- P. Laval, J.-B. Salmon and M. Joanicot, J. Crystal Growth, 2007, 303, 622 CrossRef CAS.
- O. Galkin and P. G. Vekilov, J. Phys. Chem. B, 1999, 103, 10965 CrossRef CAS.
- D. Knezic, J. Zaccaro and A. S. Myerson, J. Phys. Chem. B, 2004, 108, 10672 CrossRef CAS.
- J. Leng, B. Lonetti, P. Tabeling, M. Joanicot and A. Ajdari, Phys. Rev. Lett, 2006, 96 Search PubMed 084503.
- N. E. Chayen, J. Crystal Growth, 1999, 198, 649 CrossRef.
- J. R. Luft, J. Wolfley, I. Jurisica, J. Glasgow, S. Fortier and G. T. DeTitta, J. Crystal Growth, 2001, 232, 591 CrossRef CAS.
- N. E. Chayen, P. D. S. Stewart and D. M. Blow, J. Crystal Growth, 1992, 122, 176 CrossRef CAS.
- R. Hui and A. Edwards, J. Struct. Biol., 2003, 142, 154 CrossRef CAS.
- T. S. Walter, J. M. Diprose, M. Pickford, R. J. Owens, D. I. Stuart and K. Harlos, J. Appl. Cryst, 2003, 36, 308 CrossRef CAS.
- R. C. Stevens, Curr. Opin. Struct. Biol., 2000, 10, 558 CrossRef CAS.
- J. R. Luft and G. T. DeTitta, Methods in enzymology, 1997, 276, 110 CAS.
- C. L. Hansen, E. Skordalakes, J. M. Berger and S. R. Quake, Proc. Natl. Acad. Sci. USA, 2002, 99, 16531 CrossRef CAS.
- B. Zheng, L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 11170 CrossRef CAS.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem. Int. Ed, 2004, 43, 2508 CrossRef CAS.
- B. Zheng and R. F. Ismagilov, Angew. Chem. Int. Ed, 2005, 44, 2520 CrossRef CAS.
- L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice and R. F. Ismagilov, Proc. Natl. Acad. Sci. USA, 2006, 103, 19243 CrossRef CAS.
- P. Garstecki, M. J. Fuerstman, H. A. Stone and G. M. Whitesides, Lab Chip, 2006, 6, 437 RSC.
- P. Guillot, A. Colin, A. S. Utada and A. Ajdari, Phys. Rev. Lett., 2007, 99, 104502 CrossRef.
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785 CrossRef CAS.
- G. Juárez-Martínez, P. Steinmann, A. W. Roszak, N. W. Isaacs and J. M. Cooper, Anal. Chem., 2002, 74, 3505 CrossRef CAS.
- X. Zhou, L. Lau, W. W. Lam, S. W. Au and B. Zheng, Anal. Chem., 2007, 79, 4924 CrossRef CAS.
- C. L. Hansen, M. O. Sommer and S. R. Quake, Proc. Natl. Acad. Sci U.S. A, 2004, 101, 14431 CrossRef CAS.
- H.-P. Chou, M. A. Unger and S. R. Quake, Biomed. Microdevices, 2001, 3, 323 CrossRef.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580 CrossRef CAS.
- M. J. Anderson, C. L. Hansen and S. R. Quake, Proc. Natl. Acad. Sci. USA, 2006, 103, 16746 CrossRef CAS.
- M. O. Sommer and S. Larsen, J Synchrotron Radiat, 2005, 12, 779 CrossRef CAS.
- A. Stradner, H. Sedgwick, F. Cardinaux, W. C. Poon, S. U. Egelhaaf and P. Schurtenberger, Nature, 2004, 432, 492 CrossRef CAS.
- J. de Jong, R. G. Lammertink and M. Wessling, Lab Chip, 2006, 6, 1125 RSC.
- E. Favre, P. Schaetzel, Q. T. Nguygen, R. Clément and J. Néel, Journal of membranes science, 1994, 92, 169 Search PubMed.
- M. Lounaci, P. Rigolet, G. V. Casquillas, H. W. Huang and Y. Chen, Microelectron. Eng, 2006, 83, 1673 CrossRef CAS.
- M. Lounaci, P. Rigolet, C. Abraham, M. L. Berre and Y. Chen, Microelectron. Eng, 2007, 84, 1758 CrossRef CAS.
- J. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. Jia, K. Piattelli and S. Fraden, J. Am. Chem. Soc., 2007, 129, 8825 CrossRef CAS.
- J. Shim, G. Cristobal, D. R. Link, T. Thorsen and S. Fraden, Crystal Growth & Design, 2007, 7, 2192 CrossRef CAS.
- C. L. Hansen, S. Classen, J. M. Berger and S. R. Quake, J. Am. Chem. Soc., 2006, 128, 3142 CrossRef CAS.
- B. T. Lau, C. A. Baitz, X. P. Dong and C. L. Hansen, J. Am. Chem. Soc., 2007, 129, 454 CrossRef CAS.
- C. J. Gerdts, V. Tereshko, M. K. Yadav, I. Dementieva, F. Collart, A. Joachimiak, R. C. Stevens, P. Kuhn, A. Kossiakoff and R. F. Ismagilov, Angew. Chem. Int. Ed, 2006, 45, 8156 CrossRef CAS.
- S. Talreja, D. Y. Kim, A. Y. Mirarefi, C. F. Zukoski and P. J. A. Kenis, J. Appl. Cryst, 2005, 38, 988 CrossRef CAS.
- S. Talreja, P. J. A. Kenis and C. F. Zukoski, Langmuir, 2007, 23, 4516 CrossRef CAS.
- E. D. Greaves and A. Manz, Lab Chip, 2005, 5, 382 RSC.
- R. Barrett, M. Faucon, J. Lopez, G. Cristobal, F. Destremaut, A. Dodge, P. Guillot, P. Laval, C. Masselon and J.-B. Salmon, Lab Chip, 2006, 6, 494 RSC.
- M. K. Yadav, C. J. Gerdts, R. Sanishvili, W. W. Smith, L. S. Roach, R. F. Ismagilov, P. Kuhn and R. C. Stevens, J Appl. Crystallogr, 2005, 38, 900 CrossRef CAS.
- T. E. Paxton, A. Sambanis and R. W. Rousseau, Langmuir, 2001, 17, 3076 CrossRef CAS.
- A. Sanjoh and T. Tsukihara, J., Crystal Growth, 1999, 196, 691 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |