Dropspots: a picoliter array in a microfluidic device†
Christian H. J.
Schmitz‡
,
Amy C.
Rowat‡
*,
Sarah
Köster
and
David A.
Weitz
*
Department of Physics/School of Engineering and Applied Sciences, Harvard University, 29 Oxford Street, Cambridge, MA 02138, USA. E-mail: rowat@seas.harvard.edu; weitz@seas.harvard.edu; Fax: +1 617 496 9564
First published on 28th October 2008
Abstract
We present a simple microfluidic device that uses an array of well-defined chambers to immobilize thousands of femtoliter- to picoliter-scale aqueous drops suspended in inert carrier oil. This device enables timelapse studies of large numbers of individual drops, while simultaneously enabling subsequent drop recovery.
Introduction
Studying temporal processes on small scales is essential for understanding phenomena ranging from nucleation to single enzyme kinetics to cellular heterogeneity. For example, a population of mixed or even genetically identical cells exhibits phenotypic variation that can have implications for how cells respond to stress or drugs.1–4 To elucidate the mechanisms underlying this variation, it is essential to study these dynamic processes by tracking individual cells over time. However, conventional methods that measure large numbers of cells can only provide an average over the population as a whole, or a distribution of the population at a single time point. Following the timecourse of individual cells can be achieved by imaging; however, cells divide over time and some types grow in suspension, making it difficult to acquire data for large numbers of independent samples. The need to obtain kinetic information for single cells demands a technique to isolate cells in small volumes, to fix them in position, and to monitor the temporal evolution of each one of thousands of individual samples to generate good statistics. Moreover, the potential must exist for samples to be subsequently recovered for growth or more detailed study. These requirements have been accomplished by confining samples in nanoliter-volume microwells lithographically fabricated on a plate5 or in chambers of a microfluidic device.6 Such nanoliter-chambers are separated by pneumatic valves, and can compartmentalize thousands of independent reactions; this technology has enabled single genome amplification,6 as well as gene expression at the single cell level.7 However, precision control of the filling and retrieval of the contents of the individual wells requires on-chip valves which necessitate more complex multilayer device fabrication. Thus, fabricating devices with reduced chamber volumes or with an increased number of chambers on-chip becomes very challenging. One way to overcome these restrictions in scale is to encapsulate the reactants in aqueous drops in an inert carrier fluid: monodisperse, femtoliter- to nanoliter-scale drops can be generated in a microfluidic device at kHz frequencies.8–12 Timecourse studies can be performed by monitoring drop contents as a function of position as drops flow past a detector;13–15 this provides statistical information on the population. To obtain kinetic information at the level of specific drops, individual drops can be monitored over time by storing them in a capillary,16 or by collecting them off-chip for imaging.17 However, the position of each drop must be carefully monitored and recorded, which limits both the number of individual drops that can be studied as well as the duration of the experiment. To acquire timelapse data for large numbers of single drops, it is more practical to immobilize the drops. For example, drops could be fixed in position by trapping them in a gel matrix. For better integration, the storage device could be incorporated directly onto the microfluidic chip, using surface tension to trap the drops in pots.18 However, with these methods drops cannot be retrieved on demand. Instead, a more useful technique should allow drops to be stored, individually monitored, and then recovered and ultimately even sorted. Moreover, the ideal device should be easy to fabricate and simple to use. Such a drop storage device would be a functional tool, as it could easily interface with other microfluidic modules for drop coalescence,10 or sorting based on some optical assay such as fluorescence intensity.19Here we present a device to immobilize and store drops. We call this the ‘Dropspots’ device. It is a picoliter array that can be used to monitor thousands of individual, picoliter-scale samples over time. Drops flow through an array of round chambers connected by narrow constrictions: when there is flow, drops squeeze through the constrictions; in the absence of flow, surface tension drives the drops to their lowest energy shape and ensures a single drop per chamber. Using this simple concept, the device allows drops to be immobilized during an experiment and subsequently recovered.
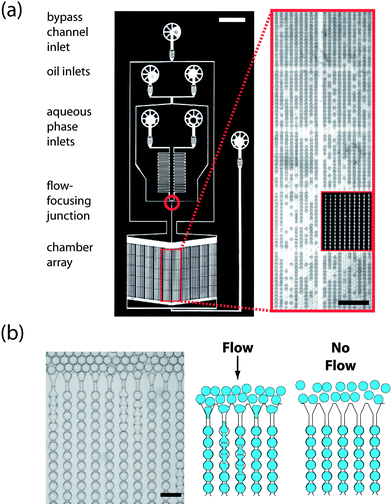 | ||
| Fig. 1 The ‘Dropspots’ device. (a) A Dropspots device that stores 8,000 drops in a 5 × 10 connected 160-chamber array within a 7 mm2 area. The device can be imaged using a microarray scanner, as illustrated in the white on black images where the channels are filled with fluorinated (FC40) oil containing rhodamine B. Scale, 1 mm. Inset: brightfield image showing ten arrays partly filled with drops. White on black image shows details of the microarray scan. Scale, 500 µm. (b) Brightfield image and schematic diagram that illustrate the loading of the Dropspots device. Scale, 100 µm. | ||
Device concept and operation
Our devices are fabricated out of polydimethylsiloxane (PDMS) using soft lithography.20 In the devices used here, drops are directly generated on-chip,8 and then flowed into an array of chambers for drop storage (Fig. 1a). The proportion of drops entering the storage array is controlled by adjusting the pressure applied to a bypass channel inlet as shown in Fig. 1a. This enables the array to be filled with drops whereupon the tubing is unplugged to stop drop-making; the bypass channel dampens the abrupt pressure change that results and the array filled with drops remains unperturbed. The resistance to flow in the bypass channel is adjusted to that of the array channels so that when drops are in the array channels, some drops continue to flow into them while the majority of drops flow out into the bypass channel. By applying pressure to the bypass or outlet channel, more drops can be forced into the array and better filling efficiency is achieved.This method of drop storage is very stable: drops remain fixed in their array positions even when the device is moved to be heated in an incubator or on a hot plate, or imaged. The large surface-area-to-volume ratio of the drops, as well as the permeability of PDMS to gases,21 makes the Dropspots device amenable to cell culture under controlled environmental conditions. For imaging drops, the device can be adapted for a microarray scanner or inverted microscope (Fig. 1a). After the experiment, drops are easily recovered by injecting oil into the device and applying pressure (ESI Fig. 1b–c†). Drops can then be reinjected into microfluidic devices with other functions or broken to retrieve the encapsulated contents (ESI Fig. 1d†).
Successful operation of the device depends on the generation of stable drops to fill the channels. This is accomplished by flow-focusing with typical flow rates ranging from 50 to 300 µL/hr. We use a fluorosurfactant that maintains drop stability during formation at volume fractions up to 80%, during incubation at elevated temperatures, and during collection and reinjection where drops are exposed to larger shear forces.22 Such drop stability is critical for the use of the Dropspots device where drops are manipulated at high volume fractions, and must deform through narrow constrictions that are one half their own diameter. These surfactants generate stable drops for incubation in the Dropspots chambers that contain a variety of inner phases ranging from water to cell media to a variety of cells such as yeast and chlamydamonas. Drop surface tension is another important parameter that must not be too large nor too small: drops must be able to deform through the constrictions, but surface tension must also be sufficient for the drops to remain in the chambers. For a typical aqueous phase and fluorinated oil-surfactant system, the interfacial tension is about 5 mN/m.22
For efficient trapping, we also optimize the channel dimensions. The chamber diameter must be at least twice the width of the constriction; otherwise drops squeeze through the constrictions and do not remain immobilized over the experimental timecourse. Moreover, constrictions that are too small can result in drops splitting. The length of a single channel is 4.5 mm, and consists of 80 individual chambers. Longer channels that contain ∼500 chambers result in a resistance to flow that exceeds our capabilities of device operation: using compliant PDMS and polyethylene tubings, we are not able to achieve sufficient pressure to ensure flow through the entire channel. This results in inadequate surface treatment, and drops that remain stuck in long channels. However, by altering the device materials, the higher pressures needed to force the drops through longer channels could be achieved.
Filling the chambers with drops requires a pressure drop across the array of chambers in order for the drops to deform and pass through the narrow constrictions. In our device, each channel has a length, l = 4.5 mm, and height, h = 25 µm, with 80 chambers of diameter 40 µm and constriction width 20 µm. For a drop to move through a channel, it must deform enough to squeeze through the constriction. To squeeze through a 20 µm constriction, a spherical drop of initial radius, r1 = 20 µm, trapped in a chamber of diameter 40 µm must increase its curvature to achieve a local radius of r2 = r1/2 = 10 µm. Assuming surfactants are distributed evenly over the interface, the pressure needed to deform a drop to these dimensions is given by Laplace's law and is approximately,
| ΔPmin ≈ γ/r2 ≈ 500 Pa. |
To attain flow in a channel, the drops must move in series. Thus, the total pressure drop over the full channel, ΔPtot, is estimated as the sum of the pressure drops over individual droplets,
| ΔPtot = 80ΔPmin = 4 × 104 Pa. |
This pressure must be applied at the entrance of each channel to cause the drops to move. As an approximate test of this simple prediction, we compare the flow rate needed to achieve this pressure with those typically used to fill the device. Upon filling the channels with drops, most of the flow is diverted through the two bypass channels of length, l = 15 mm; total width, w = 100 µm; and height, h = 25 µm. In these channels, drops are dispersed in oil at a volume fraction of less than 50%; thus the viscosity is approximately the viscosity of the fluorinated oil, η = 3.4 mPa·s. The flow rate through the bypass channels is given by the Hagen-Poiseuille relation for a rectangular channel,
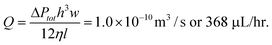
Results and discussion
To demonstrate the capability to perform timelapse measurements using the Dropspots device, we monitor growth rates of a population of cells. We encapsulate yeast cells in drops of a water-in-fluorocarbon emulsion. By adjusting the density of the cell suspension, we ensure that the vast majority of drops have at most a single cell; the resultant filling is well described by a Poisson distribution.12,14 We incubate drops overnight in the Dropspots device while repeatedly acquiring images (Fig. 2). The total number of cells increases with time, exhibiting the typical sigmoidal growth curve observed for bulk culture (ESI Fig. 2a†). However, we can monitor individual cells by following the timecourse of single drops, allowing a true distribution to be measured. At the level of individual drops, doubling times vary significantly. This observed variation may be attributed to cell cycle asynchrony as well as other factors contributing to cellular heterogeneity. We also observe that doubling times increase with the number of cells initially encapsulated (ESI Fig. 2b†). Such behavior could be a consequence of the high cell density of an encapsulated cell: the volume per cell of even a single cell in a drop of 40 µm diameter is equivalent to 108cells/mL, which is near the density of a saturated culture of yeast cells. Interestingly, the total volume of drops containing cells decreases over time (ESI Fig. 3†). The ability to maintain the identity of individual drops containing cells and to monitor the growth of encapsulated cells enables fitness assays at the single cell level; this type of kinetic screening is crucial for toxicity and drug resistance assays, as well as studies of the dynamic behavior of a library of individual cells.23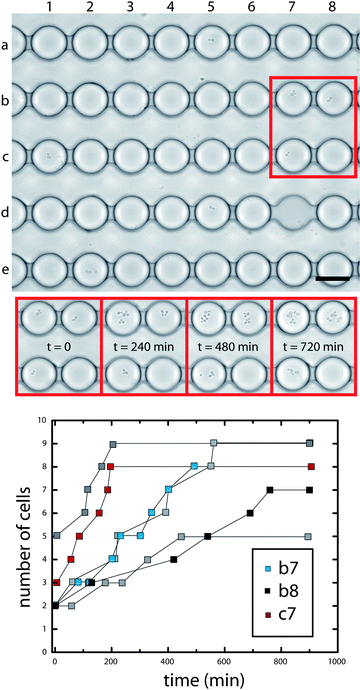 | ||
| Fig. 2 Monitoring growth rates of single cells. An array of chambers in a Dropspots device is filled with drops containing yeast cells. The number of cells in individual drops is tracked over time and is observed to increase over the 15 hour incubation period. Shown here is a small area of the filled array and growth trajectories for 6 individual representative drops. Color plots represent drops identified in the image. Scale, 40 µm. | ||
We demonstrate the utility of the Dropspots device for timelapse fluorescence experiments by monitoring enzyme levels in a population of single cells using a fluorogenic assay. Enzyme levels are measured by the rate of increase in fluorescence intensity over time due to substrate turnover; this requires monitoring each individual drop from a population of thousands at repeated time intervals. Enzymes are commonly used as reporters for gene expression,7 however, cells must be permeabilized for the fluorogenic substrate to access the enzymes, and the fluorescent product is expelled by the cells into the surrounding medium. Consequently, the signal from single cells would get mixed for cells in suspension. In contrast, isolating cells in microfluidic chambers successfully contains single cells, their enzymes, and fluorescent products,7 but detectable levels of fluorescent molecules must first accumulate in the chamber volume before analysis can begin. By encapsulating cells in picoliter drops, high concentrations of secreted molecules are rapidly attained. We stimulate cells that have a pheromone-regulated lacZ fusion (FUS2), encapsulate them in drops, and then monitor beta-galactosidase activity with a fluorogenic assay by repeated image acquisition. The fluorescence intensity in drops containing cells increases as the enzyme cleaves its fluorogenic substrate, fluorescein-di-beta-D-galactopyranoside (FDG), to produce fluorescein (Fig. 3a). Analysis of the slope in the linear regime of the enzyme progress curves for individual drops shows that reaction rates depend on the number of cells per drop, but vary even for drops containing the same number of cells. This observed variation in reaction rates reflects heterogeneity in gene expression in a population of individual, genetically identical cells (Fig. 3b,c) and highlights the importance of truly single cell assays.13 Our results demonstrate the potential of drops for kinetic enzyme assays with single cells or molecules, as well as for the study of low-copy number proteins by enzymatic amplification at the single cell level. The ability to use picoliter volumes for array studies represents a major reduction in volume for plate assays that typically require microliters of reagents per well.24 Moreover, these picoliter microvessels enable studies of single cells at high cell densities that cannot be attained using larger wells, and ensure that large concentrations of secreted molecules can accumulate rapidly.
 | ||
| Fig. 3 Detecting enzyme levels in drops by kinetic analysis. The fluorescence intensity of drops containing cells producing beta-galactosidase increases over time. (a) Brightfield image and color map gradient of a fluorescence image at time = 45 min. (b) Temporal changes in drop fluorescence show that the reaction rate depends on the number of cells per drop. Plots for 9 individual representative drops containing 1 (black), 2 (cyan), and 3 (red) cells. (c) Reaction rates for 265 drops containing cells are quantified by slope analysis in the linear regime, and displayed as a function of number of cells per drop in this histogram. Note that a total of over 2000 drops are analyzed; data is only displayed here for drops that contain cells. Scale, 40 µm. | ||
In addition to its function as a picoliter array for single cell assays, the Dropspots device provides a simple way to monitor any femtoliter- or picoliter-scale reaction that evolves over time. Statistics on hundreds to thousands of samples can be easily collected, and the device can be scaled up to study tens of thousands of drops; while we show here devices with up to 8,000 chambers, we have fabricated devices with as many as 64,000 chambers. After an experiment, drops can be retrieved from the Dropspots device.
Conclusions
Here we show a simple device that immbolizes thousands of drops, is easy to fabricate, and simple to use. In the current device design, the channels of the chamber array are fabricated in parallel; however, they could be arranged in series to preserve drop order upon retrieval for sorting based on reaction rate criterion by coupling the device to a drop sorter.19,25 In addition to coupling with existing microfluidic modules, the Dropspots device also has potential to interface to the macroscopic world for recovering and transferring drops to wells of larger volumes for cell culture or further analysis. As the layout of the device can easily be altered, the spacing of the chamber array can be designed to match that of an array of needles that could pierce through the PDMS. Single cell colonies could also be retrieved by injecting oil to increase the spacing between drops upon removing the drops from the array. Moreover, the Dropspots device could be used to create arrays of any deformable cell or vesicle whose minimal free energy shape is a sphere. This device thus offers a very simple and convenient solution for the study of large numbers of individual cells or other systems.Methods
Microfluidic device fabrication
Soft lithography is used to fabricate microfluidic channels in polydimethylsiloxane (PDMS, Sylgard 184 Silicone Elastomer, Dow Corning, Midland, MI).20 In brief, the desired design is printed onto a transparency with features no smaller than 10 μm (CAD/Art Services, Inc., Bandon, OR). SU8-2025 (Microchem, Newton, MA, USA) is spincoated onto a cleaned silicon wafer to a final thickness of 25 μm following the protocol described by the manufacturer. Exposure to UV light (200–250 mJ, OAI, San Jose, CA) crosslinks the exposed pattern, and the non-exposed photoresist is removed using propylene glycol monomethyl ether acetate (PGMEA). Channel height is measured to be 27 μm using a stylus profilometer (DekTak, Veeco Instruments Inc., Plainview, NY). PDMS with 10% (w/w) crosslinking agent is thoroughly degassed and then poured onto the SU8-mold. Small glass coverslips are cut using a diamond scribe and are placed directly above the storage areas of the device by submerging in the liquid PDMS. After heating at 65 °C for 1 hour, the structure is carefully peeled off the mold, plasma-treated, and bonded to a 25 × 75 mm glass slide of 1 mm thickness. Holes connecting to the channels are formed using biopsy punches (0.75 mm diameter Harris Uni-Core, Ted Pella, Inc., Redding, CA). Before use, channels are treated with Aquapel (PPG Industries, Pittsburgh, PA) followed by a flush with air. This treatment ensures that the oil carrier phase, and not the aqueous phase, wets the surface.During timecourse experiments, we observe that drop volume decreases over time as the PDMS absorbs water.18, 26 To counter this problem, we place a glass coverslip above the array of chambers before curing the PDMS. A flexible polymer vapor barrier can alternatively be embedded in the PDMS27 to prevent evaporation. Additionally, we fabricate a channel surrounding the entire storage array and fill it with water to saturate the surrounding PDMS. These modifications enable us to prevent drop shrinkage over several days. Drop shrinkage can alternatively be prevented by saturating the PDMS with water to allow for maximal gas permeability that is necessary for certain cell culture applications.
Yeast cell culture
Yeast cell cultures [S. cerevisiae, EY0986: MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 (S288C, ATCC 201388)] are inoculated from a single colony and incubated while shaking for 6 hours in yeast extract/peptone/dextrose (YPD) media at 30 °C. An aliquot of this stock culture is diluted in 10 mL of YPD and cultured overnight to a density of OD 600 ∼ 0.2–0.6 (1 mm path length, NanoDrop ND-1000, Wilmington, DE). Cells are washed twice by centrifugation, resuspended in fresh YPD, and then encapsulated.Enzymatic assay
We use a strain with a lacZ insertion in the FUS2 gene (105A-1)28 (gift from S. Erdman). As a negative control, we use the MATa derivative of the background strain of the transposon screening strain (YSE21). Cells of both control and reporter strain were cultured as described above, and induced with 5 μg/mL alpha-factor (Sigma, St. Louis, MO) 3 hours prior to encapsulation. The fluorogenic substrate fluorescein-di-beta-D-galactopyranoside (FDG) (Invitrogen, Carlsbad, CA) is added to the aqueous cell suspension at a final concentration of 200 μM in 8:1:1 water:DMSO:ethanol (v/v/v) just prior to encapsulation. Since the cell wall is a barrier for influx of the fluorogenic enzyme substrate, cells were washed and resuspended in 1× TBS saturated with chloroform to permeabilize the cells. To reduce photobleaching, n-propyl gallate (Sigma, St. Louis, MO) is added to the cell suspension (1 mM final concentration).Cell encapsulation and retrieval
The cell suspension is loaded into 1 mL plastic syringes fitted with a 27½ gauge Luer-lok needle (Becton-Dickinson, Franklin Lakes, NJ). Polyethylene tubing (PE-20, Intramedic, Becton-Dickinson, Franklin Lakes, NJ) is inserted onto the needle, and the tubing is plugged into the excised hole on the PDMS device. To encapsulate single cells in drops, cell suspensions are flowed together with fluorinated oil (FC-40 Sigma, St. Louis, MO) containing 1.8% (w/w) fluorinated surfactant (Holtze et al., submitted) using flow-focusing geometry8 to generate monodisperse drops of a water-in-oil emulsion. Syringe pumps (Harvard Apparatus, Holliston, MA) are used to control flow rates: 100 μl/hr for the aqueous phase and 300 μl/hr for the oil phase typically results in drops of ∼40 µm diameter. However, flow rates may vary due to factors including trapped air and leakage, and thus need to be adjusted for independent experiments so the drop size matches that of the wells.To recover cells from drops, the emulsion is diluted with 10× its fluid volume of fresh media and 15% its volume of drop release reagent is added (RainDance Technologies, Inc., Lexington, MA). The mixture is centrifuged at 1000 g for 30 s. The supernatant containing the cells is plated onto a YPD agar plate, and spread with glass beads. After incubation at 30 °C for several days, colony growth is observed and images are acquired using a stereomicroscope.
Imaging
To image using a microarray scanner, devices are placed in the 4-slide holder of a microarray scanner (LS400, Tecan, Hombrechtikon, Switzerland) and imaged at 4 μm resolution with 543 nm laser excitation. For time-lapse experiments of yeast cells, the Dropspots device is imaged on an inverted microscope (Leica DMIRBE) with a 20× objective and images are acquired every 10 minutes with a CCD color video camera (Exwave HAD, Sony). Fluorescence images are acquired on an inverted epifluorescent microscope (Nikon Eclipse TE2000-E) equipped with a 10×/0.30 Plan Fluor objective (Nikon), X-cite series 120 lamp (EXFO, Mississauga, Canada), and a CCD camera (Coolsnap HQ2). Images are acquired at 10 minute intervals with exposure times of 1 ms for brightfield, and 30 ms with 4× gain for fluorescence images. The mean intensity in a region of interest within each drop is determined using ImageJ and plotted as a function of time. Slopes were measured using the first four time points in the linear regime. Calibration of pixel intensity to fluorescein concentration is obtained by imaging drops containing determined fluorescein concentrations ranging from 0–30 μM in chloroform-saturated TBS buffer using the same image acquisition parameters. The image displayed in Fig. 3 was processed by converting a 16-bit grayscale image to RGB format, and then applying a color gradient map using the following levels: blue (0), cyan (1), green (4), yellow (8), red (11), pink (14). The legend is determined by applying the same color map gradient to images of drops containing known concentrations of fluorescein.Acknowledgements
This work was supported by the NSF (DMR-0602684 and DBI-0649865) and the Harvard MRSEC (DMR-080820484) and the Human Frontiers Science Program (RGP0004/2005-C102 and Cross-Disciplinary Fellowship to ACR). CHJS is a Leopoldina Research Fellow of the German Academy of Sciences funded by the Federal Ministry of Education and Research (BMBF-LPD 9901/8-150). SK is supported by the Deutsche Forschungsgemeinschaft (DFG, KO 3572/1). We thank Scott Erdman for the gift of his cell strain, Jeremy Agresti for critique and discussions, and Azadeh Samadani for access to her fluorescence microscope.References
- J. M. Raser and E. K. O'Shea, Science, 2005, 309, 2010–2013 CrossRef CAS.
- S. V. Avery, Nat Rev Microbiol, 2006, 4, 577–587 Search PubMed.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS.
- A. L. Bishop, F. A. Rab, E. R. Sumner and S. V. Avery, Mol. Microbiol., 2007, 63, 507–20 CrossRef CAS.
- J. C. Love, J. L. Ronan, G. M. Grotenbreg, A. G. van der Veen and H. L. Ploegh, Nat Biotechnol, 2006, 24, 703–707 CrossRef CAS.
- Y. Marcy, T. Ishoey, R. S. Lasken, T. B. Stockwell, B. P. Walenz, A. L. Halpern, K. Y. Beeson, S. M. Goldberg and S. R. Quake, PLoS Genet, 2007, 3, e155 Search PubMed.
- L. Cai, N. Friedman and X. S. Xie, Nature, 2006, 440, 358–362 CrossRef CAS.
- S. L. Anna, N. Bontoux and H. A. Stone, Applied Physics Letters, 2003, 82, 364–366 CrossRef CAS.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew Chem Int Ed Engl, 2006, 45, 7336–7356 CrossRef CAS.
- K. Ahn, J. J. Agresti, H. Chong, M. Marquez and D. A. Weitz, Applied Physics Letters, 2006, 88, 264105 CrossRef.
- J. Clausell-Tormos, D. Lieber, J. C. Baret, A. El-Harrak, O. J. Miller, L. Frenz, J. Blouwolff, K. J. Humphry, S. Köster, H. Duan, C. Holtze, D. A. Weitz, A. D. Griffiths and C. A. Merten, Chem Biol, 2008, 15, 427–437 CrossRef CAS.
- S. Köster, F. E. Angilè, H. Duan, J. J. Agresti, A. Wintner, C. Schmitz, A. C. Rowat, C. A. Merten, D. Pisignano, A. D. Griffiths and D. A. Weitz, Lab Chip, 2008, 8, 1110–1115 RSC.
- A. Huebner, L. F. Olguin, D. Bratton, G. Whyte, W. T. Huck, A. J. de Mello, J. B. Edel, C. Abell and F. Hollfelder, Anal Chem, 2008, 80, 3890–3896 CrossRef CAS.
- A. Huebner, M. Srisa-Art, D. Holt, C. Abell, F. Hollfelder, A. J. deMello and J. B. Edel, Chem Commun (Camb), 2007, 1218–1220 RSC.
- F. Courtois, L. F. Olguin, G. Whyte, D. Bratton, W. T. Huck, C. Abell and F. Hollfelder, Chembiochem, 2008, 9, 439–446 CrossRef CAS.
- L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice and R. F. Ismagilov, Proc Natl Acad Sci U S A, 2006, 103, 19243–19248 CrossRef CAS.
- M. Y. He, J. S. Edgar, G. D. M. Jeffries, R. M. Lorenz, J. P. Shelby and D. T. Chiu, Analytical Chemistry, 2005, 77, 1539–1544 Search PubMed.
- J. U. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. Jia, K. Piattelli and S. Fraden, J Am Chem Soc, 2007, 129, 8825–8835 CrossRef CAS.
- L. M. Fidalgo, G. Whyte, D. Bratton, C. F. Kaminski, C. Abell and W. T. Huck, Angew Chem Int Ed Engl, 2008, 47, 2042–2045 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS.
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, Journal of Polymer Science: Part B: Polymer Physics, 2000, 38, 415–434 Search PubMed.
- C. Holtze, A. C. Rowat, J. J. Agresti, B. Hutchison, F. E. Angilè, C. Schmitz, S. Köster, H. Duan, K. J. Humphry, D. Pisignano and D. A. Weitz, Lab on a Chip, 2008, 8(10), 1632–1639 RSC.
- Y. Suzuki and F. P. Roth, Mol Syst Biol, 2006, 2, 48.
- J. Inglese, R. L. Johnson, A. Simeonov, M. Xia, W. Zheng, C. P. Austin and D. S. Auld, Nat Chem Biol, 2007, 3, 466–479 CrossRef CAS.
- K. Ahn, C. Kerbage, T. P. Hunt, R. M. Westervelt, D. R. Link and D. A. Weitz, Applied Physics Letters, 2006, 88 Search PubMed.
- E. Verneuil, A. Buguin and P. Silberzan, Europhys. Lett., 2004, 68, 412–418.
- A. Ranjit Prakash, S. Adamia, V. Sieben, P. Pilarski, L. M. Pilarski and C. J. Backhouse, Sensors & Actuators: B. Chemical, 2006, 113, 398–409.
- S. Erdman, L. Lin, M. Malczynski and M. Snyder, J CellBiol, 1998, 140, 461–483..
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and movie. See DOI: 10.1039/b809670h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2009 |