Fabrication of a multichannel PDMS/glass analytical microsystem with integrated electrodes for amperometric detection†
Ney Henrique
Moreira
a,
André Luis
de Jesus de Almeida
a,
Maria Helena
de Oliveira Piazzeta
a,
Dosil Pereira
de Jesus
b,
Ariane
Deblire
b,
Ângelo Luiz
Gobbi
a and
José Alberto
Fracassi da Silva
*b
aLaboratory of Microfabrication, Brazilian Synchrotron Light Laboratory, Campinas, Brazil. E-mail: gobbi@lnls.br; Fax: +55 19 3512 1004; Tel: +55 19 3512 1164
bInstitute of Chemistry, University of Campinas, UNICAMP, 13083-970, Campinas, SP, Brazil. E-mail: fracassi@iqm.unicamp.br; Fax: +55 19 3521 3023; Tel: +55 19 3521 3124
First published on 22nd October 2008
Abstract
The fabrication process of novel multichannel microfluidic devices with integrated electrodes for amperometric detection is described. Soft-lithography, lift-off and O2 plasma surface activation sealing techniques were employed for rapid prototyping of cost effective PDMS/glass microchips. The capabilities of the proposed microdevices were demonstrated by the electrooxidation of hydroquinone and N-acetyl-p-aminophenol (APAP) on a Au working electrode at +800 mV and +700 mV, respectively, against a Au pseudo reference electrode, and of thiocyanate on a Cu working electrode at +700 mV against a Ag/AgCl (KCl saturated) reference electrode. Linear response over the range up to 1.0 mmol L−1 for APAP and up to 4.0 mmol L−1 for hydroquinone and thiocyanate were verified through calibration curves with correlation coefficients greater than 0.97 (minimum of five data points). The sensitivities for hydroquinone, thiocyanate, and APAP were 28, 19, and 78 μA mol−1 L, respectively. Under the experimental conditions used, the estimated limits of detection were 0.21, 0.95, and 0.12 mmol L−1 for hydroquinone, thiocyanate and APAP, respectively. The geometries of the devices were designed to allow fast calibration procedures and reliable results for in-field applications. Exerting a strong influence over the device performance, the sealing process was greatly enhanced by depositing auxiliary TiSiO2 thin-films. The general performance of the system was verified by amperometric assays of N-acetyl-p-aminophenol standard solutions, and the influences exerted by the present fabrication methods regarding reproducibility and reliability are addressed. The proposed device was successfully applied in the determination of the concentration of APAP in two commercial formulations.
Introduction
Miniaturization of analytical systems can reduce costs, increase portability, and provide faster analysis with lower consumption of reagents and samples.1–5There are several issues associated with the calibration of conventional single channel micro total analysis systems (μ-TAS). These include the long times required to clean-up the microchannel between consecutive sample injections and also the difficulty of making robust systems, capable of keeping their chemical environment unchanged during multi-injection analytical procedures.6–8 Since the actual volumes involved are so small, even trace amounts of contaminants or small changes in pH can induce significant modifications in the chemical environment, resulting in significant deviations of the analytical results.
Parallel analysis is very attractive and feasible using multichannel designs in microfluidic platforms.9 Advantages of this approach were effectively demonstrated by Emrich and co-workers,10 where the emitted fluorescences of 384 distinct channels were simultaneously monitored and genetic analysis performed. Many other successful applications of parallel sample processing and analysis have been reported and the advantages of these systems, such as high-throughput, are evident. Other examples include sample mixing and dilution,11 separation and detection,12,13 multi-injection,14 enzymatic assays,15 combinatorial chemistry,16 single cell lysis17 and even polymer electrosynthesis.18
Electrochemical detectors meet the requirements of μ-TAS for in-field uses, since they are intrinsically portable, can be easily integrated with other components and are cost effective.19 Amperometric detection is suitable for microfluidic analytical devices because of the low limits of detection (LOD) that can be achieved with reasonable selectivity and because of its compatibility with microfabrication techniques. Amperometric detection has been widely used in electrophoresis microdevices, despite interferences from the separation electric field.20–22 This detection mode has also been successfully used in many other microfluidic devices, such as liquid chromatography,23microdialysis,24,25 immunoassay,26 and determination of compounds such as peroxide,27morphine,28pesticides,29 and nucleic acids.30 As a non-absolute method, amperometric detection often requires a calibration step for the quantification of analytes. However, calibration may contribute significantly to an increase in analysis time. Thus, fast and easily calibrated microfluidic analytical systems are critically needed. Generally, a single electrochemical cell is used in analytical microdevices with amperometric detection. Consequently, during the calibration and determination steps the electrochemical cell has to be washed and filled with the standard or sample solutions many times. Finally, when using multiple isolated electrodes, multi-potentiostatic control must be employed and, in this case, the current acquisition circuitry can offer additional complexity.
Among the several materials used in microfabrication, poly(dimethylsiloxane) (PDMS) has been widely used in microfluidics. PDMS can be bonded to itself or to glass via O2 plasma surface activation, yielding low-cost irreversibly sealed microfluidic devices.31–34 PDMS has become the first choice for rapid prototyping of disposable microchips, due to its high processability, disposability, low cost and ability to promote irreversible bonds with glass.
In this work, the fabrication and characterization of a PDMS/glass multichannel microfluidic chip, with fully-integrated electrodes for amperometric detection is described. Our design is based on a single working electrode, which is shared by an array of five microchannels, allowing fast and reliable analytical procedures. This multichannel/single working electrode measurement concept was validated and it was demonstrated that it can be easily applied to obtain fast and reliable in-field quantitative analytical assays.
Experimental
Reagents and solutions
All solutions were prepared using deionized water from a Milli-Q purification system (Millipore, Bedford, MA, USA) with resistivity greater than 18 MΩ cm. All chemicals were of analytical grade and used as received. Acetone, hydroquinone (HQ), KSCN, NaNO3, NaOH, HF, HCl, and H2SO4 were purchased from Labsynth (Diadema, SP, Brazil). N-acetyl-p-aminophenol (APAP, acetaminophen) was from Fluka (purchased from Sigma-Aldrich, St. Louis, MO, USA). 0.1 mol L−1 stock solutions of APAP, HQ, and thiocyanate were prepared in deionized water and standard solutions of these analytes were prepared by appropriate dilution of stock solutions with the supporting electrolyte to the desired concentrations.Device fabrication
Conventional UV photolithographic methods were employed to pattern the replication masters and electrodes used in this work. The materials and experimental details are specified in Table 1. All mask layouts were drawn using AUTOCAD 2004 software and photo-plotted with 8000 dpi resolution. The UV exposures were carried out in a MJB-3 UV300 contact mask aligner (Karl-Suss, Garching, Germany), with a 350 W mercury source emitting from 280 nm to 350 nm.| Lithographic process | Electrode building | Master building |
|---|---|---|
| Photoresist | Shipley Microposit S1811 (Marlborough, MA, USA) | MicroChem SU-8 25 (Newton, MA, USA) |
| Spin coating | 6000 rpm (30 s) | 1000 rpm (30 s) |
| Thickness | ∼1 μm | ∼50 μm |
| Pre-exposure bake | 95 °C, 5 min | 65 °C, 5 min |
| 95 °C, 15 min | ||
| 65 °C, 5 min | ||
| Exposure time | 6 s | 70 s |
| Post-exposure bake | — | 95 °C, 15 min |
| 65 °C, 3 min | ||
| Developer | Clariant AZ351 Developer (Summerville, NJ, USA) | MicroChem SU-8 Developer (Newton, MA, USA) |
PDMS microchannels were cast by soft-lithography.32,35 The replication masters were patterned with a thick SU-8 photoresist layer over square optical glass plates (60 mm side Kodak 1A High Resolution Glass) and mounted in an appropriate holder. The material used as precursor of PDMS layers was the Sylgard 184 Silicone Elastomer Kit (Dow Corning, Midland, MI, USA). Sylgard curing agent and prepolymer base were mixed in a 1 : 10 weight ratio. The mixture was poured onto a replication master and degassed in a desiccator at 40–50 mtorr (5.3–6.7 Pa) for one hour to eliminate air bubbles. The polymer curing process was carried out for one hour on a hot plate at 100 °C, and was followed by peeling off the PDMS layers. The external access to the microfluidic arrays were obtained by drilling holes in the PDMS layers.36
The electrodes were patterned using the lift-off technique. The photoresist layers were patterned onto square optical glass plates (60 mm side Kodak 1A High Resolution Glass) by the lithographic protocol described in Table 1. Titanium-gold (TiAu) and titanium-gold-titanium (TiAuTi) thin-films, deposited with a Leybold Univex 300 e-beam evaporator (Cologne, Germany) using the lift-off technique, were tested as electrode materials. After thin-film depositions, the devices were immersed in acetone to remove the photoresist layer and excess metal, leaving the patterned electrodes on the glass surface. Substrates with TiAuTi films were fully covered with thin silica films (SiO2), which were deposited in a PECVD/RIE system (Vacutec, Cheshire, UK). Microchips with copper electrodes were also prepared using the same technique.
The sealing process was carried out by oxidizing PDMS surfaces and cleaning glass substrates through RF (radio frequency) O2 plasma oxidation using a PLAB SE80 plasma cleaner (Plasma Technology, Wrington, England). Before the plasma treatment, PDMS layers were immersed in a 10% (v/v) HCl solution for 8 min, and electrodes were dipped in a 10% (v/v) H2SO4 (v/v) solution for 10 s. The plasma working parameters were obtained from Jo and co-workers,34 and were 120 mtorr (16 Pa) O2, 70 W RF power, and 20 s exposition. After plasma oxidation, the PDMS layers and the substrates containing the electrodes were manually aligned, faced against each other and allowed to stand for two hours.
In the case of devices where electrodes were covered by silica, an additional etching step was necessary. With the sealed device, silica was removed by injecting a 10% (v/v) HF solution through the channels for a pre-established period of time, that ranged from 30 to 120 s. Due to the hazardous nature of HF, its manipulation was carried out by suction with a PIAB a7 mini vacuum pump (PIAB, Hingham, MA, USA).
Microchip designs and operation
In order to evaluate the capability of detection with a single working electrode in multiple channels, two microchip designs were used, as depicted in Fig. 1. In the first approach (design 1), a common outlet reservoir was used for positioning auxiliary and reference electrodes, while the working electrode was allowed to cross five channels at a position near the outlet reservoir. In the second approach (design 2) all channels were completely isolated and the three electrodes crossed the channels. | ||
| Fig. 1 Microchip designs. (a) Design 1: Single outlet reservoir where auxiliary and reference electrodes are placed; (b) Design 2: Five independent channels–all three electrodes are positioned on-channel. Electrode and channel widths are 50 μm and 100 μm, respectively, and the channel height is 50 μm. WE, RE, and CE stands for working, reference, and auxiliary electrodes, respectively. | ||
The operation of the microdevice is quite simple and is exemplified in Fig. 2. Before analysis, all channels and the reservoir are filled with supporting electrolyte and the potentiostat is turned on (Fig. 2a). Under this condition, no electron transfer will take place at the electrode. In the next step, the supporting electrolyte in channel 1 is replaced by the standard solution (Fig. 2b) using a mini vacuum pump while the current is monitored. Keeping the first standard solution in channel 1, the supporting electrolyte is now replaced by the second standard solution in channel 2, and so on up to the last channel. If one or more solutions inserted in the microchannels were standard solutions, a calibration can be performed. During the process, one or more channels can also be filled with sample solutions, in order to perform quantitative determinations.
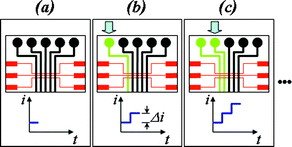 | ||
| Fig. 2 Scheme for analysis using a single working electrode and the multiple channels design. For simplicity, part of design 2 were chosen, but similar procedures were adopted for design 1. (a) All channels are filled with supporting electrolyte and the potentiostat is turned on; (b) The electrolyte solution in the first channel is replaced by a solution containing an electroative analyte. A rise in current is observed; (c) The electrolyte solution in the second channel is replaced and a new increase in current is observed. | ||
Reutilization of the devices took place only after careful cleaning of the channels and reservoir with supporting electrolyte.
Amperometric detection
Electrochemical experiments were conducted on a Model 400 potentiostat (EG & G Princeton Applied Research, Oak Ridge, TN, USA) using a three electrode cell configuration. In the case of gold working electrodes, reference and auxiliary electrodes were also gold electrodes, built in during the fabrication process. In the case of a copper working electrode, external Ag/AgCl (KCl saturated) reference and platinum auxiliary electrodes were necessary and this was only possible using microchip design 1. The potentiostat was interfaced with a conventional desktop personal computer through a PCL711B interface card (Advantech, Taipei, Taiwan). Data acquisition was performed with an in-house software program. The current was monitored at a constant potential of +700 mV and +800 mV against a pseudo-reference gold electrode for the detection of APAP and HQ, respectively, at the Au working electrode. For the detection of SCN− at the Cu working electrode, +700 mV against Ag/AgCl (saturated KCl) reference electrode was used.Results and discussion
Under conditions of controlled potential, and considering an electrochemical cell composed of a planar working electrode of area A, filled with supporting electrolyte containing an electroactive analyte at concentration C∞, the electrode current i can be written as:37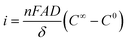 | (1) |
where n is the number of electrons involved in the reaction, F is the Faraday constant (96485 C mol−1), D is the diffusion coefficient of the analyte, δ is the length of the Nernst layer (or diffusion layer), and C0 is the analyte concentration at the surface of the electrode. With sufficient potential applied to the electrode, and after a relatively short period of time, eqn (1) can be approximated by:
| i = kC∞ | (2) |
with k = (nFAD/δ).
If we now consider the active area of the electrode distributed on different electrochemical cells, electrically connected through a conductive media, the current developed on each of these active areas will be proportional to the effective concentration of the analyte present in the cell. As the potentiostat will measure the total current developed and, assuming that the active area is equal in all electrochemical cells, the total current can be described by:
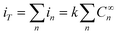 | (3) |
To evaluate the proportionality of the current, the first approach was to construct a microchip with design 1 (Fig. 1a).
Since electron transfer occurs only at the surface of the electrode, it is possible to define separated active areas over the same working electrode in order to use them as distinct electrodes. Based on this concept, we have projected an integrated amperometric detection system with a single WE shared by five microchannels, in order to build microchips suitable for fast calibration procedures.
Precise control over fabrication processes must also be taken into account in order to obtain good analytical results. In electrochemical microdevices, the fabrication process plays a central role in setting the active limits of the electrodes, exerting an influence on the behavior of the systems. Among all the fabrication steps, sealing is one of the most critical due to two major factors: (i) portability requirements: permanently sealed microdevices are highly preferable since they do not require any active alignment and can be easily transported without risk of damage; (ii) the sealing step determines the active area of the electrodes. As a result, the analytical reliability of a device is under the direct influence of its sealing step reliability, since this is the most fragile and least controllable step of the whole fabrication process.
As can be seen in Fig. 3a, when the supporting electrolyte is replaced by the solution containing the electroactive species, a current increase is observed. Note that this current depends on the transport of material to the electrode surface. Consequently, higher currents are observed under flow conditions. When pressure is removed, the current decays to a stable level determined by diffusion of the electroactive species to the surface of the electrode. Under this diffusional condition, a linear analytical curve is obtained, as shown in Fig. 3b. Using this approach, it is possible to sequentially use several channels, for example, one or more for calibration and the others for the injection of solutions of unknown concentration, in a procedure that resembles external calibration. Alternatively, standard solutions could be added to the sample in order to perform standard addition calibration.
 | ||
| Fig. 3 (a) Representative current response for the oxidation of 1.0 mmol L−1 hydroquinone (HQ) in 0.1 mol L−1 NaNO3 using a microchip with design 1. To obtain this current profile, channels were first filled with 0.1 mol L−1 NaNO3 electrolyte. The supporting electrolyte was then sequentially replaced by solutions containing HQ. (b) Current differences between injections (quadruplicate), measured at the diffusion condition, present a linear relationship in the range of 1.0 to 4.0 mmol L−1 of HQ. Other conditions: three Au electrode cell; Potential: +800 mV against Au pseudo-reference. | ||
The same design was tested for the detection of thiocyanate at a copper electrode using 0.1 mol L−1NaOH as supporting electrolyte. In contrast to devices built with gold electrodes, copper can not be used as the pseudo-reference and auxiliary electrodes. In order to overcome this drawback, platinum and Ag/AgCl (saturated KCl) auxiliary and reference electrodes, respectively, were placed at the common outlet reservoir. The results obtained for the sequential injection of 1.0 mmol L−1potassium thiocyanate solution are listed in Table 2. Copper promoted a very good irreversible seal to the PDMS layer and no leakages were observed for these devices. In order to obtain good device performance, leaking in the region between the PDMS and the gold electrodes must be minimized. Sealing deficiencies in these regions result in an increase in the active area of the electrode, leading to instabilities in the current profiles. This problem can be partially solved by applying negative pressure at the outlet reservoir, but in the case of microchips with design 1 this might be difficult because a common reservoir is used.
| Device/electrode | Analyte a | Range/mmol L−1 | Fit parametersb | R | N c |
|---|---|---|---|---|---|
| a Detection potentials: HQ: +800 mV against a Au pseudo-reference electrode; Thiocyanate: +700 mV against a Ag/AgCl (saturated KCl); Acetaminophen: +700 mV against a Au pseudo-reference electrode. b Current in μA, concentration in mmol L−1; Values are for triplicate determinations for hydroquinone, single determination for thiocyanate, and quadruplicate determinations for acetaminophen. c N is the number of data points for each analytical curve. | |||||
| Design 1/Au | Hydroquinone (HQ) | 1.0–4.0 | Δi = (0.000 ± 0.002) + (0.028 ± 0.001) [HQ] | > 0.99 | 5 |
| Design 1/Cu | Thiocyanate | 1.0–4.0 | Δi = (0.006 ± 0.006) + (0.019 ± 0.003) [SCN−] | > 0.97 | 5 |
| Design 2/Au-SiO2 | Acetaminophen (APAP) | 0.5–2.5 | Δi = (−0.002 ± 0.003) + (0.078 ± 0.001) [APAP] | > 0.999 | 6 |
Although good results were obtained with microchips using design 1, back-flow interference led us to further improve the performance of the device. Back-flow can cause the transport of material by convection, inducing baseline instabilities. In contrast, using the design 2 (Fig. 1b), all three gold electrodes were placed inside the channel array, with the working electrode positioned at the center. Microchips with design 2 were used for the oxidation of APAP, as can be seen in Fig. 4. This second design allowed negative pressure to be applied at the outlet, preventing leakages and channel deformations. So, APAP solutions were inserted into the channels by applying negative pressure at outlet reservoirs and releasing the pressure after the solution in a given channel was replaced. After a transient response period, the current reaches a stable value, which corresponds to the diffusion of the analyte to the surface of the electrode, as illustrated for the oxidation of APAP in Fig. 4.
 | ||
| Fig. 4 Quadruplicate full analytical procedure carried out by sequential injections of 1 mmol L−1APAP standard solutions in a microchip with design 2 containing TiAu electrode: (a) first, (b) second, (c) third and (d) fourth replicates. | ||
Role of the sealing process
Although devices with design 2 effectively eliminated the limitations due to back-flow, the desired analytical performance could not be achieved due to sealing defects at the TiAu electrodes after a few analytical procedures. Fig. 4 shows four consecutive full analytical procedures, carried out with TiAu electrode devices and performed in quadruplicate.The first two experiments revealed acceptable amperometric profiles (Fig. 4a and b). However, the analytical signal decreases in the third replicate (Fig. 4c) and then vanishes in the fourth replica (Fig. 4d). In addition, it is possible to note some exponential-like current decays at the end of some transient pulses, instead of a sharp decay. Such increases in the stabilization times can be associated with solution leakage between the PDMS layer and Au electrode surfaces. Unlike glass, Au is inert to the O2 plasma and does not have enough bonding sites to react with oxidized PDMS. When positive pressure is applied at the inlet reservoirs, leakage can be clearly observed in the region of the electrodes. Therefore, the incomplete sealing obtained over the Au surfaces degrades under stress. With the seal broken and leakages taking place onto the Au surface, the oxidation currents are no longer reproducible, since control over the dimensions of the active area of the working electrode is lost.
To overcome this drawback, SiO2 thin-films were deposited over glass plates previously patterned with TiAuTi electrodes to give a TiSiO2 bi-layer. Just like glass, SiO2 surfaces present reactive silanol groups that can react with oxidized PDMS surfaces providing good seals between the electrodes and PDMS layer. However, in order to expose the active areas after the sealing step, it is necessary to remove the TiSiO2 bi-layer from the electrode surfaces in the channel region. This was done using HF buffer, which etches the SiO2 and the Ti layers. This procedure must be carefully controlled since HF attacks the glass electrode support and even the sealing layer under the PDMS if the etching time is relatively long (see Fig. 5).
 | ||
| Fig. 5 SiO2 auxiliary sealing layer being removed by 10% (v/v) HF etching solution for: (a) 45 s, (b) 90 s, (c) 115 s, and (d) 145 s. Black arrows illustrate some etched areas under the PDMS layer. | ||
The addition of TiSiO2 to the TiAu electrodes has improved the sealing and enhanced the reproducibility of analytical procedures carried out in the microdevices. These effects are verified in the quadruplicate analytical assay shown in Fig. 6, where the experiments were performed with 1.0 mmol L−1APAP standard solutions.
 | ||
| Fig. 6 Quadruplicate full analytical procedure carried out by sequential injections of 1.0 mmol L−1APAP standard solutions in microchip with design 2 and TiAu electrode covered by an auxiliary TiSiO2 sealing layer: (a) first, (b) second, (c) third and (d) fourth replicate. The use of the sealing layer promotes maintenance of amperometric profiles throughout the whole assay. | ||
In contrast to the results obtained from devices without the TiSiO2 layer, the additional layer showed good amperometric profiles in all runs without significant decrease in sensitivity. We tested devices with TiSiO2 layer for over three months and no noticeable degradation in device performance was observed.
Analytical behavior
The reliability of the analytical responses achieved with the microchip can be verified by plotting the absolute (cumulative) oxidation current between injections (Fig. 7). Each plot concerns quadruplicate analyses carried out on the same device. In order to investigate the role of the TiSiO2etching time, devices were submitted to HF treatment for (a) 90 s and (b) 45 s. Although we obtained comparable results for both devices, it can be noticed that in Fig. 7a the error bars increase systematically with the number of filled channels. This cumulative error during the analytical procedure is attributed to over-etched regions generated at the microchannel edges close to the working electrode when the longer HF treatment was used (Fig. 5). Decreasing the HF etching time reduced the over-etched regions, resulting in significant reliability of the device response (see Fig. 7b).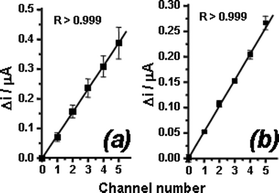 | ||
| Fig. 7 Effect of TiSiO2 removal time on measurement variabilities. The assays were performed in microchips with design 2 treated with 10% (v/v) HF for: (a) 90 s and (b) 45 s. Results obtained for the oxidation of APAP solutions on Au electrodes at +700 mV against Au pseudo-reference electrodes. Injected solution: 1.0 mmol L−1APAP standard solution. | ||
Using the same data set used to generate Fig. 7, but taking only the differences between consecutive non-injecting current levels (Δi)—instead of the absolute current values—it is possible to compare the relative responses given by distinct working electrode active areas, both in the same and in different devices (Fig. 8). Again, the effect of the SiO2etching times on measurement variability becomes evident due to the differences in error bars, which are smaller for the device submitted to 45 s of HF treatment. However, it is also interesting to note that both plots show the same oscillating pattern around the mean value. We attribute this behavior to slight differences at the edges of the active areas of each working electrode, as a result of the resolution limitations of the fabrication mask.
 | ||
| Fig. 8 Current variations recorded per filled microchannel for devices treated with 10% (v/v) HF for (a) 45 s and (b) 90 s. Results obtained for the oxidation of APAP solutions on Au electrodes at +700 mV against Au pseudo-reference electrode. Injected solution: 1.0 mmol L−1APAP standard solution. | ||
Such differences must be taken into account in quantitative analysis, since they are systematic error sources and may lead to significant deviation in the response of the microchips. For instance, an analytical curve using APAP standard solutions at 0.25, 0.5, 0.75, and 1.0 mmol L−1 injected into the first, second, third and fourth microchannels, respectively, and taking the current increase due to each injection as the analytical signal can be seen in the electronic supplementary information.† However, even though this result compares well with similar attempts to perform quantitative assays carried out in μ-TAS, it can be improved when the data are divided by a reference standard data set (electronic supplementary information).† In this case, the reference set was obtained from the responses of each working electrode active area to the injection of a APAP standard solution at 1.0 mmol L−1. This correction eliminates the analytical signal dependence on the actual active area of the working electrode and ensures more reliable analyses, as indicated by the improvement in the correlation factor.
Application to commercial formulations
The proposed microchip was applied in the determination of two commercial formulations of APAP containing 750 mg per tablet (sample 1, mean weight 0.84 ± 0.05 g) and 500 mg per tablet (sample 2, mean weight 0.6146 ± 0.009 g). For this, four tablets were ground to powder and a mass of sample sufficient to prepare 100 mL of a nominal 5.0 mmol L−1APAP solution was weighted and dissolved in supporting electrolyte, giving the stock solutions of the samples. These solutions were ten-fold diluted, filtered with 0.22 μm syringe filters (Millipore, Bedford, MA, USA) and injected using the procedure described for the standard solutions. For each analysis, 0.5 mmol L−1APAP standard solution were injected in three channels, while the other two channels were filled with the sample solution. The results obtained using the calibration curve were 0.70 ± 0.12 g per tablet and 0.43 ± 0.08 g per tablet for sample 1 and 2, respectively. Although these results agree with the nominal content per tablet, the analysis of sample 1 presented a greater deviation, probably due to the variability in the weight of the tablet.Conclusions
The fabrication process of a novel multichannel μ-TAS based on PDMS and glass materials and with fully-integrated electrodes for amperometric detection has been described. Our results suggest that the complete independent channel design is more suitable due to the absence of back-flow problems associated with devices with a common outlet reservoir. In addition, the results indicate that the performance of the device is affected by the sealing quality, and that O2 plasma sealing efficiency can be strongly enhanced by covering electrodes with thin-films of SiO2, deposited by PECVD.The concept of amperometric measurements with a single shared working electrode has been demonstrated to be functional, leading to fast calibration and reliable analytical procedures. The resolution of the masks used in the photolithographic step is responsible for small variations in the active area of the working electrodes and leads to some analytical errors. These errors can be corrected using the relationship between the raw analytical signal and a standard reference sample.
Under the experimental conditions, the sensitivity of the devices ranged from 19 (thiocyanate) to 78 μA mol−1 L and the selectivity could be improved by the modification of the electrodes or by the use of a separation technique coupled to the detector. Moreover, different modifications could be made in each of the active areas in order to promote specific responses per channel.
Acknowledgements
Financial support for this project was provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). The authors thank Dr Ayrton A. Bernussi, Dr Carol H. Collins, and Wendell K. T. Coltro for manuscript revision.References
- V. Dolník, S. Liu and S. Jovanovich, Electrophoresis, 2000, 21, 41 CrossRef CAS.
- P. Auroux, D. Iossifidis, D. R. Reyes and A. Manz, Anal. Chem., 2002, 74, 2637 CrossRef CAS.
- T. Vilkner, D. Janasek and A. Manz, Anal. Chem., 2004, 76, 3373 CrossRef CAS.
- P. S. Dittrich, K. Tachikawa and A. Manz, Anal. Chem., 2006, 78, 3887 CrossRef CAS.
- P. S. Dittrich and A. Manz, Nat. Rev. Drug Discovery, 2006, 5, 210 CrossRef CAS.
- D. P. de Jesus, J. G. A. Brito-Neto, E. M. Richter, L. Angnes, I. G. R. Gutz and C. L. do Lago, Anal. Chem., 2005, 77, 607 CrossRef CAS.
- M. Macka, P. Anderson and P. R. Haddad, Anal. Chem., 1998, 70, 743 CrossRef CAS.
- R. R. Fuller and J. V. Sweedler, Anal. Chem., 1999, 71, 4014 CrossRef CAS.
- T. L. Paxon and A. G. Ewing, Adv. Chromatogr., 2006, 44, 1 CAS.
- C. A. Emrich, H. Tian, I. L. Medintz and R. A. Mathies, Anal. Chem., 2002, 74, 5076 CrossRef CAS.
- S. C. Jacobson, T. E. McKnight and M. Ramsey, Anal. Chem., 1999, 71, 4455 CrossRef CAS.
- H. Shapour, M. L. Hupert, D. Patterson, C. Liu, M. Galloway, W. Stryjewski, J. Goettert and S. A. Soper, Anal. Chem., 2007, 79, 870 CAS.
- S. B. Cheng, C. D. Skinner, J. Taylor, S. Attiya, W. E. Lee, G. Picelli and D. J. Harrison, Anal. Chem., 2001, 73, 1472 CrossRef CAS.
- Z. Wu, H. Jensen, J. Gamby, X. Bai and H. H. Girault, Lab Chip, 2004, 4, 512 RSC.
- H. Xu and A. G. Ewing, Electrophoresis, 2005, 26, 4711 CrossRef CAS.
- R. A. Potyrailo and V. M. Mirsky, Chem. Rev., 2008, 108, 770 CrossRef CAS.
- N. R. Munce, J. Li, P. R. Herman and L. Lilge, Anal. Chem., 2004, 76, 4983 CrossRef CAS.
- Y. Xiang, and D. LaVan, Proceedings of the 2nd IEEE/ASME International Conference on Mechatronic and Embedded Systems and Applications, The Institute of Electrical and Electronics Engineers, Inc., Piscataway, page 1, 2006.
- C. Yi, Q. Zhang, C. W. Li, J. Yang, J. Zhao and M. Yang, Anal. Bioanal. Chem., 2006, 384, 1259 CrossRef CAS.
- N. A. Lacher, K. E. Garrison, R. S. Martin and S. M. Lunte, Electrophoresis, 2001, 22, 2526 CrossRef CAS.
- W. R. Vandaveer IV, S. A. Pasas-Farmer, D. J. Fischer, C. N. Frankenfeld and S. M. Lunte, Electrophoresis, 2004, 25, 3528 CrossRef.
- J. Wang, Electroanalysis, 2005, 17, 1133 CrossRef CAS.
- M. McEnery, A. Tan, J. Alderman, J. Patterson, S. C. O'Mathuna and J. D. Glennon, Analyst, 2000, 125, 25 RSC.
- D. G. Pijanowska, A. J. Sprenkels, W. Olthuis and P. Bergveld, Sens. Actuators, B, 2003, 91, 98 CrossRef.
- S. Gáspár, X. Wang, H. Suzuki and E. Csöregi, Anal. Chim. Acta, 2004, 525, 75 CrossRef CAS.
- Y. Jang, S. Y. Oh and J. Park, Enzyme Microb. Technol., 2006, 39, 1122 CrossRef CAS.
- A. V. Krylov, H. Adamzig, A. D. Walter, B. Löchel, E. Kurth, O. Pulz, J. Szeponik, F. Wegerich and F. Lisdat, Sens. Actuators, B, 2006, 119, 118 CrossRef.
- C. Weng, W. Yeh, K. Ho and G. Lee, Sens. Actuators, B, 2007, 121, 576 CrossRef.
- X. Llopis, N. Ibánez-Garcia, S. Alegret and J. Alonso, Anal. Chem., 2007, 79, 3662 CrossRef CAS.
- S. Kwakye, V. N. Goral and A. J. Baeumner, Biosens. Bioelectron., 2006, 21, 2217 CrossRef CAS.
- V. I. Vullev, J. Wan, V. Heinrich, P. Landsman, P. E. Bower, B. Xia, B. Millare and G. Jones, J. Am. Chem. Soc., 2006, 128, 16062 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS.
- J. A. Vickers, M. M. Caulum and C. S. Henry, Anal. Chem., 2006, 78, 7446 CrossRef CAS.
- B. H. Jo, L. M. Van Leberghe, K. M. Motsegood and D. J. Beebe, J. Microeletromech. Sys., 2000, 9, 76 Search PubMed.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27 CrossRef CAS.
- C. K. Fredrickson and Z. H. Fan, Lab Chip, 2004, 4, 526 RSC.
- A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, New York, 1980 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical curves obtained with APAP standard solutions. See DOI: 10.1039/b807409g |
| This journal is © The Royal Society of Chemistry 2009 |