The influence of oligo(ethylene glycol) side chains on the self-assembly of benzene-1,3,5-tricarboxamides in the solid state and in solution
Patrick J. M.
Stals
,
Jan F.
Haveman
,
Rafael
Martín-Rapún
,
Carel F. C.
Fitié
,
Anja R. A.
Palmans
* and
E. W.
Meijer
*
Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl; Fax: +31 (0)40 2451036; Tel: +31 (0)40 2473101
First published on 6th November 2008
Abstract
Substituted benzene-1,3,5-tricarboxamides (BTAs) 1–4 comprising polar tetraethyleneglycol (tetraEG) and/or apolar (R)-3,7-dimethyloctyl side chains were synthesised and their self-assembly in the solid state and in solution was investigated. While BTA 1 (comprising 3 apolar side chains) shows helical columnar packing via threefold α-helical type intermolecular hydrogen bonding in the solid state and up to high dilutions in alkane solution (10−5 M), helical columnar order is only preserved for asymmetric BTA 2 (comprising 1 polar and 2 apolar side chains) in the solid state and in a concentrated alkane solution (10−2 M). The association constant Kass is reduced by a factor of 107 by introducing one polar tetraEG chain into the BTA. A further increase in the number of polar tetraEG chains attached to BTA core results in the complete loss of intermolecular hydrogen bond formation in the solid state and in solution. Moreover, for the polar BTAs 3–4, comprising 2 or 3 polar tetraEG chains, no self-assembly in water occurs because of the lack of hydrophobic shielding. We propose that tetraEG side chains interfere with the intermolecular hydrogen bonds, weakening the stacking behaviour of these asymmetric derivatives and drastically lowering the association constant due to competing intramolecular hydrogen bonding interactions. In contrast, one methoxyethyl unit does not affect the stability of the aggregation of BTAs (Kass = 3 × 107 M−1) showing that more than one EG unit is required to disrupt the self-assembly of BTAs.
1. Introduction
The application of intermolecular hydrogen bonding as a directional and ordering interaction in (macro)organic molecules and supramolecular polymers has attracted considerable interest both in academic research and in industry.1Hydrogen bonding motifs such as bisureas,2 ureidopyrimidinones,3 cyclohexane-1,3,5-tricarboxamides4 and benzene-1,3,5-tricarboxamides5 have been studied in detail as gelators, hard phases in thermoplastic elastomers, nanostructured materials, and as liquid crystals. Especially the benzene-1,3,5-tricarboxamide (BTA) unit is of particular interest; the amide groups are involved in strong, threefold α-helix type intermolecular hydrogen bonding between neighbour molecules leading to well-defined, helical, 1D columnar aggregates.6 These supramolecular polymers behave as organogels in a variety of solvents7 and are also stable in dilute alkane solutions (c = 10−5 M).8Nowadays, supramolecular assemblies that display structure and properties in water become increasingly important in view of their potential in biomedical applications such as drug delivery systems, hydrogels and tissue engineering scaffolds.9 Non-covalent interactions such as hydrogen bonding, π–π stacking or ion pairing are well known to induce order between molecules in (aqueous) solution.10 Short oligo(ethylene glycol) (oligoEG) chains are frequently introduced to render the supramolecular assembly compatible with water.11 However, when the dominant interaction of the self-assembling system is hydrogen bonding, a hydrophobic pocket around the hydrogen bonding units is required for finding a balance between water compatibility/solubility and the formation of ordered aggregates. The success of this approach was illustrated inter alia for bisureas,2e,11acyclohexane-1,3,5-tricarboxamide based hydrogelators,4b,cureido-triazines12 and bipyridine based BTAs.13
We recently reported on the impact of attaching oligoEG chains on the strength of hydrogen bonding interactions in apolar solvents.14 In ureidopyrimidinones, the dimerisation constant dropped by a factor of 1000 when an oligoEG chain was attached while in BTAs the association constant was reduced by a factor of 107 upon introducing one tetraEG chain. This paper is a continuation of our work on the effect of oligoEG chains on the self-assembly of BTAs. Although BTAs directly functionalised with oligoEG chains are known, their self-assembly in organic solvents or water was not discussed.15 We here discuss the synthesis and systematic study of BTAs 1–4 (Scheme 1) comprising 0, 1, 2 or 3 tetraEG chains. The influence of polar oligoEG chains on the thermotropic liquid crystallinity of BTAs is evaluated, as well as the self-assembly of compounds 1–4 in alkane solvents and in water. As a reference, compound 5 and its chiral analogue 6 comprising two alkyl and one methoxyethyl side chain were prepared. A short methoxyethyl unit is selected for comparison with the tetraEG unit since backfolding of the oxygen and concomitant interference with the intermolecular hydrogen bonds is unlikely. In addition, the association constant Kass is determined for compounds 5–6 and compared to the Kass previously found for compounds 1 and 2.
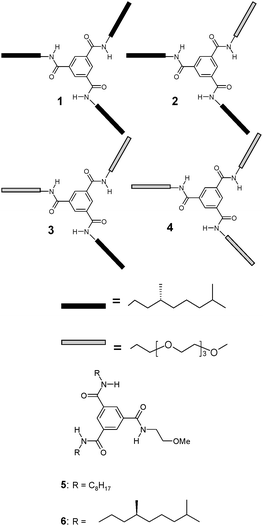 | ||
| Scheme 1 Chemical structures of compounds 1–6. | ||
2. Experimental
2.1 Materials and methods
CD and UV measurements were recorded on a Jasco J-815 spectrometer at room temperature using spectroscopic grade solvents. Cells with an optical path length of 1 cm (10−5 M solutions), 1 mm (10−4 M solutions), 0.1 mm (10−3 M solutions) and 0.01 mm (10−2 M solutions) were employed. The association constant Kass of compound 5 was determined by performing a sergeants-and-soldiers experiment at T = 25 °C as previously described by us.16 To a solution of achiral compound 5, small aliquots of chiral 6 were added via a syringe and the increase in CD effect was probed at λ = 223 nm. The normalised CD effect was plotted as a function of chiral 6 added and the best fit was obtained with Kass = 3 × 107 M−1 and s = 60 (see ref. 16 for details). 1H-NMR measurements were conducted on a Varian Mercury 200 MHz. Maldi-TOF-MS were acquired using a Perserptive Biosystem Voyager-DE PRO spectrometer. In all cases, α-cyano-4-hydroxycinnamic acid was employed as the matrix material. Elemental analysis was performed on a Perkin Elmer 2400 series II CHNS/O analyser. IR spectra were recorded at room temperature on a Perkin Elmer spectrum 1 using a universal ATR. Polarisation optical microscopy measurements were done using a Jenaval polarisation microscope equipped with a Linkam THMS 600 heating device, with crossed polarisers. The thermal transitions of compounds 1–6 were determined with a Perkin-Elmer Pyris 1 DSC apparatus under a nitrogen atmosphere with heating and cooling rates of 10 K/min. The Tg was determined with heating and cooling rates of 40 K/min. Simultaneous WAXS and SAXS measurements for compound 2 were performed using an in-house setup with a rotating anode X-ray generator (Rigaku RU-H300, 18 kW) equipped with two parabolic multilayer mirrors (Bruker, Karlsruhe), which produced a highly parallel beam (divergence about 0.012°) of monochromatic Cu Kα radiation (λ = 0.154 nm). The SAXS intensity was collected with a two-dimensional gas-filled wire detector (Bruker Hi-Star). A semitransparent beamstop placed in front of the area detector allowed monitoring of the intensity of the direct beam. The WAXS intensity was recorded with a linear position sensitive detector (PSD-50M, M. Braun, Germany), which could be rotated around the beam path. The SAXS and WAXS intensities were corrected by subtracting a background that was normalized to the intensity of the direct beam. The powder samples were prepared between two pieces of Kapton foil (5 mm diameter, 20 µm thickness) spaced with an O-ring (Viton, 5 × 1 mm) in a custom made brass sample holder. A Linkam THMS600 temperature-controlled system was employed as sample stage. The glass windows were replaced by thin Kapton foil (20 µm). The XRD patterns for compound 1 and 5 were obtained with a pinhole camera (Anton-Paar) which operates with a point-focused Ni-filtered Cu-Kα beam. In this setup, the samples were held in Lindemann glass capillaries (0.9 mm diameter). The capillary axis was perpendicular to the X-ray beam and the pattern was collected on a flat photographic film perpendicular to the beam. Spacings were obtained via Bragg's law. 2-{2-[2-(2-Methoxyethoxy)-ethoxy]-ethoxy}-ethyl amine was synthesised according to previously described procedures.17(R)-(+)-Citronellal (95%) was purchased from ABCR and converted to (R)-3,7-dimethyloctylamine while18(S)-citronellol was obtained from Takasago and converted to (S)-3,7-dimethyloctylamine.19Ethylene glycol dimethyl ether was distilled prior to use.2.2 Synthesis of compounds 1–4
A 100 mL two-necked round bottom flask was charged with a solution of 2-{2-[2-(2-methoxyethoxy)-ethoxy]-ethoxy}-ethyl amine (2.505 g, 12.09 mmol), triethyl amine (2.523 g, 24.93 mmol) and (R)-3,7-dimethyl-octylamine (1.915 g, 12.17 mmol) in 25 mL of dry chloroform (amylene stabilised), and stirred under inert atmosphere. To this mixture, a solution of 1,3,5-benzenetricarboxylic acid chloride (1.924 g, 7.247 mmol) in 10 mL of dry chloroform (amylene stabilized), was added dropwise. After the addition was complete, the reaction mixture was stirred overnight at room temperature and under inert atmosphere. To the reaction mixture, 20 mL of chloroform was added, and the solution was washed with 25 mL 1 M HCl (check for acidity). The organic layer was collected and evaporated to yield 5.50 g of the crude product as a yellowish/orange oil, which also contained solid product. Compounds 1–4 were isolated by gradient column chromatography. Ethylene glycol dimethyl ether/n-heptane, 3/7, was applied to afford compound 1. After compound 1 eluted from the column (checked with TLC), the eluent was changed to ethylene glycol dimethyl ether/n-heptane (7/3) to obtain compound 2 (checked with TLC). After this, compound 3 was obtained after changing the eluent to THF and the last product fraction was obtained by flushing the column with THF (or alternatively with 5% MeOH in THF). The product fractions (4.2 g in total) were collected and checked with 1H-NMR and Maldi-TOF-MS after evaporationin vacuo.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1563 (amide II). Maldi-TOF Calcd. [M + Na+] = 650.53 Da; Obs. [M + Na+] = 650.51 Da. Anal. Calcd for C39H69N3O6 (MW = 627.97 g/mol): C, 74.59; H, 11.07; N, 6.69. Found: C, 74.05; H, 11.20; N, 6.46%.
O), 1556 (amide II). Maldi-TOF Calcd. [M + Na+] = 700.50 Da; Obs. [M + Na+] = 700.47 Da. Anal. Calcd for C38H67N3O7 (MW = 677.97 g/mol): C, 67.32; H, 9.96; N, 6.20. Found: C, 67.34; H, 10.18; N, 6.16%.
O), 1553 (amide II). Maldi-TOF Calcd. [M + Na+] = 750.46 Da; Obs. [M + Na+] = 750.45 Da.
O), 1533 (amide II). Maldi-TOF Calcd. [M + Na+] = 800.43 Da; Obs. [M + Na+] = 800.41 Da.
O), 1563 (amide II). Maldi-TOF Calcd. [M + Na+] = 650.53 Da; Obs. [M + Na+] = 650.51 Da. Anal. Calcd for C39H69N3O6 (MW = 627.97 g/mol): C, 74.59; H, 11.07; N, 6.69. Found: C, 74.05; H, 11.20; N, 6.46%.
O), 1556 (amide II). Maldi-TOF Calcd. [M + Na+] = 700.50 Da; Obs. [M + Na+] = 700.47 Da. Anal. Calcd for C38H67N3O7 (MW = 677.97 g/mol): C, 67.32; H, 9.96; N, 6.20. Found: C, 67.34; H, 10.18; N, 6.16%.
O), 1553 (amide II). Maldi-TOF Calcd. [M + Na+] = 750.46 Da; Obs. [M + Na+] = 750.45 Da.
O), 1533 (amide II). Maldi-TOF Calcd. [M + Na+] = 800.43 Da; Obs. [M + Na+] = 800.41 Da.
2.3 Synthesis of N-(2-methoxyethyl)-N′,N″-di(n-octyl)-benzene-1,3,5-tricarboxamide (5) and N-(2-methoxyethyl)-N′,N″-di((S)-3,7-dimethyloctyl)-benzene-1,3,5-tricarboxamide (6)
The synthesis and isolation of compounds 5 and 6 was done in analogy to compound 2 using 2-methoxyethylamine and octylamine or (S)-3,7-dimethyloctylamine for 5 and 6, respectively. Compound 5: 1H-NMR (CDCl3): δ = 8.32 (m, 3H, Ar–H), 6.94 (t, 1H, N–H), 6.62 (t, 2H, N–H), 3.43–3.70 (m, 8H, O–CH2, NH–CH2), 3.39 (s, 3H, O–CH3), 1.29–1.61(m, 24H, CH2), 0.88 (m, 6H, CH3). IR ν (cm−1) 3234 (N–H stretch), 1639 (CO), 1557 (amide II). Anal. Calcd for (MW = 489.69 g/mol): C, 68.67; H, 9.67; N, 8.59. Found: C, 68.69; H, 9.91; N, 8.44%. Compound 6: 1H-NMR (CDCl3): δ = 8.29 (m, 3H, Ar–H), 6.96 (t, 1H, N–H), 6.62 (t, 2H, N–H), 3.40–3.66 (m, 8H, O–CH2, NH–CH2), 3.37 (s, 3H, O–CH3), 1.29–1.61(m, 20H, CH, CH2), 0.92 (d, 6H, CH3), 0.88 (d, 12H, CH3). IR ν (cm−1) 3235 (N–H stretch), 1635 (CO), 1556 (amide II). Maldi-TOF Calcd. [M + Na+] = 568.42 Da; Obs. [M + Na+] = 568.31 Da.
3. Results and discussion
3.1 Synthesis of compounds 1–6
Asymmetric benzene-1,3,5-tricarboxamides (BTAs) that comprise both apolar and polar side chains (Scheme 1) are easily prepared via a simple one-pot procedure by reacting benzene-1,3,5-tricarboxylic acid chloride with a 1/1 mix of polar and apolar amines followed by purification with column chromatography. We selected 2-{2-[2-(2-methoxyethoxy)-ethoxy]-ethoxy}-ethyl amine and (R)-3,7-dimethyloctylamine as the reactants; the latter because its chirality provides a convenient handle to study the self-assembly of the different derivatives with circular dichroism (CD) spectroscopy. Both amines are accessible via a straightforward synthesis and ample difference in their polarity ensures easy separation of compounds 1–4 with gradient column chromatography. As expected, a statistical mixture was obtained for compounds 1–4 after column chromatography, which indicates that both amines displayed similar reactivity. Reference compounds 5 and 6 were synthesised and isolated in analogy to compound 2 with the exception that methoxyethylamine and n-octylamine or (S)-3,7-dimethyloctylamine were employed as the amine mixture. The purity of all compounds was confirmed with NMR and Maldi-ToF-MS or elemental analysis.3.2 Properties of compounds 1–6 in the solid state
The physical appearance of compounds 1–6 differs dramatically. At room temperature, compounds 1, 5 and 6 are solids, compound 2 is a sticky solid and compounds 3–4 are oils. We investigated the thermal behaviour of all compounds 1–6 with polarisation optical microscopy (POM) and differential scanning calorimetry (DSC). If liquid crystallinity was present, the nature of the mesophase was confirmed with WAXS and SAXS measurements. In addition, we measured the infrared (IR) spectra of compounds 1–6 to assess the presence of intermolecular hydrogen bonding. All data are summarised in Tables 1 and 2.| Compound | ν (NH) [cm−1] | ν (CO) [cm−1] |
ν (C–N) [cm−1] | Tg [°C] | Tm [°C] ΔH [kJ/mol] | Tcl [°C] ΔH [kJ/mol] |
|---|---|---|---|---|---|---|
| a Data obtained from reference 8a. | ||||||
| 1 | 3225 | 1636 | 1563 | — | 109a | 236a |
| 16.0 | 21.0 | |||||
| 2 | 3238 | 1637 | 1556 | — | — | 134 |
| 10.2 | ||||||
| 3 | 3331 | 1655 | 1533 | −40 | — | — |
| 4 | 3334 | 1656 | 1537 | — | — | — |
| 5 | 3234 | 1639 | 1557 | — | 83 | 145 |
| 1.9 | 9.7 | |||||
| 6 | 3235 | 1635 | 1556 | — | 84 | 181 |
| 2.4 | 9.7 |
Compound 1 was previously reported to be liquid crystalline (LC) in enantiopure (S)-form between 109 °C (ΔH = 16 kJ/mol) and 236 °C (ΔH = 21 kJ/mol) and the mesophase was tentatively assigned as a columnar mesophase.8a WAXS and SAXS measurements on a sample of (R)-1 now unambiguously confirm a hexagonally ordered, columnar mesophase (Colho) phase for the compound (Table 2). At 140 °C two features are detected in the wide-angle region: first, a sharp reflection that corresponds to a distance of 3.5 Å and is characteristic of the stacking of disc-like molecules; second, a broad diffuse halo associated to a distance of about 4.7 Å that arises from short-range interactions between the aliphatic chains as is typically observed in liquid crystalline phases. In the small-angle region, a set of three reflections with spacings in the reciprocal ratio 1:√3:2 is consistent with a hexagonal packing of the columns. From these maxima we can deduce an intercolumnar distance of 19.9 Å.
Under crossed polarisers, compound 2 shows a birefringent texture at room temperature and becomes isotropic at around 140 °C. Slow cooling induces the growth of a pseudo-focal-conic texture with large homeotropic areas, which points to a Colho mesophase (Fig. 1A). The DSC trace of compound 2 shows only one large transition at 134 °C (ΔH = 10.2 kJ/mol) in agreement with POM observations. The assignment of the mesophase is confirmed by performing SAXS and WAXS measurements (Table 2): the pattern is analogous to that of 1, and is consistent with a Colho mesophase with 19.6 Å intercolumnar distance.
 | ||
| Fig. 1 POM images under crossed polarisers of the texture of compound 2 at 138 °C (A) and of the texture of compound 5 growing from the isotropic liquid (B). | ||
Compounds 3 and 4 show no birefringence under crossed polarizers at room temperature, which is indicative of the absence of long range ordering. The DSC trace of compound 3 shows a single glass transition temperature at −40 °C while no transitions are observed for compound 4 within the measured temperature range from −100 °C to room temperature.
Compounds 5 and 6 (Scheme 1) are both obtained as solid compounds at room temperature and show solid state properties that closely resemble those of compounds 1 and 2. An LC phase is observed between 83 and 145 °C for 5 and the textures grown for the mesophase suggest a Colho phase (Fig. 1B). More evidence for the presence of a Colho phase in 5 was obtained from SAXS and WAXS measurements (Table 2). The intercolumnar distance in this case is 16.9 Å. For compound 6, the temperature range in which LC behaviour is observed ranges from 84 to 181 °C (Table 1). Upon cooling from the isotropic melt, textures are obtained that are indicative for a Colho phase.
Evidently, replacing one alkyl chain by one tetraEG chain drastically lowers the stability of the mesophase as evidenced by the lowering of the clearing temperature from 236 °C in compound 1 to 134 °C in compound 2. Also in compounds 5 and 6 the clearing temperatures are reduced by ∼50 °C compared to those of the symmetrical analogues (the corresponding N,N′,N″-trioctylbenzene-1,3,5-tricarboxamide has a clearing temperature of 204 °C).5a The reduction of the clearing temperature upon the introduction of EG chains to discotic compounds has been observed previously and is related to the greater conformational flexibility of EG chains compared to alkyl chains.20
The influence of the polar side chains on the α-helix type intermolecular hydrogen bonding was further investigated by infrared (IR) spectroscopy. Brunsveld et al. reported that the N–H stretch vibration of (S)-1 in the solid state occurred at 3223 cm−1 and the CO stretch at 1640 cm−1.8a These values were attributed to the intermolecular hydrogen bonds between N–H and CO. In tetrachloromethane, where (S)-1 is molecularly dissolved and no intermolecular hydrogen bonding is present, the values for the N–H and CO stretch shifted to 3458 cm−1 and 1665 cm−1, respectively.
Fig. 2 shows the solid state IR spectra of compounds 1–4 measured at room temperature, the data are collected in Table 1. For compound (R)-1, we observe the N–H stretch vibration at 3225 cm−1 and a CO stretch at 1636 cm−1 which is in good agreement with the reported values.8a The positions of the N–H stretch at 3238 cm−1 and CO stretch at 1637 cm−1 in compound 2 indicate that the intermolecular hydrogen bonds are retained, despite the presence of one polar side chain. In contrast, the N–H stretch at 3331 and 3334 cm−1 and CO stretch at 1655 and 1656 cm−1 for compounds 3 and 4, respectively, suggests the loss of the threefold α-helical type intermolecular hydrogen bonds in compounds 3 and 4 in the bulk; this observation explains the lack of mesophase order in these compounds at room temperature. The IR spectra of compounds 5 and 6 show vibrations at positions characteristic for a threefold, intermolecularly hydrogen bonded structure (Table 1).
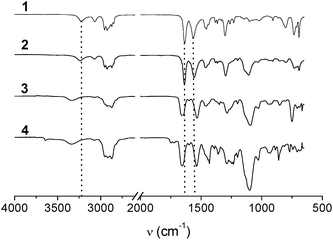 | ||
| Fig. 2 IR spectra of compounds 1–4 taken at room temperature. | ||
3.3 Self-assembly of compounds 1–6 in alkanes and water
The influence of polar side chains on the self-assembly of BTAs in solution was investigated by UV and CD measurements in n-heptane or methylcyclohexane. Compound 1 is known to helically stack in diluted alkane solvents; this is reflected by a λmax at 193 nm in UV and a strong CD effect at 223 nm (Δε = 44 L.mol−1.cm−1).8 In polar solvents such as acetonitrile, the CD effect of 1 disappears and in UV the λmax shifts to 208 nm, indicative of molecularly dissolved species.8b We recently showed that concentrated solutions of compound 2 in decalin also exhibit a CD effect (c = 48 mM, Δε = 37 L.mol−1.cm−1 at λ = 225 nm).14 The Havinga model,16 derived for bipyridine based BTAs, allowed for the estimation of Kass for compounds 1 and 2 in alkane solvents. In this model, chiral and achiral derivatives are mixed in different ratios (the “sergeants-and-soldiers” experiment21) and the chiroptical response is measured by CD spectroscopy as a function of the amount of chiral compound added. If amplification of chirality is present, a non-linear relationship is observed between the chiroptical response and the amount of chiral compound added. Comparing the measured data to values predicted by the Havinga model allows for an estimation of Kass. In this way, we previously estimated a Kass of 5 × 108 M−1 for compound 1 in n-heptane at 20 °C.22 For compound 2 a Kass of 21 M−1 was estimated in decalin at 20 °C.23We prepared solutions of compounds 1–3 in n-heptane at a concentration of 6.5 × 10−5 M and measured the UV and CD spectra at 20 °C (Fig. 3).24Fig. 3A shows that the λmax in UV shifts to 208 nm upon introduction of one tetraEG moiety, which is indicative of molecularly dissolved species. In addition, the CD effect at 225 nm collapses when going from compound 1 to 2 and 3, respectively (Fig. 3B). The loss of the CD effect of 2 at lower concentrations is a direct consequence of the low Kass of 2 in alkane solvents. Since compound 3 shows no thermotropic LC behaviour, increasing its concentration in decalin was not expected to induce aggregation. As a result, we can conclude that compounds 2 and 3 are molecularly dissolved at the concentration of 6.5 × 10−5 M.
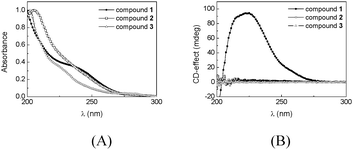 | ||
| Fig. 3 (A) UV and (B) CD spectra of compounds 1–3 in n-heptane; c = 6.5 × 10−5 M. | ||
In contrast, the UV and CD spectra of compound 6 measured in methylcyclohexane at a 3 × 10−5 M (Δε = −48 L.mol−1.cm−1 at λ = 223 nm) closely resemble those of 1. Since the configurations of the alkyl side chains differ, the CD spectra of 1 and 6 are, as expected, mirror images of each other (Fig. 4A). This suggests that one methoxyethyl unit does not dramatically affect the strength of association in BTAs. Performing a “sergeants-and-soldiers” experiment using chiral analogue 6 as the sergeant allowed us to determine a Kass of 3 × 107 M−1 in methylcyclohexane at 25 °C. Fig. 4B clearly shows the non-linearity when the normalized CD effect is plotted as a function of the amount of chiral 6 added to a solution of achiral 5. Although Kass for compounds 5–6 is a factor of 10 lower than the Kass of 5 × 108 M−1 reported for compound 1, it clearly shows that one methoxyethyl unit does not have a significant impact on the self-assembly of BTAs.
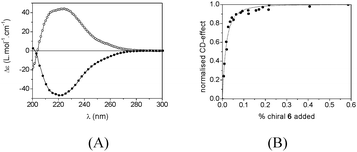 | ||
| Fig. 4 (A) CD spectra of compound (R)-1 (open circles) in heptane (c = 6.5 × 10−5 M) and (S)-6 (closed circles) in methylcyclohexane (c = 3 × 10−5 M). (B) Development of the normalised CD effect as a function of chiral 6 added to a solution of achiral 5 (c = 3 × 10−5 M in methylcyclohexane, T = 25 °C). The dotted line represents the best fit of the data to the model where Kass = 3 × 107 M−1. | ||
Finally, we evaluated the aggregation of compounds 1–6 in water. Compounds 1, 2, 5 and 6 are, unsurprisingly, not compatible with water. Compounds 3 and 4, on the other hand, are. We measured the CD and UV spectra of 3 in water in concentrations from 10−4 to 10−2 M. None of the investigated concentrations show a CD effect and all UV spectra (Fig. 5) suggest that 3 is molecularly dissolved in water, even at high concentrations. Also compound 4, even more hydrophilic, is completely soluble in water at all concentrations investigated.
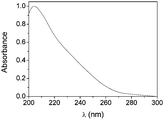 | ||
| Fig. 5 UV spectrum of compound 3 in water; c = 60 mM. | ||
3.4 Influence of oligoEG chains on intermolecular hydrogen bonding in BTAs in the solid state and in solution
The introduction of one polar oligoEG side chain impacts the strength of association in BTAs as evidenced by the significant reduction of Kass from 5 × 108 M−1 in 1 to 21 M−1 in 2. Such a large reduction in Kass is not observed in the case of a methoxyethyl unit (compounds 5–6) where Kass = 3 × 107 M−1. Also in the solid state, oligoEG side chains eventually lead to loss of columnar order.We propose that the reduction of Kass in 2 results from folding back of the ethylene glycol residues. Competitive hydrogen bonding interactions between the EG oxygen and the amide N–H of the tricarboxamide core are possible as tentatively depicted in Fig. 6. In compounds 5 and 6 on the other hand, a 5-membered ring is required for intramolecular hydrogen bonding between the EG oxygen an the amide N–H which is less likely to form. Interestingly, at high concentrations and in bulk the threefold α-helical type hydrogen bonding in 2 is present as evidenced by IR spectroscopy.14
 | ||
| Fig. 6 Proposed folding back of ethylene glycol residues in compounds 2 and 5. | ||
In water, no self-assembly is observed for the hydrophilic BTAs 3 and 4. Evidently, the hydrophobic shielding in oligoEG-substituted BTAs is insufficient for inducing aggregate formation in water.
4. Conclusions
We described a straightforward and accessible synthesis to obtain asymmetrical BTAs. An increase in the number of polar side chains attached to the benzene-1,3,5-tricarboxamide core decreases the strength of intermolecular hydrogen bonding and results in the loss of columnar packing in compounds 3 and 4 in the solid state. While compound 1 shows helical columnar packing up to high dilutions in alkane solution (10−5 M), helical columnar order is only preserved for compound 2 in concentrated alkane solution (10−2 M) and in the solid state. We propose that the tetraEG side chain interferes with the intermolecular hydrogen bonds, weakening the stacking behavior of these asymmetric derivatives and drastically lowering the association constant due to competing interactions. In contrast, one methoxyethyl unit does not affect the stability of the aggregation of BTAs suggesting that more than one EG unit is required. The polar BTAs 3 and 4 do not show self-assembly in water because of the lack of hydrophobic shielding.The possibility of backfolding of oligoEG chains and resulting competitive hydrogen bonds may also be responsible for low association constants in other supramolecular systems after introducing oligoEG residues. Therefore, when preparing water-compatible small molecules programmed to self-assemble into complex structures, care should be taken when designing the molecules: hydrogen bonding groups must be shielded from the oligoEG units by linker units and sufficiently strong hydrophobic, π–π stacking or other non-covalent interactions must be built into the system. Eventually, we aim to elucidate the structure–properties relationships in molecules intended to self-assemble in water in order to achieve our goal of design driven non-covalent synthesis of complex, functional supramolecular assemblies.
Acknowledgements
R.M.-R. acknowledges support from an EU Marie Curie Intra-European Fellowship (Project MEIF-CT-2006-042044) within the EC Sixth Framework Programme. The authors would like to thank Ivo Filot for providing the art work, NWO for funding and Dr Dmytro Byelov of AMOLF in Amsterdam for his help with the SAXS/WAXS measurements. Dr J. Barberá is gratefully acknowledged for providing us access to the powder X-ray diffraction equipment at the University of Zaragoza (ES).References
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS; (b) A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Materials Today, 2004, 34 CrossRef CAS.
- (a) L. Bouteiller, O. Colombani, F. Lortie and P. Terech, J. Am. Chem. Soc., 2005, 127, 8893 CrossRef CAS; (b) O. Colombani, C. Barioz, L. Bouteiller, C. Chaneac, L. Fomperie, F. Lortie and H. Montes, Macromolecules, 2005, 38, 1752 CrossRef CAS; (c) R. M. Versteegen, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2005, 38, 3176 CrossRef CAS; (d) R. A. Koevoets, R. M. Versteegen, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 2999 CrossRef CAS; (e) N. Chebotareva, P. H. H. Bomans, P. M. Frederik, N. A. J. M. Sommerdijk and R. P. Sijbesma, Chem. Commun., 2005, 4967 RSC; (f) E. Wisse, L. E. Govaert, H. E. H. Meijer and E. W. Meijer, Macromolecules, 2006, 39, 7425 CrossRef CAS; (g) K. Yabuuchi, E. Marfo-Owusu and T. Kato, Org. Biol. Chem., 2003, 1, 3464 Search PubMed; (h) L. A. Estroff and A. D. Hamilton, Angew. Chem. Int. Ed., 2000, 39, 3447 CrossRef CAS.
- (a) R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS; (b) H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265 CrossRef CAS.
- (a) K. Hanabusa, A. Kawakami, M. Kimura and H. Shirai, Chem. Lett., 1997, 191 CrossRef CAS; (b) A. Friggeri, C. van der Pol, K. J. C. van Bommel, A. Heeres, M. C. A. Stuart, B. L. Feringa and J. van Esch, Chem. Eur. J., 2005, 11, 5353 CrossRef CAS; (c) A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. van Esch, Angew. Chem. Int. Ed., 2008, 47, 1 CrossRef; (d) M. de Loos, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron, 2007, 63, 7285 CrossRef CAS; (e) K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem. Int. Ed., 2004, 43, 1663 CrossRef CAS.
- (a) Y. Matsunaga, N. Miyajima, Y. Nakayasu, S. Sakai and M. Yonenaga, Bull. Chem. Soc. Jpn., 1988, 61, 207; (b) S. J. Lee, C. R. Park and J. Y. Chang, Langmuir, 2004, 20, 9513 CrossRef CAS; (c) A. Sakamoto, D. Ogata, T. Shikata and K. Hanabusa, Macromolecules, 2005, 38, 8983 CrossRef CAS; (d) C. F. C. Fitié, I. Tomatsu, D. Byelov, W. H. de Jeu and R. P. Sijbesma, Chem. Mater., 2008, 20, 2394 CrossRef CAS; (e) J. Roosma, T. Mes, Ph. Leclere, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 1120 CrossRef CAS; (f) K. P. van den Hout, R. Martín-Rapún, J. A. J. M. Vekemans and E. W. Meijer, Chem. Eur. J., 2007, 13, 8111 CrossRef CAS; (g) I. Paraschiv, M. Giesbers, B. Van Lagen, F. C. Grozema, R. D. Abellon, L. D. A. Siebbeles, A. T. M. Marcelis, H. Zuilhof and E. J. R. Sudhölter, Chem. Mater., 2006, 18, 968 CrossRef CAS; (h) D. K. Kumar, D. A. Jose, P. Dastidar and A. Das, Chem. Mater., 2004, 16, 2332 CrossRef CAS; (i) S. Y. Ryu, S. Kim, J. Seo, Y.-W. Kim, O.-H. Kwon, D.-J. Jang and S. Y. Park, Chem. Commun., 2004, 70 RSC; (j) A. J. Wilson, M. Masuda, R. P. Sijbesma and E. W. Meijer, Angew. Chem. Int. Ed., 2005, 44, 2275 CrossRef CAS.
- (a) M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. W. Warren, Chem. Commun., 1999, 1945 RSC; (b) P. P. Bose, M. G. B. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196 RSC.
- (a) K. Hanabusa, C. Koto, M. Kimura, H. Shirai and A. Kakehi, Chem. Lett., 1997, 5, 429 CrossRef; (b) Y. Yasuda, E. Iishi, H. Inada and Y. Shirota, Chem. Lett., 1996, 7, 575.
- (a) L. Brunsveld, A. P. H. J. Schenning, M. A. C. Broeren, H. M. Janssen, J. A. J. M. Vekemans and E. W. Meijer, Chem. Lett., 2000, 292 CrossRef CAS; (b) M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606 CrossRef CAS.
- (a) G. A. Silva, C. Czeisler, K. L Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I Stupp, Science, 2004, 303, 1352 CrossRef CAS; (b) S. R. Bull, M. O. Guler, R. E. Bras, T. J. Meade and S. I. Stupp, Nanoletters, 2005, 5, 1 CrossRef CAS; (c) K. J. C. van Bommel, M. C. A. Stuart, B. L. Feringa and J. van Esch, Org. Biol. Chem., 2005, 3, 2917 Search PubMed; (d) S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070 CrossRef CAS.
- For a recent review see: T. Rehm and C. Schmuck, Chem. Commun., 2008, 801 Search PubMed and references cited therein.
- (a) E. Obert, M. Bellot, L. Bouteiller, F. Andrioletti, C. Lehen-Ferrenbach and F. Boué, J. Am. Chem. Soc., 2007, 129, 15601 CrossRef CAS; (b) I. Yoshikawa, J. Sawayama and K. Araki, Angew. Chem. Int. Ed., 2008, 47, 1038 CrossRef CAS; (c) C. Schmuck and W. Wienand, J. Am. Chem. Soc., 2003, 125, 452 CrossRef CAS; (d) P. Rzepecki, K. Hochdoerffer, T. Schaller, J. Zienan, K. Harms, Ch. Ochsenfeld, X. Xie and T. Schrader, J. Am. Chem. Soc., 2008, 130, 586 CrossRef CAS.
- (a) J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef CAS; (b) L. Brunsveld, J. A. J. M. Vekemans, J. H. K. K. Hirschberg, R. P. Sijbesma and E. W. Meijer, PNAS, 2002, 99, 4977 Search PubMed.
- (a) L. Brunsveld, H. Zhang, M. Glasbeek, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 6175 CrossRef CAS; (b) L. Brunsveld, B. G. G. Lohmeijer, J. A. J. M. Vekemans and E. W. Meijer, J. Incl. Phenomena Macrocyclic Chem., 2001, 41, 61 Search PubMed.
- T. F. A. de Greef, M. M. L. Nieuwenhuizen, P. J. M. Stals, C. F. C. Fitié, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2008, 4306 RSC.
- M. Akiyama, A. Katoh and T. Ogawa, J. Chem. Soc., Perkin. Trans. II, 1989, 1213 RSC.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem. Int. Ed., 1997, 36, 2648 CrossRef CAS.
- O. A. Sherman, G. B. W. L. Lighthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, PNAS, 2006, 203, 11850 Search PubMed.
- M. Fontana, H. Chanzy, W. R. Caseri, P. Smith, A. P. H. J. Schenning, E. W. Meijer and F. Gröhn, Chem. Mater., 2002, 14, 1730 CrossRef CAS.
- G. Koeckelberghs, L. De Cremer, W. Vanormelingen, W. Dehaen, T. Verbiest, A. Persoons and C. Samyna, Tetrahedron, 2005, 61, 687 CrossRef CAS.
- (a) R. A. Cormier and B. A. Gregg, Chem. Mater., 1998, 10, 1309 CrossRef CAS; (b) L. Brunsveld, J. A. J. M. Vekemans, H. M. Janssen and E. W. Meijer, Mol. Cryst. Liq. Cryst., 1999, 331, 449 CrossRef; (c) N. B. McKeown and J. Painter, J. Mater. Chem., 1994, 4, 1153 RSC.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Wilson, J. Am. Chem. Soc., 1989, 111, 6452 CrossRef.
- L. Brunsveld: Supramolecular chirality: from molecules to helical assemblies in polar media PhD Thesis, Eindhoven University of Technology, The Netherlands, 2001, p. 22. See http://library.tue.nl/catalog/TUEPublicationNew.csp?Language=dut&Type=dissertation&Sort=year&Level = 2^2001 Search PubMed.
- The solubility of 2 in heptane is not sufficient at higher concentrations.
- Compound 4 is omitted in the discussion since it is molecularly dissolved in all solvents investigated.
| This journal is © The Royal Society of Chemistry 2009 |