DOI:
10.1039/B815202K
(Paper)
J. Mater. Chem., 2009,
19, 111-123
The stabilization and bio-functionalization of iron oxide nanoparticles using heterotelechelic polymers†
Received
1st September 2008
, Accepted 30th September 2008
First published on 11th November 2008
Abstract
Iron oxide nanoparticles (IONPs) are important tools for nanobiotechnology applications. However, aqueous instability and non-specific biodistribution problems limit the applications of IONPs. Considering this, α-phosphonic acid, ω-dithiopyridine functionalized polymers were synthesized via the reversible addition–fragmentation chain transfer (RAFT) polymerization and used for stabilizing and biofunctionalizing IONPs. A new trithiocarbonate RAFT agent bearing dimethyl phosphonate group was utilized in the synthesis of well-defined telechelic polymers of styrene, oligoethylene glycol acrylate (OEG-A) and N-isopropylacrylamide (NIPAAm). IONPs were grafted with α-phosphonic acid, ω-dithiopyridine functionalized poly(OEG-A) through the α-chain end of the polymer, as evidenced by FTIR-ATR, XPS and zeta potential measurements. Using TGA results, the grafting density of the polymer chains was calculated between 0.12 and 0.23 chains/nm2 particle depending on the molecular weight of the polymer. DLS measurements indicated that the particles grafted with poly(OEG-A) larger than 10 000 g/mol were stable in water for several days and the mean diameter of the particles was between 40 and 130 nm depending on the molecular weight of the polymer. Moreover, particles stabilized with poly(OEG-A) with a Mn = 62 000 g/mol were stable in phosphate buffer (pH 6.5, 0.1 M) containing varying concentrations of BSA. Polymer-stabilized IONPs were successfully functionalized with two different peptides, i.e. reduced glutathione as a model peptide and NGR motif as a tumor-targeting peptide through the ω-dithiopyridine functionality of the polymer, as measured by XPS and zeta potential analysis. Poly(OEG-A)-stabilized IONPs were also found to be resistant to protein adsorption.
Introduction
Magnetic nanoparticles have become leading tools for nanobiotechnology applications.1–4 In particular, iron oxide nanoparticles (IONPs), because of their superparamagnetic properties and general low toxicity,5 have been increasingly used as contrast agents for magnetic resonance imaging,6–9 as vectors for gene therapy,10,11 in non-invasive local drug delivery,12,13 clinical diagnosis14 and bioseparations.15 In most biomedical applications, iron oxide nanoparticles are required to have uniform size below a critical value which is typically below 50 nm,16 good dispersibility and biocompatibility in physiological medium, and at the same time to be (multi)functionalizable with biological molecules like targeting agents, affinity ligands and drug molecules.17
Besides small molecular surfactants,18–20 biocompatible and hydrophilic polymers that can stabilize the particles in aqueous media and also provide functional groups for conjugation of biological molecules have been attractive materials. Polymers used for this purpose are required to have more than one functional group for both interaction with IONPs and biofunctionalization. The portfolio of polymers used to date has been limited mostly to dextran and other poly(saccharide)s, poly(vinyl alcohol), telechelic PEGs, poly(hydroxyethyl methacrylate) and poly(acrylic acid).21–26 The widespread utilization of these polymers arises not only from their hydrophilicity, biocompatibility25 and possession of functional groups, but also from their readily-availability to nanotechnologists and biomedical researchers through commercial sources. Thus there is a need to enlarge the portfolio of IONP stabilizing and functionalizing polymers especially with vinyl polymers which are relatively easy to generate under laboratory conditions commonly accessible to polymer non-specialists. In addition, commercially available polymers used to date in general do not possess reactive groups (besides the groups binding to the iron oxide surface) necessary for more sophisticated bioconjugations with high selectivity and reactivity. For example, most polymers used to date contain hydroxyl, amino or carboxylic acid groups, for both interaction with iron oxide and conjugation with biological moieties such as cell targeting molecules, bioaffinity ligands, etc. To incorporate functional groups with higher reactivity and selectivity to polymer coated IONPs, additional modification steps are inevitable. Also, the common functional groups (i.e. carboxylic acid, hydroxyl, amine)27–34 that the commercially available polymers possess may not provide strong attachment between the polymer coating and iron oxide, resulting in the leaching of the polymer chains from IONPs under physiological conditions. To ensure that the polymer coating on IONPs does not leach, polymers with functional groups providing stronger chelating with iron oxide need to be generated.
In this context, living radical polymerization (LRP) techniques,35–38 such as reversible addition–fragmentation chain transfer polymerization (RAFT), offer an excellent platform for the fabrication of polymer coatings on IONPs via grafting-from and -to approaches. It is possible to generate a variety of functional and water-soluble polymers with well-defined properties such as designed end-group functionalities, controlled molecular weights and narrow molecular weight distributions with these techniques.34 Among these techniques, RAFT does not require special reagents and conditions for efficient polymerizations except the use of chain transfer agents,39–42 tolerant to functional groups and impurities to some extent,43–46 and provides well-defined hetero-functional telechelic polymers which eliminates the need for post-polymerization end-group modifications.47–54 Nevertheless, the potential of the RAFT technique in stabilization and biofunctionalization of IONPs has yet to be fully explored.
Herein, we report a new method for efficient stabilization and biofunctionalization of IONPs using hetero-telechelic polymers generated by RAFT polymerization. A new trithiocarbonate RAFT agent bearing a dimethyl phosphonate group was utilized in the synthesis of a number of telechelic polymers including water soluble polymers, poly(oligoethyleneglycol acrylate) and poly(N-isopropyl acrylamide). Following the post-polymerization modifications, IONPs were grafted with α-phosphonic acid, ω-dithiopyridine functionalized water-soluble polymers to yield nanoparticles stable in aqueous solutions and reactive toward thiol groups. While the phosphonic acid end group provides strong iron oxide chelation properties for stabilization of the particles,32,55 the pyridyl disulfide end group makes the site-selective conjugation of biomolecules to the nanoparticles possible via selective thiol–pyridyldisulfide exchange reaction.56,57 We also show the results of the surface adsorption of heterotelechelic polymers to IONPs, physicochemical properties of the polymer coated particles and biofunctionalization with two different peptides, i.e. reduced glutathione as a model peptide and NGR motif,58,59 being a potential tumor-targeting peptide.
Experimental
Material
All reagents were purchased from Aldrich at the highest available purity, and used without further purification unless indicated. N-Isopropylacrylamide (NIPAAm) (97%) was recrystallized twice from hexane prior to use (mp = 64 °C). 2,2′-Azobisisobutyronitrile (AIBN) was recrystallized twice from methanol prior to use. Oligoethylene glycol acrylate (OEG-A) (number average molecular weight Mn = 450 g/mol, PDI = 1.04, Aldrich) was filtered through an alumina column to remove the inhibitor prior to use. Styrene was distilled over CaH2 prior to use. The characteristics of the reagents and materials used are given in the ESI.†Iron oxide nanoparticles in the form of maghemite were synthesized rapidly via a one-step flame spray pyrolysis (FSP) as similarly reported in our previous publications.60,61 Briefly, a liquid precursor consisting of 0.5 M of iron 2-ethylhexanote (52% in mineral spirit, Alfa Aeser) in xylene (Riedel de Haen) was delivered to the FSP nozzle at 5 and 10 ml min−1 where it was dispersed with 5 L min−1 and ignited by a supporting oxy-methane flame. Additional 5 L min−1 O2 was issued as sheath gas.
NGR
peptide (sequence: GNGRGC, purity 95%) was purchased from AUSPEP company (PO Box 806, Parkville 3052, Australia). Bovine serum albumin (BSA, purity 90%), Bradford assay and reduced glutathione (95%) were purchased from Aldrich.
RAFT agent synthesis
The synthesis of the RAFT agent is described in ESI.†
RAFT polymerizations
Styrene
polymerization
.
RAFT agent (50.0 mg, 9.7 × 10−2 mmol) (bearing phosphonate and carboxylic groups) and styrene (1.04 g, 1 mmol) were dissolved in acetonitrile (8 mL) in septa sealed vials. The vials were placed in an ice bath and the solutions were purged with nitrogen for 30 minutes. Deoxygenated solutions of the initiator (i.e. AIBN, 1.2 mL from 15.2 mM in acetonitrile (1.8 × 10−2 mmol)) were introduced into the reaction mixture at 0 °C via a syringe. The final concentrations of the monomer, RAFT agent and the initiator in the polymerization medium were: 1.00, 9.7 × 10−3, 1.78 × 10−3 M, respectively ([Monomer]0:[RAFT]0:[Initiator]0 = 103.3:1.0:0.2). The solutions were further deoxygenated at 0 °C for 15 min and then placed in an oil bath at 70 °C. Aliquots (0.5 ml) were taken at predetermined time intervals and quenched via rapid cooling and exposure to oxygen. Two or three drops of hydroquinone solution (5%-wt) were added. These samples were directly analyzed by 1H NMR and THF GPC to determine the monomer conversion and the molecular weight. The polymer was concentrated by partial evaporation of acetonitrile, and the polymer was precipitated in cold methanol (in an ice bath) two times to remove the non-reacted monomer and RAFT agent. Post-filtration, the solid was dried in vacuum at 40 °C for 4 hrs to give a yellow-orange solid. The samples were further analyzed by THF GPC and 1H, 31P NMR.
NIPAAm and OEG-A polymerizations.
Similar procedures were followed for the polymerizations of NIPAAm and OEG-A except for changes in the concentration of the components used. For polymerizations with NIPAAm, the final concentrations of NIPAAm, RAFT agent and the initiator were 1.0, 10.09 × 10−3, 2.02 × 10−3 M, respectively, yielding a ratio of [Monomer]0:[RAFT]0:[Initiator]0 of 100.0:1.0:0.2. For polymerizations with OEG-A, the final concentrations of OEG-A, RAFT agent and the initiator were 0.51, 20.27 × 10−3, 4.06 × 10−3 M, respectively, yielding a ratio of [monomer]0:[RAFT]0:[Initiator]0 of 20.0:1.0:0.2. Both NIPAAm and OEG-A polymerizations were performed at 65 °C. The samples obtained during the polymerizations of NIPAAm and OEG-A were directly analyzed by 1H NMR and DMAc GPC to determine the monomer conversions and the molecular weights. The polymer samples were concentrated by partial evaporation of acetonitrile, and the poly(NIPAAm) was precipitated in cold diethyl ether (in an ice bath) two times to remove the non-reacted NIPAAm and the RAFT agent, while the poly(OEG-A) was dialyzed using a dialysis membrane with a molecular weight cutoff (MWCO) of 3 500 Da in a mixture of ethanol and water (50/50 v/v). After filtration, the solids were dried in vacuum at 40 °C for 24 hrs to give a yellow-orange solid. The purified products were further analyzed by 1H NMR, DMAc GPC and by FT-IR. The molecular weights assessed by DMAc GPC for poly(NIPAAm) and poly(OEG-A) were 9 000 g/mol (PDI = 1.16) and 6 500 g/mol (PDI = 1.22), respectively.
Cleavage of dimethylphosphonate into phosphonic acid by bromosilane.
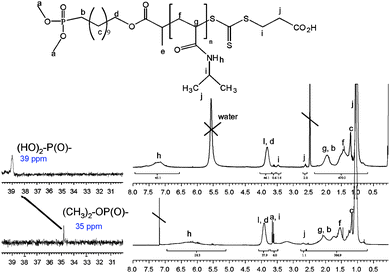
A similar procedure was used for all polymers. For example, poly(styrene) (0.36 g, 5.0 × 10−5 mol) (Mn, THF GPC = 7 200 g/mol) was dissolved in dichloromethane (5 mL) in a vial at room temperature. The solution was degassed for 2 mins to remove traces of moisture and the flask was sealed by a septum. A solution of bromosilane (0.5 g (3.2 × 10−3 mol) in 1 mL) was slowly added at 0 °C. The solution was stirred for 4 hrs at room temperature. At the end of the reaction, the large excesses of bromosilane and dichloromethane were removed by evaporation under low pressure. After the total elimination of bromosilane, ethanol (50 mL) was added to the flask. The solution was stirred at room temperature for 1 hr to generate phosphonic acid and ethanol was removed under vacuum. The product was analyzed by 1H and 31P NMRs, by ESI-MS (for low molecular weights), and by FT-IR. The yield was evaluated by 31P NMR, indicating 100%, consistent with a total shift of signal at 35 ppm to 40 ppm.
Chemical modification of trithiocarbonate into pyridyl disulfide group.
The trithiocarbonate was removed by aminolysis to generate a thiol. The thiol was modified in-situ to give a pyridyl disulfide group in the presence of dithiopyridine. A typical reaction is described. Poly(NIPAAm) (0.9 g, 10−4 mol) (i.e. Mn, DMAc GPC = 9 000 g/mol) and dithiopyridine (0.1 g, 4.5 × 10−4 mol) were dissolved in dichloromethane (10 mL) in a reaction flask. The flask was sealed by a septum and the solution was purged with nitrogen for 30 minutes to remove traces of oxygen. A degassed solution of aminoethoxyethanol (0.025 g, 2.4 × 10−4 mol diluted in 1 mL of dichloromethane) was added slowly. The solution was stirred at room temperature for 4 hrs, undergoing a color change from yellow-orange to yellow-green. Dichloromethane was removed. The product was dissolved in ethanol and dialyzed (MWCO = 1 000 Da) for 3 days in a mixture of water/ethanol (50/50 v/v) to remove the by-product pyridithione and the excess of dithiopyridine and aminoethoxyethanol. The solvent was evaporated to leave a colorless product (yield 80%). The product was analyzed by 1H NMR, UV-visible, ESI-MS (for the low molecular weights) and by DMAc GPC.
Grafting of α-phosphonic acid, ω-dithiopyridine functionalized poly(OEG-A) to the iron oxide nanoparticles.
Iron oxide nanoparticles (10 mg), (diameter 8 nm) were dispersed in water (9 mL). The solution was sonicated for 1 min at 12 watts with a sonicator 3000 Misonix. Poly(OEG-A) (20 mg), (Mn DMAc GPC = 6 200 g/mol, PDI = 1.22) was dissolved in water (1 mL). The solution of poly(OEG-A) was added slowly to the dispersion of iron oxide nanoparticles, and the resulting solution/dispersion was sonicated for 2 mins (power = 30 W). The solutions were filtered and centrifuged (using a Eppendorf Centrifuge 5804) for 10 mins (1 112 relative centrifugal force) to remove the unstabilized particles. Subsequently, the particles were again centrifuged at high speed for 1 h at 4 468 rcf/min to separate the particles from the supernatant. The iron oxide nanoparticles were isolated from the base of the centrifuge tube. The solutions were decanted and the particles were again dispersed in 20 mL of ethanol. The solutions were placed in the centrifuge one more time and the process was repeated 3 times. Finally, the iron oxide nanoparticles were dialyzed (MWCO = 50 000 Da) against water for 2 days and then freeze-dried (8 mg of product). Iron oxide nanoparticles were subsequently analyzed by TGA, XPS, TEM and DLS.
Conjugation of model peptides (glutathione and NGR peptide) to α-phosphonic acid, ω-dithiopyridine functionalized poly(OEG-A).
Reduced glutathione and NGR peptide were conjugated to iron oxide nanoparticles coated with polymer (e.g.α-phosphonic acid, ω-dithiopyridine functionalized poly(OEG-A) (Mn: 5 100 g/mol by NMR and 6 200 g/mol by GPC, PDI: 1.22, conversion 70%). In a typical conjugation experiment, iron oxide nanoparticles (5 mg) were incubated with the peptide (3.44 × 10−6 mol) in a phosphate buffer (5 mL) at pH 7 for 6 hours at room temperature. Purifications of the peptide conjugated nanoparticles were performed by centrifugation, as described previously. The particles were also purified viadialysis, over 3 days, against water, using a membrane with a MWCO of 50 000 Da. Peptide conjugation was quantified by monitoring the release of pyridine-2-thione (present in the solution after centrifugation) via a UV-vis spectrophotometer at 343 nm (ε = 8080 M−1cm−1 in water).62,63 The purified nanoparticle samples were further characterized by XPS, zeta potential measurements and TEM.
Protein adsorption.
Magnetic particles (1.0 g/L) were suspended in a series of phosphate buffers (pH = 6.5, similar results were obtained at 7.4) with initial bovine serum albumin (BSA) concentrations from 0.05 g/L to 16.00 g/L. The samples were then placed on a shaker at room temperature for 3 h to reach adsorption equilibrium. Iron oxide nanoparticles were removed by using a magnet and the supernatant analyzed. The particles were redispersed in a buffer solution at pH = 6.5. The supernatant BSA concentrations were determined following the Bradford method64 using a UV-visible spectrometer (Varian Cary 300 scan). Supernatant (10 µL) was added to Bradford's reactant (3 mL), and mixed for 2 mins at room temperature (the solution becomes blue). The concentration of BSA was measured by adsorption at 595 nm. The test was repeated three times for each sample. The average of the replicate measures was taken to determine the BSA concentration at equilibrium. The amount of BSA adsorbed was calculated by mass balance.
| [BSA]adsor. = [BSA]0 − [BSA]eq. |
where [BSA]0 and [BSA]eq. correspond to BSA concentrations measured by Bradford's assay without iron oxide nanoparticles and BSA concentrations in the presence of iron oxide nanoparticles at equilibrium, respectively.
Results and discussion
Synthesis of α-phosphonate, ω-carboxylic acid RAFT agent
A new RAFT agent bearing a dialkyl phosphonate group, i.e. dimethyl phosphonate or diethylphosphonate, and a carboxylic acid group as the R- and Z-fragments, respectively, was synthesized and characterized (Scheme 1 and see ESI†). The introduction of a phosphonate group to the RAFT agent was achieved using radical addition of dialkyl hydrogenophosphonate groups onto the double bond of 11-undecen-1-ol at 135 °C. This approach has been described previously for radical addition to 11-undecenoic acid, resulting in a selective monoaddition,65,66 as the P–H bond is easily cleavable by radicals.67 However, in the present case of 11-undecen-1-ol, a large quantity of by-products was observed by THF GPC, i.e. diadduct and higher adducts. To minimize the formation of these by-products, an excess of dialkyl hydrogenophosphonate was used. Nonetheless, trace amounts of diadduct and triadduct were still detected by ESI-MS and by THF GPC. To avoid the formation of these by-products, the hydroxyl group was capped by acetyl chloride. Indeed, it is known that the addition of alcohols onto enes can occur in radical medium and by Michael’s addition catalyzed by the presence of phosphine. The elimination of these by-products was achieved using a silica column. 1H and 31P NMR confirmed the successful synthesis of 11-dimethylphophonate decan-1-ol. Subsequently, the hydroxyl function was reacted with bromopropionic bromide introducing a bromine atom. A new NMR signal (CH–Br) appears at 4.1 ppm, while the signal of CH2–O is shifted from 3.5 to 4.0 ppm after esterification. Finally, the terminal bromine atom was exploited to introduce a trithiocarbonate group by nucleophilic substitution by a dithiocarboxysulfanyl propionic acid salt. The NMR signal of CH- is shifted from 4.1 ppm to 4.8 ppm (attributed to CH–S–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–) (Fig. S1 in the ESI†).
S)–) (Fig. S1 in the ESI†).
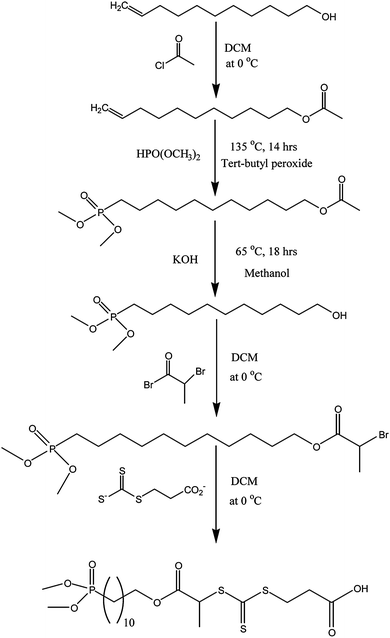 |
| | Scheme 1 Synthesis of the trithiocarbonate RAFT agent bearing a di(methyl)phosphonate and a carboxylic acid group at R- and Z-fragments, respectively. | |
Polymerizations of OEG-A, NIPAAm and styrene
The new RAFT agent was used to mediate the radical polymerizations of styrene, N-isopropylacrylamide (NIPAAm) and oligo(ethylene glycol)-acrylate (OEG-A) in the presence of AIBN (Scheme 2). NIPAAm and OEG-A were used as hydrophilic monomers with potential utility for stabilizing iron oxide nanoparticles. Styrene was used, as a conventional, model monomer, to ensure the living nature of the RAFT polymerization and to study the subsequent chemical transformation of the dialkyl phosphonate group to phosphonic acid. Different polymerizations were performed with a typical [RAFT]0/[Initiator]0 ratio, i.e. 5/1, to confirm the efficacy of the new RAFT agent. The experimental conditions and results obtained for the polymerizations are summarized in Table 1. The targeted and experimental molecular weights were in good agreement for all three monomers, confirming the utility of this RAFT agent.
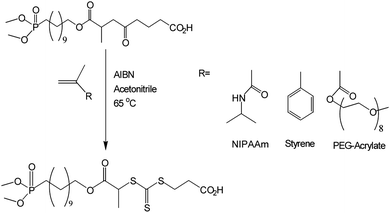 |
| | Scheme 2 RAFT-mediated synthesis of heterotelechelic polymers. | |
| Run |
Monomer |
T (°C) |
[M] (M) |
[M]0/[RAFT]0/[I]0 |
Conv.(%) |
M
n (g/mol) |
PDI |
f
|
| Theoretical |
GPC
|
NMR
|
|
Note: [M], [RAFT], [I] are the feed concentrations of monomer, RAFT agent and initiator, respectively. Conversion and PDI are monomer conversion and polydispersity index, respectively. All polymerizations were performed in acetonitrile. Theoretical molecular weight was calculated by Mn = Conversion (%) × [M]0/[RAFT]0 × MW(monomer) + MWRAFT. GPC molecular weights were determined by GPC (mobile phases: DMAc for OEG-A and NIPAAm; THF for styrene), f is the ratio between dimethyl phosphonate group and carboxylic group.
|
| 1 |
Styrene
|
80 |
1.00 |
100.0/1.0/0.2 |
70 |
7 500 |
7 500 |
8100 |
1.12 |
0.90 |
| 2 |
OEG-A
|
60 |
1.00 |
20.0/1.0/0.2 |
55 |
4 800 |
6 000 |
— |
1.22 |
0.90 |
| 3 |
OEG-A
|
60 |
1.00 |
50.0/1.0/0.2 |
65 |
14 000 |
13 000 |
— |
1.21 |
0.85 |
| 4 |
OEG-A
|
60 |
1.00 |
100.0/1.0/0.2 |
72 |
32 000 |
36 000 |
— |
1.22 |
— |
| 5 |
OEG-A
|
60 |
1.00 |
200.0/1.0/0.2 |
70 |
63 000 |
62 000 |
— |
1.24 |
— |
| 5 |
NIPAAm
|
60 |
1.02 |
20.0/1.0/0.2 |
65 |
1 700 |
3 200 |
3600 |
1.08 |
0.95 |
| 6 |
NIPAAm
|
60 |
1.02 |
50.0/1.0/0.2 |
74 |
4 900 |
4 500 |
4400 |
1.12 |
0.90 |
| 7 |
NIPAAm
|
60 |
1.02 |
100.0/1.0/0.2 |
70 |
8 900 |
7 500 |
8500 |
1.08 |
— |
For all three monomers, the pseudo first-order kinetic plots are linear, indicative of constant radical concentrations. Short inhibition periods were observed. Inhibition periods in RAFT are commonplace and have been ascribed to a number of possible origins including slow fragmentation,68 initiation69,70 and impurities.71 Any of these explanations could be applied here. As expected, styrene polymerization displays the slowest rate, in contrast to NIPAAm and OEG-A.
The linear evolution of molecular weights (determined by GPC) with increasing monomer conversions indicates behaviour consistent with the known features of living radical polymerization. The attainment of low polydispersities up to 70% conversion (Fig. 1 for OEG-A) furnishes evidence that the di(methyl)phosphonate functionality is benign in radical polymerizations.
![Polymerization results of oligo(ethylene glycol)-acrylate (OEG-A) in the presence of RAFT agent bearing a di(methyl)phosphonate group and a carboxylic acid group. A: evolution of Ln ([M]0/[M]) with polymerization time, B: evolution of Mn and PDI with OEG-acrylate conversion, C: evolution of DMAc GPC distribution with OEG-A conversion. Experimental conditions: (red symbol) [M]/[RAFT]:[I] = 100/1/0.2 and (blue symbol) [M]/[RAFT]:[I] = 50/1/0.2.](/image/article/2009/JM/b815202k/b815202k-f1.gif) |
| | Fig. 1
Polymerization results of oligo(ethylene glycol)-acrylate (OEG-A) in the presence of RAFT agent bearing a di(methyl)phosphonate group and a carboxylic acid group. A: evolution of Ln ([M]0/[M]) with polymerization time, B: evolution of Mn and PDI with OEG-acrylate conversion, C: evolution of DMAc GPC distribution with OEG-A conversion. Experimental conditions: (red symbol) [M]/[RAFT]:[I] = 100/1/0.2 and (blue symbol) [M]/[RAFT]:[I] = 50/1/0.2. | |
Experimental conditions: [OEG-A]0/[RAFT]0/[Initiator]0 = 50.0/1.0/0.2 and 100.0/1.0/0.2, initiated by AIBN at 60 °C.
Subsequent 1H NMR analyses of the purified polymers confirmed the presence of the dimethyl phosphonate and carboxylic acid groups on the polymer chains by signals at 3.60 ppm and 2.60 ppm (see Fig. 2 for poly(NIPAAm), NMR). 31P NMR provided additional confirmation of the presence of phosphonate group, with a characteristic signal at 35 ppm. Molecular weights calculated from 1H NMR, assuming one di(methyl)phosphonate group per polymer chain (Mn, 1H NMR = ∫CHat 4.0 ppm/∫(CH3at 3.6 ppm)/6), were also in good agreement with the theoretical molecular weights in the case of poly(NIPAAm) (Table 1). The telechelic stoichiometry (i.e. mole ratio of the ω-carboxylic acid to the α-dimethyl phosphonate end-group) for NIPAAm polymers (up to 70% conversion), calculated from 1H NMR spectra, was above 0.85–0.90 (Table 1), consistent with the generation of well-defined heterotelechelic polymers. As expected, the stoichiometry decreases slowly with monomer conversion (probably the result of conventional radical termination and transfer processes). The theoretical proportion of non-bifunctional polymer can be estimated by f = [RAFT]0/([RAFT]0 + [Init.]0 × (1 − exp(kd.t))), where [RAFT]0 and [Init.]0 are the initial RAFT and initiator concentrations, kd and t are the initiator decomposition constant and polymerization time respectively.
Additional evidence from ESI-MS was accrued to confirm the successful synthesis of telechelic polymers. Short-chain poly(NIPAAm)s were synthesized by RAFT polymerization and the ESI-MS analyses confirmed the expected heterotelechelic polymer structure as the principal population (centered at 906.5 m/z) (data not shown). As expected, polymer chains terminated with AIBN fragments were also present as a sub-population.
Cleavage of the dimethyl phosphonate into phosphonic acid group
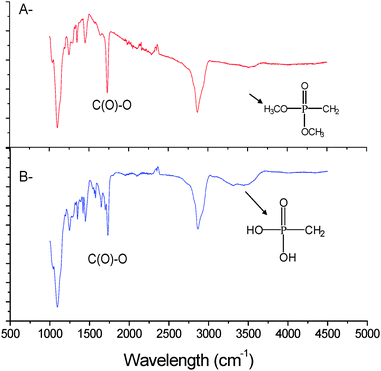
Two established methods are known to facilitate the cleavage of dimethyl phosphonate: the first method uses sodium iodide at 60 °C in acetone for 14 hrs and the second method uses bromosilane at room temperature for a couple of hours. These two methods result in different product structures. The use of sodium iodide generates a monophosphonic acid,65 whereas the use of bromosilane results in a diphosphonic acid.72 As our intention was to use the phosphonic acid to stabilize iron oxide particles we decided to maximise the phosphonic acid functionality by adopting bromosilane to effect the total cleavage of dialkyl phosphonate. 1H NMR and 31P NMR spectroscopy both confirmed a total conversion into phosphonic acid by the disappearance of the CH3 signal at 3.6 ppm and by a shift of the P signal from 35 ppm to 39 ppm. Moreover, the presence of a signal at 2.6 ppm after treatment (which corresponds to CH2–COH) confirmed that the bromosilane treatment did not alter the integrity of the RAFT functionality. However, we caution that a prolonged reaction in the presence of bromosilane (>14 hrs) was observed to cause some degradation to the RAFT agent, characterized by a colour change from yellow-orange to dark orange. Finally, the presence of a new broad absorbance band at 3100 cm−1 observed by ATR confirms the formation of phosphonic acid (P–OH) for both poly(OEG-A) (Fig. 3) and poly(NIPAAm).
Chemical transformation of the trithiocarbonate functionality
The trithiocarbonate functionality is retained in the polymer after the RAFT polymerisation. This functionality is often used to control subsequent block copolymerizations. However, in an atom-efficient synthetic approach, it can also be used to introduce additional functionality. Aminolysis of the trithiocarbonate group can be exploited to generate a thiol, as described by others.73–78 Optimization of this reaction, to avoid oxidation of the thiol to disulfide, requires protection of the thiol by using DTT (DL-Dithiothreitol) and TCEP (Tris(2-carboxyethyl)phosphine hydrochloride). The thiol can be easily protected by reaction with dithiopyridyl disulfide (Scheme 3).
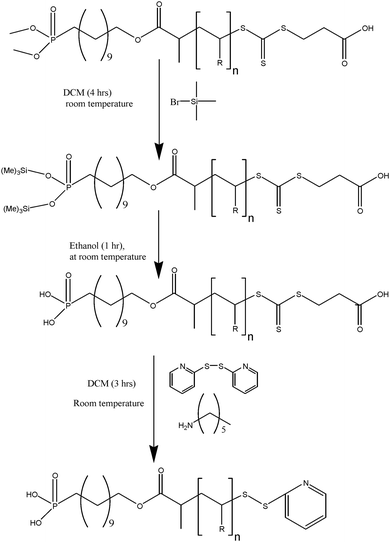 |
| | Scheme 3 Synthesis of α,ω-hetero functional polymer by modification of di(methyl)phosphonate group into phosphonic acid group and trithiocarbonate into ethylpyridyl disulfide end-group. | |
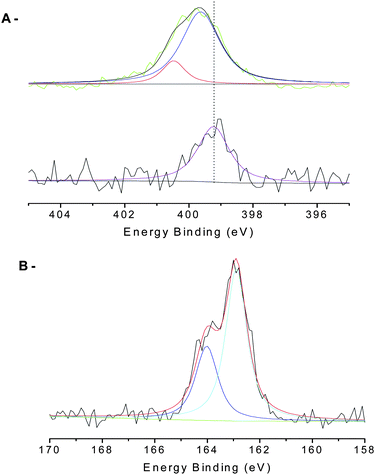
This synthetic approach was adopted for different poly(OEG-A) and poly(NIPAAm) polymers. The presence of pyridyl disulfide was confirmed by 1H NMR spectroscopy, with the appearance of signals at 8.4, 7.6 and 7.2 ppm (dithiopyridyl ring), and the disappearance of the signal at 2.6 ppm. Moreover, ESI-MS spectra confirmed the expected product structure for poly(NIPAAm) (data not shown). XPS also showed the disappearance of the trithiocarbonate group (signal at 162–163.2 eV from CS bond and at 163.6–164.8 eV for C–S bonds) and the appearance of a new signal attributed to disulfide bond at 163.4–164.6 eV54 (Fig. S2 in ESI†).
To confirm the presence and reactivity of the pyridyl disulfide functionality we performed a model reaction with thioglycolic acid using poly(OEG-A) and poly(NIPAAm). UV-visible spectroscopic analysis of pyridine-2-thione release indicated a reaction yield of 80% for both polymers (data not shown).
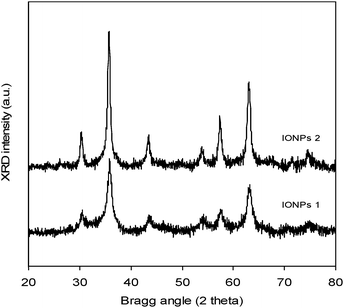
Two different iron oxide nanoparticles in the form of maghemite (γ-Fe2O3) having diameter of 8 and 13 nm (Table 2 summarizes the physical characteristics of the iron oxide particles) were synthesized in a rapid one-step flame spray pyrolysis.60,61 Flame spray pyrolysis (FSP) of 0.5 M iron 2-ethylhexanoate in mineral spirit/xylene mixture resulted in IONPs consisting of γ-Fe2O3 maghemite phase (Fig. 4). This is in agreement with our earlier findings using an acetylacetonate based precursor.60,61 During the one-step FSP, increasing the liquid feed flow rates from 5 to 10 ml min−1 directly increases the rate of combustion enthalpy as well as the metal concentrations in the flame. The former in turn resulted in higher flame temperature as well as its residence time (longer flame). These conditions including the higher metal concentrations are favorable for the coalescence and sintering of the particles whilst in the flame.79,80 Based on this, IONPs of two different sizes were synthesized. This is clearly evident from Fig. 4 where Bragg peak broadening was observed for IONPs synthesized at 5 ml min−1 liquid feed flow rate (IONPs 1) as compared to those at 10 ml min−1 (IONPs 2). Further estimation of the crystallite size by the Scherrer formula81 found dXRD of 9 and 15 nm, respectively. These values are in good agreement with the specific surface area derived sizes, dBET, of 8 and 13 nm, respectively. The slightly smaller (dBET<dXRD) but otherwise comparable sizes were typical of the higher surface area to volume ratio of the single crystallite and non-porous hexagonal IONP platelets.60,61
| IONPs |
d
XRD
(nm) |
BET (N2 adsorption) |
| Isotherm type |
SSA (m2/g) |
dBET (nm) |
|
Note: There is a slight hysteresis arising from pores between the non-porous particles.
|
| IONPs 1 |
9 |
II |
147 |
8 |
| IONPs 2 |
15 |
II |
94 |
13 |
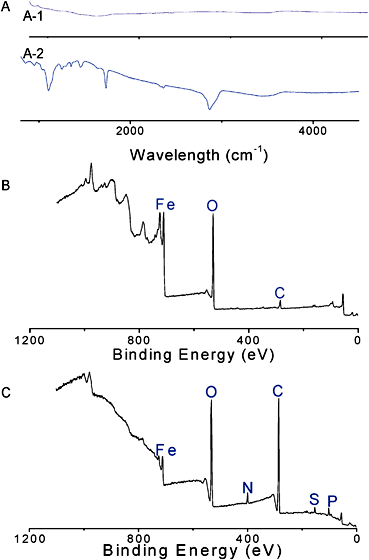
Confirmation of particle surface modification was attained via a number of analytical approaches, namely FTIR-ATR and XPS (Fig. 5) and TGA. FTIR-ATR results confirmed the presence of poly(OEG-A) on the surface by bands at 1730 cm−1 (Fig. 5A). However, ATR cannot prove polymer attachment to the surface. Therefore, XPS analysis (Fig. 5B and C) was performed confirming the presence of the P–O–Fe bond at 113.2 eV (Fig. S3 in the ESI†), while before attachment P–OH appears at 112.4 eV in agreement with a previous study82 before attachment. Moreover, characteristic signals of C and O of poly(OEG-A) were also observed (Table 3 and Fig. S4 in the ESI†). The quantification of these elements confirmed the attachment of poly(OEG-A) on the surface.
Table 3 Binding energies and assignments of the component peaks
|
Envelope
|
Binding Energy (eV) |
Assignment |
| P2p |
132.6 ± 0.1 |
–P(O)–(OH)2 |
| P2p |
133.5 ± 0.1 |
–P(O)–(O–Fe)2 |
| S2p |
162.0–163.2 |
CS |
| S2p |
163.0–164.2 |
C–S in –C–S–S–C– group |
| S2p |
163.5–164.7 |
C–S in –C–S–(CS)–S–C– group |
| C1s |
285 ± 0.1 |
Non-functional carbon, sp3, sp2 |
| C1s |
285.6 ± 0.05 |
Non-functional carbon adjacent to –COOR |
| C1s |
286.7 ± 0.1 |
Carbon linked to oxygenvia a simple bond, i.e. C–O–C |
| C1s |
288.1 ± 0.1 |
Carbon in COOR |
| N1s |
399.2 ± 0.1 |
N
pyridinyl
|
| N1s |
399.9 ± 0.1 |
N in amide group |
| N1s |
400.5 ± 0.1 |
N in imine group |
| O1s |
531.4 ± 0.1 |
Carbonyl of amide group |
| O1s |
532.2 ± 0.1 |
Carbonyl of ester group |
| O1s |
533.5 ± 0.1 |
O in C–O–C group |
Thermogravimetric analysis (TGA) was applied to determine the amount of polymer on the surface of the iron oxide. The results for different molecular weights of polymer grafted onto the surface of the iron oxide nanoparticles are shown in Fig. 6. In the absence of a polymer coating, the weight loss is less than 4% (Fig. 6), attributed to the desorption of water from the surface. The weight loss from polymer-stabilized particles is much more substantial, ranging from 10%–60%, depending on the polymer molecular weights and the nanoparticle sizes. It is possible to correlate the weight loss (see equations in the ESI†) with the number of chain per particle and thus determine the grafting density. It is noteworthy that the number of chains per particle decreases concomitantly with the molecular weight of the polymer chains (Table 4). It may be surmised that steric hindrance limits the accessibility of the end group, i.e. phosphonic acid to the surface of iron oxide. It may be also expected that mushroom type assembly of the polymer chains onto the surface is favoured with the increase in the polymer chain length, a phenomenon widely observed in polymer adsorption to surfaces via a grafting-to approach.83
| Run |
Particle diameter (nm) |
M
n (g/mol) |
PDI |
Wt-(%) |
Number of chainsa |
Densityb |
|
Number of chains per particle.
Density of chains per nm2.
|
| 0 |
8 |
0 |
— |
04.0 |
— |
— |
| 1 |
8 |
6 000 |
1.22 |
18.0 |
186 |
0.23 |
| 2 |
8 |
13 000 |
1.21 |
35.0 |
180 |
0.23 |
| 3 |
8 |
36 000 |
1.22 |
44.6 |
144 |
0.18 |
| 4 |
8 |
62 000 |
1.26 |
55.6 |
115 |
0.14 |
| 5 |
13 |
13 000 |
1.22 |
12.0 |
2200 |
0.21 |
| 6 |
13 |
36 000 |
1.21 |
17.2 |
1600 |
0.12 |
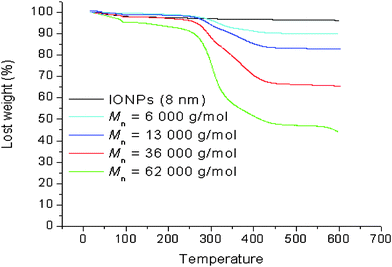 |
| | Fig. 6
TGA analysis of iron oxide nanoparticles (8 nm) coated with poly(OEG-A)s having varying molecular weights (experimental conditions: 5 °C/min under air). | |
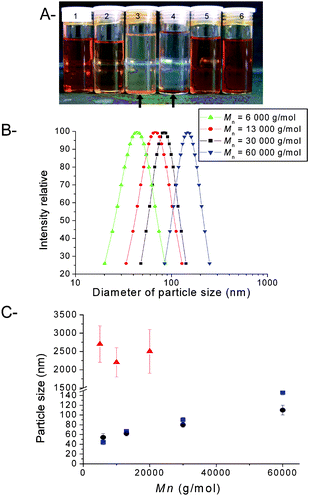 |
| | Fig. 7 Physical properties of iron oxide nanoparticles (IONPs) coated with poly(OEG-A). A: Picture of IONP solutions: 1 and 2: IONPs (13 nm) stabilized with poly(OEG-A) (Mn = 30 000 g/mol, PDI = 1.22) in water at 2 mg/mL and 0.1 mg/mL concentration, respectively; 3: IONPs (13 nm) without a polymer coating, in water; 4: IONPs (8 nm) without a polymer coating, in water; 5 and 6: IONPs (8 nm) stabilized with poly(OEG-A) (Mn = 30 000 g/mol, PDI = 1.22), in water at 0.5 mg/mL and 2 mg/mL concentration, respectively. B: Size distribution of IONPs (8 nm) stabilized with poly(OEG-A)s having varying molecular weights, assessed by DLS. C: Evolution of the size of IONPs (with two different original diameters: ● 13 nm; ■ 8 nm) with the molecular weight of poly(OEG-A); ▲ IONPs (13 nm) after treatment with a non-functional PEG at varying molecular weights. | |
Dynamic light scattering (DLS) was used to determine the size of the stabilized nanoparticles in water. The results indicated that the size of the nanoparticles increased with increasing molecular weight of the grafted polymer (Fig. 7). In the case of a non-functional PEG, the particles were unstable and formed aggregates larger than 1 μm (Fig. 7C). An unexpected result was observed, viz., for polymers of a similar molecular weight, the size of the stabilized particles was independent of the size of the original iron nanoparticles (8 nm and 13 nm).
DLS was conducted on samples as they were allowed to age, to assess the level of stability with time (Fig. 8). After 14 days in water, the average particle size changed when poly(OEG-A)s with an Mn > 10 000 g/mol were used for stabilization. In contrast, when polymers of Mn < 10 000 g/mol were used for stabilization, the particles showed some aggregation after 72 hrs. The stability of the nanoparticles was also examined in phosphate buffer (pH 6.5, 0.1 M) containing BSA (0.1 g/L) (Fig. 8B), to mimic the physiological conditions. Only the particles stabilized with poly(OEG-A) with a Mn = 62 000 g/mol were stable for 48 h under these conditions (without any precipitation and any significant variation of size determined by DLS). When incubating IONPs (stabilized with poly(OEG-A) of 62 000 g/mol) in buffer containing BSA at increasing concentrations (0.05 g/L–1.6 g/L) for 5 h, the size was not affected by BSA concentration in the studied range (Fig. 8C). The effective stability of the particles was maintained for 72 hours (Fig. 8C). However, after 7 days, the formation of a small precipitate was observed. When lower molecular weights of poly(OEG-A) were used, some precipitation occurred after 1 day, consistent with DLS measurements indicating the formation of aggregates.
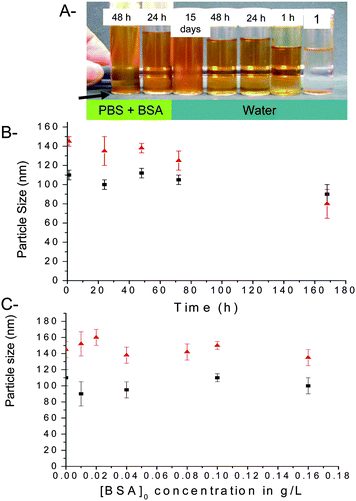 |
| | Fig. 8 Particle size and stability of iron oxide nanoparticles (IONPs) after coating with poly(OEG-A). A: Pictures of IONPs (original particle diameter: 8 nm, Mn of stabilizing poly(OEG-A): 62 000 g/mol) after incubation in phosphate buffer (concentration 0.1 M, pH = 6.5) containing BSA (0.1 g/L) for varying times. Tube 1 contains the IONPs without polymer after 15 mins. Concentration of IOPNs was fixed at 0.5 mg/mL for all the experiments. B: Evolution of the size of IONPs with time: IONPs (diameter 8 nm, Mn of stabilizing poly(OEG-A): 62 000 g/mol) ▲ in phosphate buffer containing BSA and ■ in water. Arrow shows the time when the precipitation occurs. C: Evolution of the particle size in phosphate buffer (at pH 6.5) containing BSA with increasing BSA concentrations (g/L) for ▲ IONPs (diameter 8 nm, Mn of polymer: 62 000 g/mol) and ■ IONPs (diameter 13 nm, Mn of polymer: 62 000 g/mol). | |
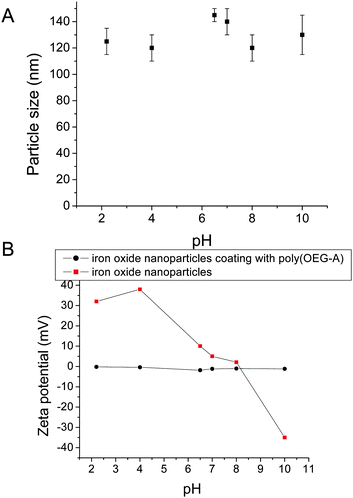
Evolution of particle sizes and zeta potentials were also measured as a function of pH as shown in Fig. 9. For IONPs without any polymer coating, the charges vary from positive (+30 mV) to negative (−30 mV) with increasing pH. In contrast, the charges of IONPs with poly(OEG-A) grafted on their surfaces remain constant, close to neutral zeta potential (Fig. 9B). This result confirms the presence of a polymer shell on the surface of IONPs, as poly(OEG-A) is an uncharged polymer. Moreover, the particles stabilized with a polymer shell retain dimensional stability, independent of pH, confirming the efficacy of the phosphonate attachment (Fig. 9A).
SEM was used to evaluate the size and shape of the polymer-stabilized nanoparticles. In the absence of polymer, the particles (sampled from aqueous solution, 0.1 mg/mL) were observed to form large aggregates (size > 1 µm) (Fig. 10-A1). In contrast, with polymer, the particle sizes were around 140 nm (Fig. 10-A2). TEM also confirmed the improved dispersion with polymer (Fig. 10-B1 and B2). TEM was also applied together with a contrast agent to stain the polymer layer. Thus, the polymer image appeared grey around the iron oxide nanoparticles (see the micrographs in Fig. 10-B3 and B4). The morphologies vary depending on the sizes of the original iron oxide nanoparticles. Several small nanoparticles (8 nm) clustered within each polymer shell (Fig. 10-B3), whereas the larger nanoparticles (13 nm) were largely isolated within each polymer shell (Fig. 10-B4). This observation is in good agreement with the DLS results and explains well why the size of the stabilized nanoparticles was observed to be independent of the size of the original iron oxide nanoparticles.
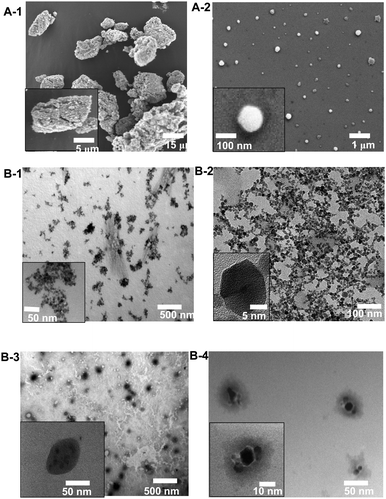 |
| | Fig. 10 Representative SEM (A) and TEM (B) images of iron oxide nanoparticles. A-1: IONPs dispersed in water without polymer coating, A-2: with a polymer coating (poly(OEG-A); Mn = 62 000 g/mol); B-1: IONPs without polymer coating, B-2: with a polymer coating (poly(OEG-A); Mn = 62 000 g/mol), B-3 and B-4: TEM images of polymer coated IONPs treated with a contrast agent (phosphotungstic acid) demonstrating the polymer coating (poly(OEG-A); Mn = 36 000 g/mol) (the polymer appears in grey) (B-3: IONPs with an original diameter of 8 nm, B-4: IONPs with an original diameter of 13 nm). | |
 |
| | Fig. 11 XPS results of IONPs before and after attachment of peptide (NGR). | |
The peptide functional nanoparticles were then analysed by DLS and zeta potential. The size of the nanoparticles remained virtually unchanged as expected. However, the charge on the nanoparticles was measured to be positive (+0.2 mV) at pH = 7, consistent with peptide functionalization. In contrast, in the absence of peptide the poly(OEG-A)-stabilized particles are uncharged or slightly negatively charged (−0.1 mV).
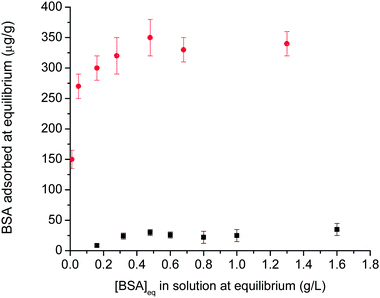
Protein adsorption studies were also performed on the polymer-stabilised IONPs. It is already known that IONPs exhibit surface charge that facilitates surface adsorption of proteins.17,88,89 The adsorption of BSA onto the particles was investigated, with the results shown in Fig. 12. In the absence of polymer stabilization, the BSA adsorption conforms to a Langmuir isotherm (as previously observed for different surfaces, such as silica, titania, zirconia, and alumina89). The saturation adsorption values were found to agree with previous results reported in the literature,60,88 indicative of a BSA monolayer. However, for polymer-stabilized iron oxide nanoparticles, low protein adsorption (<30 µg/g nanoparticles) was observed over a wide range of BSA concentration (0.05 to 10 g/L) which can be explained by the presence of a hydrophilic and neutral polymer layer on the iron oxide particle. The presence of a well-known protein-repellent polymer, OEG, on the surface lowers the protein adsorption to the iron oxide particles, making them compatible with physiological fluids.54,60,90–92
Conclusions
In this report, we described a new synthetic approach to α,ω-telechelic polymers suitable for stabilizing and biofunctionalizing iron oxide nanoparticles. We have shown that the stability of the iron oxide nanoparticles is significantly enhanced, even in conditions similar to the physiological medium. Further, we have shown that we can decorate the stabilised nanoparticles with specific peptides that potentially could be used for targeting purposes. Our future efforts will be focused on investigations of the cellular uptake of the stabilised nanoparticles.
Acknowledgements
The authors thank the Australian Research Council for Research Grants and a Federation Fellowship Award for T.P.D. We also acknowledge the Bioanalytical Mass and NMR Spectrometry Facilities at UNSW.
References
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS.
- Y.-w. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Internat. Ed., 2008, 47, 5122–5135 Search PubMed.
- T. A. Larson, J. Bankson, J. Aaron and K. Sokolov, Nanotech., 2007, 18, 5101–5108.
- E. Duguet, S. Vasseur, S. Mornet and J.-M. Devoisselle, Nanomedicine, 2006, 1, 157–168 CrossRef CAS.
- O. Veiseh, C. Sun, J. Gunn, N. Kohler, P. Gabikian, D. Lee, N. Bhattarai, R. Ellenbogen, R. Sze, A. Hallahan, J. Olson and M. Zhang, Nano Lett., 2005, 5, 1003–1008 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- M. Knauth, T. Egelhof, S. U. Roth, C. R. Wirtz and K. Sartor, Am J Neuroradiol, 2001, 22, 99–102 Search PubMed.
- J. W. Gunn and M. Zhang, Cancer Nanotech., 2007, 61–80 Search PubMed.
- Z. Medarova, W. Pham, C. Farrar, V. Petkova and A. Moore, Nature Medicine, 2006, 13, 372–377 CrossRef.
- Z. Medarova, W. Pham, Y. Kim, G. Dai and A. Moore, Inter. J. Cancer, 2006, 118, 2796–2802 Search PubMed.
- P. Barnett Brad, A. Arepally, V. Karmarkar Parag, D. Qian, D. Gilson Wesley, P. Walczak, V. Howland, L. Lawler, C. Lauzon, M. Stuber, L. Kraitchman Dara and W. M. Bulte Jeff, Nature Medicine, 2007, 13, 986–991 CrossRef CAS.
- N. Kohler, C. Sun, J. Wang and M. Zhang, Langmuir, 2005, 21, 8858–8864 CrossRef CAS.
- C. S. Chen, Nature Nanotech., 2008, 3, 13–14 Search PubMed.
- M. Lewin, N. Carlesso, C.-H. Tung, X.-W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nature Biotech., 2000, 18, 410–414 CrossRef CAS.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clinical Pharmacology & Therapeutics, 2008, 83, 761–769 Search PubMed.
- B. Samanta, H. Yan, N. O. Fischer, J. Shi, D. J. Jerryc and V. M. Rotello, J. Mater. Chem., 2008, 18, 1204–1208 RSC.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS.
- P. Majewski and B. Thierry, Critical Reviews in Solid State and Materials Sciences, 2007, 32, 203–215 Search PubMed.
- J.-H. Lee, Y.-w. Jun, S.-I. Yeon, J.-S. Shin and J. Cheon, Angew. Chem., Internat. Ed., 2006, 45, 8160–8162 Search PubMed.
- E. A. Neuwelt, P. Varallyay, A. G. Bago, L. L. Muldoon, G. Nesbit and R. Nixon, Neuropathology and Applied Neurobiology, 2004, 30, 456–471 Search PubMed.
- N. Kohler, G. E. Fryxell and M. Zhang, J. Am. Chem. Soc., 2004, 126, 7206–7211 CrossRef CAS.
- J. Wan, W. Cai, X. Mengb and E. Liu, Chem. Commun., 2007, 5004–5006 RSC.
- H. Lee, M. K. Yu, S. Park, S. Moon, J. J. Min, Y. Y. Jeong, H.-W. Kang and S. Jon, J. Am. Chem. Soc., 2007, 129, 12739–12745 CrossRef CAS.
- H. Lee, E. Lee, D. K. Kim, N. K. Jang, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2006, 128, 7383–7389 CrossRef CAS.
- A. H. Latham and M. E. Williams, Langmuir, 2006, 22, 4319–4326 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS.
- Z. Li, L. Wei, M. Gao and H. Lei, Adv. Mater., 2005, 17, 1001–1005 CrossRef CAS.
- N. Saleh, T. Phenrat, K. Sirk, B. Dufour, J. Ok, T. Sarbu, K. Matyjaszewski, R. D. Tilton and G. V. Lowry, Nano Lett., 2005, 5, 2489–2494 CrossRef CAS.
- S.-H. Yun, B.-H. Sohn, J. C. Jung, W.-C. Zin, J.-K. Lee and O. Song, Langmuir, 2005, 21, 6548–6552 CrossRef CAS.
- S. Mohapatra, N. Pramanik, S. K. Ghosh and P. Pramanik, J. Nanosci. Nanotechnol., 2006, 6, 823–829 CrossRef CAS.
- R. Narain, M. Gonzales, A. S. Hoffman, P. S. Stayton and K. M. Krishnan, Langmuir, 2007, 23, 6299–6304 CrossRef CAS.
- Q.-L. Fan, K.-G. Neoh, E.-T. Kang, B. Shuter and S.-C. Wang, Biomaterials, 2007, 28, 5426–5436 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. Boyer, P. Lacroix-Desmazes, J.-J. Robin and B. Boutevin, Macromolecules, 2006, 39, 4044–4053 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci.,Part A: Polym. Chem., 2003, 41, 365–375 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Comm., 2007, 28, 539–559 CrossRef CAS.
- G. Moad and C. Barner-Kowollik, Handbook of RAFT Polymerization, 2008, 51–104 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- L. Albertin, T. P. Davis, M. H. Stenzel, L. J. R. Foster and C. Barner-Kowollik, Macromolecules, 2004, 37, 7530–7537 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2005, 38, 9075–9084 CrossRef CAS.
- P.-E. Millard, L. Barner, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and A. H. E. Muller, Macromol. Rapid Comm., 2006, 27, 821–828 CrossRef CAS.
- S. Muthukishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 CrossRef.
-
B. Boutevin, C. Boyer, G. David and P. Lutz, in Macromolecular Engineering , 2007, vol. 2, pp. 775–812 Search PubMed.
- B. Boutevin, G. David and C. Boyer, Adv. Polym. Sci., 2007, 206, 31–135 CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Internat. Ed., 2007, 46, 3099–3103 Search PubMed.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641–5650 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Comm., 2008, 28, 3245–3247 RSC.
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci.,Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- H. M. Zareie, C. Boyer, V. Bulmus, E. Nateghi and T. P. Davis, ACS Nano, 2008, 2, 757–765 CrossRef CAS.
- D. Roberts, W. L. Zhu, C. M. Frommen and Z. Rosenzweig, J. Appl. Phys., 2000, 87, 6208–6210 CrossRef CAS.
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS.
- J. Liu, V. Bulmus, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Macromol. Rapid Comm., 2007, 28, 305–314 CrossRef CAS.
- G. Colombo, F. Curnis, G. M. S. D. Mori, A. Gasparri, C. Longoni, A. Sacchi, R. Longhi and A. Corti, J. Biolog. Chem., 2002, 277, 47891–47897 Search PubMed.
- F. Pastorino, C. Brignole, D. Marimpietri, M. Cilli, C. Gambini, D. Ribatti, R. Longhi, T. M. Allen, A. Corti and M. Ponzoni, Cancer Research, 2003, 63, 7400–7409 CAS.
- D. Li, W. Y. Teoh, C. Selomulya, R. C. Woodward, R. Amal and B. Rosche, Chem. Mater., 2006, 18, 6403–6413 CrossRef CAS.
- D. Li, W. Y. Teoh, C. Selomulya, R. C. Woodward, P. Munroe and R. Amal, J. Mater. Chem., 2007, 17, 4876–4884 RSC.
- J. Liu, V. Bulmus, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Macromol. Rapid Commun., 2007, 28, 305–314 CrossRef CAS.
- V. Bulmus, M. Woodward, M. Lin, N. P. Staton and A. Hoffman, J. Controlled Release, 2003, 93, 105–120 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- O. Senhaji, J. J. Robin, M. Achchoubi and B. Boutevin, Macromol. Chem. Phys., 2004, 205, 1039–1050 CrossRef CAS.
- O. Senhaji, J. J. Robin, M. Achoubi and B. Boutevin, Macromol. Chem. Phys., 2004, 205, 951–957 CrossRef CAS.
- S. Wolfram, K. Keiji and K. Mikiharu, Marcromolecules, 1993, 26, 1656 CrossRef CAS.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300–8306 CrossRef CAS.
- J. B. McLeary, M. P. Tonge and B. Klumperman, Macromol. Rapid Comm., 2006, 27, 1233–1240 CrossRef CAS.
- E. T. A. van den Dungen, H. Matahwa, J. B. McLeary, R. D. Sanderson and B. Klumperman, J. Polym. Sci.,Part A: Polym. Chem., 2008, 46, 2500–2509 CrossRef CAS.
- A. Favier, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Macromol. Chem. and Phys., 2004, 205, 925–936 Search PubMed.
- J. L. Meek, F. Davidso and F. W. Hobbs, J. Am. Chem. Soc., 1988, 110, 2317–2318 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J Am Chem Soc, 2002, 124, 11562–11563 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci.: Part A Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, P. Zhang, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559–5562 CrossRef CAS.
- M. Nuopponen and H. Tenhu, Langmuir, 2007, 23, 5352–5357 CrossRef CAS.
-
L. Barner and S. Perrier, inHandbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, 2008, pp. 455–482 Search PubMed.
- L. Mädler, W. J. Stark and S. E. Pratsinis, J. Mater. Res., 2002, 17, 1356 CrossRef CAS.
- W. Y. Teoh, L. Mädler, D. Beydoun, S. E. Pratsinis and R. Amal, Chem. Eng. Sci., 2005, 60, 5852 CrossRef CAS.
- P. Scherrer, Göttingen Nachrichten, 1918, 2, 98 Search PubMed.
- C. Yee, G. Kataby, A. Ulman, T. Prozorov, H. White, A. King, M. Kafailovich, J. Sokolov and A. Gedanken, Langmuir, 1999, 15, 7111–7115 CrossRef CAS.
-
Y. S. Li, L. S. and B. C. Benicewicz, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, 2008, pp. 423–453 Search PubMed.
-
G. T. Hermanson, Bioconjugates Techniques, Academic Press, New York, 1996 Search PubMed.
-
D. B. Beamson, High Resolution XPS of Organic Polymers-The Scienta ESCA 300 Data Base, Wiley, New York, 1992 Search PubMed.
- D. Cagniant, P. Magri, R. Gruber, S. Berlozecki, P. D. Salbut, J. Bimer and G. Nanse, J. Anal. Appl. Pyrolysis, 2002, 65, 1–23 CrossRef CAS.
- A. L. Vance, T. M. Willey, A. J. Nelson, T. van Buuren, C. Bostedt, L. J. Terminello and G. A. Fox, Langmuir, 2002, 18, 8123–8128 CrossRef CAS.
- K. Rezwan, L. P. Meier, M. Rezwan, J. Voeroes, M. Textor and L. J. Gauckler, Langmuir, 2004, 20, 10055–10061 CrossRef CAS.
- S. Fukuzaki, H. Urano and K. Nagata, J. Ferment. Bioeng., 1996, 81, 163–167 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10714–10721 CrossRef CAS.
- D. Schwendel, R. Dahint, S. Herrwerth, M. Schloerholz, W. Eck and M. Grunze, Langmuir, 2001, 17, 5717–5720 CrossRef CAS.
- R. Nuzzo, Nat. Mater., 2003, 2, 207–208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complement experimental part, RAFT agent synthesis, 1H NMR of RAFT agent and equations. See DOI: 10.1039/b815202k |
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.